Multiple Gene-Environment Interactions on the Angiogenesis Gene-Pathway Impact Rectal Cancer Risk and Survival

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Interview Data
2.3. Tumor Registry Data
2.4. Candidate Gene-Pathway
2.5. Environmental Variables
2.5.1. Cigarette Smoking
2.5.2. Alcohol
2.5.3. Dietary Protein
2.6. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author contributions
Conflicts of Interest
References
- Le Marchand, L.; Wilkens, L.R. Design considerations for genomic association studies: Importance of gene-environment interactions. Cancer Epidemiol. Biomark. Prev. 2008, 17, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Houlston, R.S. Architecture of inherited susceptibility to colorectal cancer: A voyage of discovery. Genes 2014, 5, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Schmit, S.L.; Jiao, S.; Edlund, C.K.; Wang, H.; Zhang, B.; Hsu, L.; Huang, S.C.; Fischer, C.P.; Harju, J.F.; et al. Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat. Commun. 2015, 6, 7138. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Bien, S.; Zubair, N. Genetic architecture of colorectal cancer. Gut 2015, 64, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Matsuda, K.; Jia, W.H.; Chang, J.; Kweon, S.S.; Xiang, Y.B.; Shin, A.; Jee, S.H.; Kim, D.H.; Zhang, B.; et al. Identification of Susceptibility Loci and Genes for Colorectal Cancer Risk. Gastroenterology 2016, 150, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D. Methods for investigating gene-environment interactions in candidate pathway and genome-wide association studies. Annu. Rev. Public Health 2010, 31, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D. Gene—Environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 2010, 11, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Elements of ‘missing heritability’. Curr. Opin. Cardiol. 2012, 27, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Angiogenesis. Successful growth of tumours. Nature 1989, 339, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Wei, C.; Ensor, J.E.; Smolenski, D.J.; Amos, C.I.; Levin, B.; Berry, D.A. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control 2013, 24, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.D. Colorectal cancer: Molecules and populations. J. Natl. Cancer Inst. 1999, 91, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Jenab, M.; Norat, T.; Moskal, A.; Slimani, N.; Olsen, A.; Tjonneland, A.; Overvad, K.; Jensen, M.K.; Boutron-Ruault, M.C.; et al. Lifetime and baseline alcohol intake and risk of colon and rectal cancers in the European prospective investigation into cancer and nutrition (EPIC). Int. J. Cancer 2007, 121, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Potter, J.D.; Friedman, G.D.; Ma, K.N.; Edwards, S. Tobacco use and colon cancer. Int. J. Cancer 1997, 70, 259–264. [Google Scholar] [CrossRef]
- Cheng, J.; Chen, Y.; Wang, X.; Wang, J.; Yan, Z.; Gong, G.; Li, G.; Li, C. Meta-analysis of prospective cohort studies of cigarette smoking and the incidence of colon and rectal cancers. Eur. J. Cancer Prev. 2015, 24, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Haile, R.W.; Siegmund, K.D.; Campbell, P.T.; Figueiredo, J.C.; Limburg, P.; Young, J.; Le Marchand, L.; Potter, J.D.; Cotterchio, M.; et al. Associations between smoking, alcohol consumption, and colorectal cancer, overall and by tumor microsatellite instability status. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2745–2750. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Hutter, C.; Baron, J.A.; Berndt, S.; Caan, B.; Campbell, P.T.; Casey, G.; Chan, A.T.; Cotterchio, M.; Fuchs, C.S.; et al. A pooled analysis of smoking and colorectal cancer: Timing of exposure and interactions with environmental factors. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1974–1985. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.K.; Giovannucci, E.; Wu, K.; Rosner, B.; Fuchs, C.S.; Willett, W.C.; Colditz, G.A. Comparison of risk factors for colon and rectal cancer. Int. J. Cancer 2004, 108, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Smith-Warner, S.A.; Ritz, J.; van den Brandt, P.A.; Colditz, G.A.; Folsom, A.R.; Freudenheim, J.L.; Giovannucci, E.; Goldbohm, R.A.; Graham, S.; et al. Alcohol intake and colorectal cancer: A pooled analysis of 8 cohort studies. Ann. Intern. Med. 2004, 140, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Tramacere, I.; Bagnardi, V.; Rota, M.; Scotti, L.; Islami, F.; Negri, E.; Straif, K.; Romieu, I.; La Vecchia, C.; et al. Alcohol drinking and colorectal cancer risk: An overall and dose-response meta-analysis of published studies. Ann. Oncol. 2011, 22, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Heeschen, C.; Chang, E.; Aicher, A.; Cooke, J.P. Endothelial progenitor cells participate in nicotine-mediated angiogenesis. J. Am. Coll. Cardiol. 2006, 48, 2553–2560. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.W.; Elam, J.; Sartin, A.; Li, W.; Roach, R.; Adair, T.H. Moderate levels of ethanol induce expression of vascular endothelial growth factor and stimulate angiogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R365–R372. [Google Scholar] [PubMed]
- Larsson, S.C.; Wolk, A. Meat consumption and risk of colorectal cancer: A meta-analysis of prospective studies. Int. J. Cancer 2006, 119, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.A.; Riboli, E. Diet and cancer prevention: Contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur. J. Cancer 2010, 46, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.; Lau, R.; Aune, D.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Red and processed meat and colorectal cancer incidence: Meta-analysis of prospective studies. PLoS ONE 2011, 6, e20456. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.D.; Miller, A.J.; Cushing, C.A.; Lowe, K.A. Processed meat and colorectal cancer: A quantitative review of prospective epidemiologic studies. Eur. J. Cancer Prev. 2010, 19, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.D.; Cushing, C.A. Red meat and colorectal cancer: A critical summary of prospective epidemiologic studies. Obes. Rev. 2011, 12, e472–e493. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Narisawa, T.; Weisburger, J.H. Effect of a diet with high levels of protein and fat on colon carcinogenesis in F344 rats treated with 1,2-dimethylhydrazine. J. Natl. Cancer Inst. 1976, 57, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, T.; Sato, S. Mutagens-carcinogens in foods. Cancer Res. 1983, 43, 2415s–2421s. [Google Scholar] [PubMed]
- Sun, Z.; Zhu, Y.; Wang, P.P.; Roebothan, B.; Zhao, J.; Zhao, J.; Dicks, E.; Cotterchio, M.; Buehler, S.; Campbell, P.T.; et al. Reported intake of selected micronutrients and risk of colorectal cancer: Results from a large population-based case-control study in Newfoundland, Labrador and Ontario, Canada. Anticancer Res. 2012, 32, 687–696. [Google Scholar] [PubMed]
- Kim, E.; Coelho, D.; Blachier, F. Review of the association between meat consumption and risk of colorectal cancer. Nutr. Res. 2013, 33, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Silvester, K.R.; Cummings, J.H. Does digestibility of meat protein help explain large bowel cancer risk? Nutr. Cancer 1995, 24, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Thun, M.J.; Connell, C.J.; McCullough, M.L.; Jacobs, E.J.; Flanders, W.D.; Rodriguez, C.; Sinha, R.; Calle, E.E. Meat consumption and risk of colorectal cancer. JAMA 2005, 293, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Kim, Y.S.; Song, J.H.; Chung, S.J.; Lee, I.H.; Hong, K.J.; Lee, E.J.; Kim, D.H.; Yim, J.Y.; Park, M.J.; et al. Dietary risk factors in relation to colorectal adenoma. Korean J. Gastroenterol. 2012, 60, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lindgren, C.M.; Luan, J.; Magi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Grandison, R.C.; Piper, M.D.; Partridge, L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 2009, 462, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Porat, R.; Keshet, E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am. J. Physiol. Cell Physiol. 2001, 280, C1367–C1374. [Google Scholar] [PubMed]
- Barral, M.C.; Jimenez-Aparicio, R.; Priego, J.L.; Royer, E.C.; Urbanos, F.A.; Amador, U. Diruthenium(II,III) Carboxylate Compounds: Existence of both Polymeric and Ionic Forms in Solution and Solid State. Inorg. Chem. 1998, 37, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Sharafeldin, N.; Slattery, M.L.; Liu, Q.; Franco-Villalobos, C.; Caan, B.J.; Potter, J.D.; Yasui, Y. A Candidate-Pathway Approach to Identify Gene-Environment Interactions: Analyses of Colon Cancer Risk and Survival. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Caan, B.J.; Benson, J.; Murtaugh, M. Energy balance and rectal cancer: An evaluation of energy intake, energy expenditure, and body mass index. Nutr. Cancer 2003, 46, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Edwards, S.L.; Caan, B.J.; Kerber, R.A.; Potter, J.D. Response rates among control subjects in case-control studies. Ann. Epidemiol. 1995, 5, 245–249. [Google Scholar] [CrossRef]
- Edwards, S.; Slattery, M.L.; Mori, M.; Berry, T.D.; Caan, B.J.P.; Palmer, P.; Potter, J.D. Objective system for interviewer performance evaluation for use in epidemiologic studies. Am. J. Epidemiol. 1994, 140, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Caan, B.J.; Duncan, D.; Berry, T.D.; Coates, A.; Kerber, R. A computerized diet history questionnaire for epidemiologic studies. J. Am. Diet Assoc. 1994, 94, 761–766. [Google Scholar] [CrossRef]
- Liu, K.; Slattery, M.; Jacobs, D., Jr.; Cutter, G.; McDonald, A.; Van Horn, L.; Hilner, J.E.; Caan, B.; Bragg, C.; Dyer, A.; et al. A study of the reliability and comparative validity of the cardia dietary history. Ethn. Dis. 1994, 4, 15–27. [Google Scholar] [PubMed]
- McDonald, A.; Van Horn, L.; Slattery, M.; Hilner, J.; Bragg, C.; Caan, B.; Jacobs, D., Jr.; Liu, K.; Hubert, H.; Gernhofer, N.; et al. The CARDIA dietary history: Development, implementation, and evaluation. J. Am. Diet Assoc. 1991, 91, 1104–1112. [Google Scholar] [PubMed]
- Young, J.L.J.; Roffers, S.D.; Ries, L.A.G.; Fritz, A.G.; Hurlbut, A.A. SEER Summary Staging Manual—2000: Codes and Coding Instructions; National Cancer Institute: Rockville, ML, USA, 2001. [Google Scholar]
- Dennis, B.; Ernst, N.; Hjortland, M.; Tillotson, J.; Grambsch, V. The NHLBI nutrition data system. J. Am. Diet Assoc. 1980, 77, 641–647. [Google Scholar] [PubMed]
- Ruczinski, I.; Kooperberg, C.; LeBlanc, M.L. Logic regression. J. Comput. Graph. Stat. 2003, 12, 475–511. [Google Scholar] [CrossRef]
- Dai, J.Y.; Kooperberg, C.; Leblanc, M.; Prentice, R.L. Two-stage testing procedures with independent filtering for genome-wide gene-environment interaction. Biometrika 2012, 99, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Siegert, S.; Hampe, J.; Schafmayer, C.; von Schonfels, W.; Egberts, J.H.; Forsti, A.; Chen, B.; Lascorz, J.; Hemminki, K.; Franke, A.; et al. Genome-wide investigation of gene-environment interactions in colorectal cancer. Hum. Genet. 2013, 132, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Hutter, C.M.; Chang-Claude, J.; Slattery, M.L.; Pflugeisen, B.M.; Lin, Y.; Duggan, D.; Nan, H.; Lemire, M.; Rangrej, J.; Figueiredo, J.C.; et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res. 2012, 72, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Lewinger, J.P.; Song, C.; Campbell, P.T.; Conti, D.V.; Edlund, C.K.; Duggan, D.J.; Rangrej, J.; Lemire, M.; Hudson, T.; et al. Genotype-environment interactions in microsatellite stable/microsatellite instability-low colorectal cancer: Results from a genome-wide association study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.D.; Hutter, C.M.; Minnier, J.; Berndt, S.I.; Brenner, H.; Caan, B.J.; Campbell, P.T.; Carlson, C.S.; Casey, G.; Chan, A.T.; et al. Gene-environment interaction involving recently identified colorectal cancer susceptibility loci. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L. Empirical evaluation of gene and environment interactions: Methods and potential. J. Natl. Cancer Inst. 2011, 103, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L.; Huang, Y.; Hinds, D.A.; Peters, U.; Pettinger, M.; Cox, D.R.; Beilharz, E.; Chlebowski, R.T.; Rossouw, J.E.; Caan, B.; et al. Variation in the FGFR2 gene and the effects of postmenopausal hormone therapy on invasive breast cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Boyle, A.P.; Wu, L.; Zhai, J.; Kawli, T.; Snyder, M. Dynamic trans-acting factor colocalization in human cells. Cell 2013, 155, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.S.; Chen, T.Y.; Giovannucci, E. Cigarette smoking and colorectal cancer incidence and mortality: Systematic review and meta-analysis. Int. J. Cancer 2009, 124, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Iodice, S.; Bagnardi, V.; Raimondi, S.; Lowenfels, A.B.; Maisonneuve, P. Smoking and colorectal cancer: A meta-analysis. JAMA 2008, 300, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Peters, U.; Berndt, S.; Bezieau, S.; Brenner, H.; Campbell, P.T.; Chan, A.T.; Chang-Claude, J.; Lemire, M.; Newcomb, P.A.; et al. Powerful Set-Based Gene-Environment Interaction Testing Framework for Complex Diseases. Genet. Epidemiol. 2015, 39, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Hutter, C.M.; Newcomb, P.A.; Ulrich, C.M.; Bien, S.A.; Campbell, P.T.; Baron, J.A.; Berndt, S.I.; Bezieau, S.; Brenner, H.; et al. Genome-Wide Interaction Analyses between Genetic Variants and Alcohol Consumption and Smoking for Risk of Colorectal Cancer. PLoS Genet. 2016, 12, e1006296. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Herrick, J.S.; Bondurant, K.L.; Wolff, R.K. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int. J. Cancer 2012, 130, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Nihon-Yanagi, Y.; Terai, K.; Murano, T.; Matsumoto, T.; Okazumi, S. Tissue expression of Toll-like receptors 2 and 4 in sporadic human colorectal cancer. Cancer Immunol. Immunother. 2012, 61, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Tchorzewski, M.; Lewkowicz, P.; Dziki, A.; Tchorzewski, H. Expression of toll-like receptors on human rectal adenocarcinoma cells. Arch. Immunol. Ther. Exp. 2014, 62, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Nunes, P.; Teixeira, A.L.; Pereira, C.; Gomes, M.; Brandao, C.; Rodrigues, C.; Goncalves, N.; Boal-Carvalho, I.; Roncon-Albuquerque, R., Jr.; Moreira-Dias, L.; et al. Functional polymorphisms of Toll-like receptors 2 and 4 alter the risk for colorectal carcinoma in Europeans. Dig. Liver Dis. 2013, 45, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Anto, R.J.; Mukhopadhyay, A.; Shishodia, S.; Gairola, C.G.; Aggarwal, B.B. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): Correlation with induction of cyclooxygenase-2. Carcinogenesis 2002, 23, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut 2014, 63, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Hsu, L.; Hutter, C.M.; Lin, Y.; Campbell, P.T.; Baron, J.A.; Berndt, S.I.; Jiao, S.; Casey, G.; Fortini, B.; et al. Genome-wide diet-gene interaction analyses for risk of colorectal cancer. PLoS Genet. 2014, 10, e1004228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinckerhoff, C.E.; Rutter, J.L.; Benbow, U. Interstitial collagenases as markers of tumor progression. Clin. Cancer Res. 2000, 6, 4823–4830. [Google Scholar] [PubMed]
- Affara, M.; Dunmore, B.J.; Sanders, D.A.; Johnson, N.; Print, C.G.; Charnock-Jones, D.S. MMP1 bimodal expression and differential response to inflammatory mediators is linked to promoter polymorphisms. BMC Genom. 2011, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.; Waltenberger, J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR). Biochem. Biophys. Res. Commun. 1998, 252, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.C.; Cruickshank, S.M.; Newton, D.J.; Wakenshaw, L.; Graham, A.; Lan, J.; Lodge, J.P.; Felsburg, P.J.; Carding, S.R. Toll-like receptor-mediated responses of primary intestinal epithelial cells during the development of colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G514–G524. [Google Scholar] [CrossRef] [PubMed]


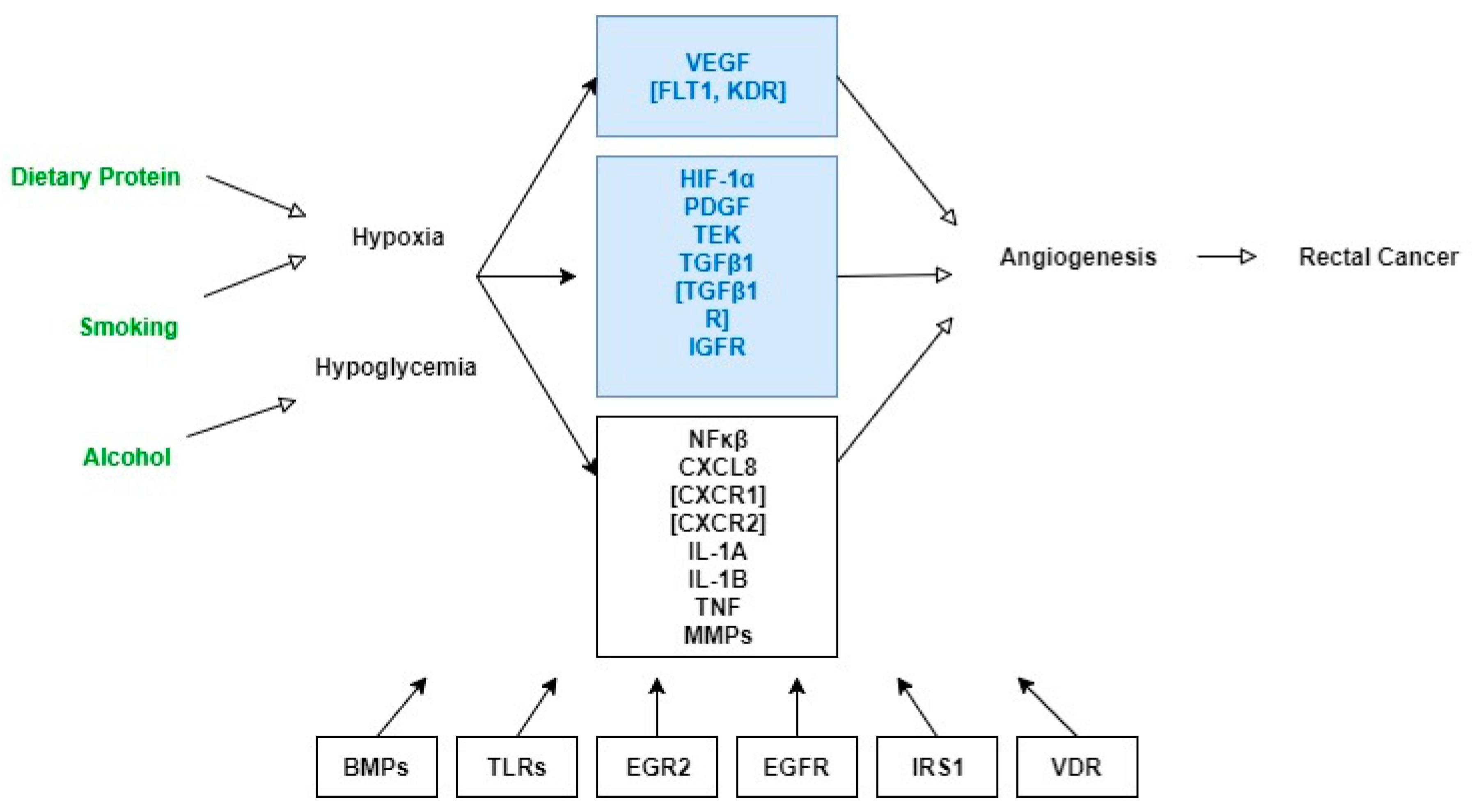{kind=link}
| Analysis Step | Interaction of Interest | Variable of Interest | Model | Specific Procedures | Product |
|---|---|---|---|---|---|
| Step 1: Summarize gene effects | SNP-set interaction within gene | SNPs on each gene separately | Logic regression with logit link/fitting exponential survival models | Cross-validation to determine optimal model size | Gene-specific trees (GSTs) |
| Step 2: Summarize pathway effects | Gene-set interaction within pathway | All GSTs on the pathway | Logic regression with logit link/fitting exponential survival models | Cross-validation to determine optimal model size | Pathway Trees |
| Step 3: Test gene-environment interaction | Gene-environment interaction within pathway | a. Sub-pathway specific GSTxE * b. Full pathway GSTxE *,§ | Logistic regression model §/Cox Proportional Hazards model ¥ | Statistical significance testing | Pathway GEIs |
| Characteristic | Rectal Cancer Cases (n = 747) | Controls (n = 956) | p-Value |
|---|---|---|---|
| Age (Mean, SD) | 61.2 (10.8) | 62.1 (10.6) | 0.09 |
| Sex | |||
| Male | 447 (59.8%) | 542 (56.7%) | |
| Female | 300 (40.2%) | 414 (43.3%) | 0.19 |
| Race | |||
| White non-Hispanic | 617 (82.6%) | 821 (85.9%) | |
| Other | 130 (17.4%) | 135 (14.1%) | 0.06 |
| Education | |||
| ≤High School | 266 (35.6%) | 324 (33.9%) | |
| >High School | 481 (64.4%) | 632 (66.1%) | 0.46 |
| Marital Status | |||
| Married | 556 (74.4%) | 730 (76.4%) | |
| Other | 191 (25.6%) | 226 (23.6%) | 0.36 |
| Annual Income | |||
| ≤30 K | 206 (29.9%) | 238 (27.4%) | |
| >30 K | 483 (70.1%) | 632 (72.6%) | 0.27 |
| Cigarette Smoking | |||
| Non-smoker | 346 (46.3%) | 485 (50.7%) | |
| ≤20 pack-years | 158 (21.2%) | 240 (25.1%) | |
| >20 pack-years | 243 (32.5%) | 231 (24.2%) | 0.001 |
| Alcohol | |||
| Non/Moderate | 556 (74.4%) | 759 (79.4%) | |
| Heavy | 191 (25.6%) | 197 (20.6%) | 0.02 |
| Animal/Vegetable Protein Ratio | |||
| Low | 242 (32.4%) | 363 (38.0%) | |
| High | 505 (67.6%) | 593 (62.0%) | 0.02 |
| Center | |||
| Utah | 270 (36.1%) | 366 (38.3%) | |
| Northern California | 477 (63.9%) | 590 (61.7%) | 0.37 |
| Cancer Stage | |||
| In-situ | 20 (2.7%) | ||
| Local | 395 (52.9%) | ||
| Regional | 255 (34.1%) | ||
| Distant | 63 (8.4%) | ||
| Unknown | 14 (1.9%) |
| GST | Gene | Chr. | Cases (%) | Control (%) | Gene OR a (95% CI) | Env. Factor | Category | N (%) b | Gene OR by Env. Factor (95% CI) | ORINT c (95% CI) | PINT d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs4821877 (CC or CT) | PDGFB | 22q13.1 | 610 (80.7%) | 746 (77.6%) | 1.21 (0.95, 1.54) | Protein e | Low | 612 (35.6%) | 0.85 (0.57, 1.26) | Ref | |
| High | 1106 (64.4%) | 1.47 (1.08, 2.00) | 1.75 (1.04, 2.92) | 0.034 | |||||||
| rs2139924 (AA) | IGF1R | 15q26.3 | 243 (30.4%) | 287 (28.5%) | 0.93 (0.77, 1.00) | Alcohol | Non/Moderate | 1396 (77.4%) | 0.82 (0.66, 1.02) | Ref | |
| Heavy | 408 (22.6%) | 1.36 (0.91, 2.05) | 1.69 (1.04, 2.72) | 0.033 | |||||||
| rs1800630 (CA or AA) | TNF | 6p21.33 | 240 (31.8%) | 267 (27.8%) | 1.19 (0.96, 1.47) | Smoking | Non | 834 (48.7%) | 0.95 (0.70, 1.30) | Ref | |
| <20 PY | 400 (23.4%) | 1.13 (0.71, 1.81) | 1.14 (0.65, 2.01) | 0.644 | |||||||
| ≥20 PY | 477 (27.9%) | 1.68 (1.11, 2.54) | 1.85 (1.10, 3.11) | 0.021 | |||||||
| rs470215 (TT or TC) | MMP1 | 11q22.2 | 715 (90.3%) | 880 (87.9%) | 1.23 (0.89, 1.69) | Protein e | Low | 640 (35.7%) | 0.67 (0.40, 1.14) | Ref | |
| High | 1153 (64.3%) | 1.78 (1.18, 2.70) | 2.44 (1.24, 4.81) | 0.010 | |||||||
| rs1927911 (CC) | TLR4 | 9q33.1 | 396 (52.4%) | 495 (51.5%) | 0.93 (0.76, 1.15) | Smoking | Non | 834 (48.7%) | 0.80 (0.59, 1.08) | Ref | |
| <20 PY | 400 (23.4%) | 0.65 (0.41, 1.04) | 0.99 (0.57, 1.74) | 0.980 | |||||||
| rs11536889 (GG) | 546 (72.2%) | 684 (71.1%) | ≥20 PY | 477 (27.9%) | 1.33 (0.90, 1.98) | 2.34 (1.38, 3.98) | 0.002 | ||||
| rs1927911 (CT or TT) | TLR4 | 9q33.1 | 360 (47.6%) | 467 (48.5%) | 1.07 (0.87, 1.32) | Alcohol | Non/Moderate | 1326 (77.2%) | 0.95 (0.75, 1.21) | Ref | |
| rs11536889 (GC or CC) | 210 (27.8%) | 278 (28.9%) | Heavy | 391 (22.8%) | 1.58 (1.01, 2.47) | 2.10 (1.22, 3.60) | 0.007 | ||||
| rs2295814 (GA or AA) | EGR2 | 10q21.3 | 106 (14.0%) | 115 (12.0%) | 1.11 (0.83, 1.49) | Smoking | Non | 834 (48.7%) | 0.90 (0.58, 1.37) | Ref | |
| <20 PY | 400 (23.4%) | 1.21 (0.65, 2.28) | 1.84 (0.83, 4.09) | 0.130 | |||||||
| ≥20 PY | 477 (27.9%) | 1.53 (0.89, 2.65) | 2.23 (1.04, 4.78) | 0.040 | |||||||
| rs2295814 (GG) | EGR2 | 10q21.3 | 650 (86.0%) | 847 (88.0%) | 0.90 (0.67, 1.20) | Alcohol | Non/Moderate | 1326 (77.2%) | 0.81 (0.57, 1.14) | Ref | |
| Heavy | 391 (22.8%) | 1.21 (0.69, 2.11) | 2.12 (1.01, 4.46) | 0.048 |
| GST | Gene | Chr. | Cases (%) | Gene HR a (95% CI) | Env. Factor | Category | N (%) b | Gene OR by Env. Factor a (95% CI) | HRINT c (95% CI) | PINT d |
|---|---|---|---|---|---|---|---|---|---|---|
| rs6838752 (TT or TC) | KDR | 4q12 | 705 (93.6%) | 0.89 (0.55, 1.45) | Protein e | Low | 258 (32.4%) | 0.44 (0.21, 0.91) | Ref | |
| High | 538 (67.6%) | 1.43 (0.73, 2.83) | 4.12 (1.52, 11.13) | 0.005 | ||||||
| rs1008562 (GG) | CXCR1 | 2q35 | 211 (27.9%) | 1.17 (0.89, 1.53) | Smoking | Non | 348 (46.2%) | 1.04 (0.68, 1.60) | Ref | |
| <20 PY | 160 (21.2%) | 0.88 (0.44, 1.75) | 0.96 (0.46, 1.98) | 0.905 | ||||||
| ≥20 PY | 245 (32.5%) | 1.88 (1.20, 2.95) | 2.05 (1.12, 3.76) | 0.019 | ||||||
| rs7656411 (GG) | TLR2 | 4q31.3 | 61 (8.1%) | 0.83 (0.48, 1.44) | Protein e | Low | 244 (32.3%) | 0.13 (0.02, 0.98) | Ref | |
| High | 512 (67.7%) | 1.33 (0.74, 2.38) | 8.69 (1.09, 69.12) | 0.041 | ||||||
| rs224082 (GA or AA) | EGR2 | 10q21.3 | 455 (60.2%) | 0.72 (0.56, 0.92) | Protein e | Low | 244 (32.3%) | 0.39 (0.25, 0.62) | Ref | |
| High | 512 (67.7%) | 0.93 (0.68,1.23) | 2.41 (1.40, 4.15) | 0.002 | ||||||
| rs17151957 (AA) | EGFR | 7p11.2 | 41 (6.5%) | 1. 82 (1.16, 2.88) | Protein e | Low | 244 (32.3%) | 0.54 (0.19, 1.53) | Ref | |
| High | 512 (67.7%) | 3.37 (1.95, 5.82) | 5.84 (1.80, 18.94) | 0.003 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharafeldin, N.; Slattery, M.L.; Liu, Q.; Franco-Villalobos, C.; Caan, B.J.; Potter, J.D.; Yasui, Y. Multiple Gene-Environment Interactions on the Angiogenesis Gene-Pathway Impact Rectal Cancer Risk and Survival. Int. J. Environ. Res. Public Health 2017, 14, 1146. https://doi.org/10.3390/ijerph14101146
Sharafeldin N, Slattery ML, Liu Q, Franco-Villalobos C, Caan BJ, Potter JD, Yasui Y. Multiple Gene-Environment Interactions on the Angiogenesis Gene-Pathway Impact Rectal Cancer Risk and Survival. International Journal of Environmental Research and Public Health. 2017; 14(10):1146. https://doi.org/10.3390/ijerph14101146
Chicago/Turabian StyleSharafeldin, Noha, Martha L. Slattery, Qi Liu, Conrado Franco-Villalobos, Bette J. Caan, John D. Potter, and Yutaka Yasui. 2017. "Multiple Gene-Environment Interactions on the Angiogenesis Gene-Pathway Impact Rectal Cancer Risk and Survival" International Journal of Environmental Research and Public Health 14, no. 10: 1146. https://doi.org/10.3390/ijerph14101146