

Novel Pathogenic Mutations Identified from Whole-Genome Sequencing in Unsolved Cases of Patients Affected with Inherited Retinal Diseases
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Whole-Genome Sequencing of IRD Patients
2.3. In Vitro Validation of Novel Intronic Variants
3. Results
3.1. Validation of Novel Intronic Cryptic Splicing Variants
3.2. Patients Carrying Novel Coding Pathogenic Mutations
3.3. Patient with Reported Pathogenic Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, X.F.; Huang, F.; Wu, K.C.; Wu, J.; Chen, J.; Pang, C.P.; Lu, F.; Qu, J.; Jin, Z.B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PloS ONE 2013, 8, e78496. [Google Scholar] [CrossRef]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef]
- Hu, M.L.; Edwards, T.L.; O’Hare, F.; Hickey, D.G.; Wang, J.H.; Liu, Z.; Ayton, L.N. Gene therapy for inherited retinal diseases: Progress and possibilities. Clin. Exp. Optom. 2021, 104, 444–454. [Google Scholar] [CrossRef]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2015, 17, 253–261. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef]
- Tehreem, R.; Chen, I.; Shah, M.R.; Li, Y.; Khan, M.A.; Afshan, K.; Chen, R.; Firasat, S. Exome Sequencing Identified Molecular Determinants of Retinal Dystrophies in Nine Consanguineous Pakistani Families. Genes 2022, 13, 1630. [Google Scholar] [CrossRef]
- Stavropoulos, D.J.; Merico, D.; Jobling, R.; Bowdin, S.; Monfared, N.; Thiruvahindrapuram, B.; Nalpathamkalam, T.; Pellecchia, G.; Yuen, R.K.C.; Szego, M.J.; et al. Whole Genome Sequencing Expands Diagnostic Utility and Improves Clinical Management in Pediatric Medicine. NPJ Genom. Med. 2016, 1, 15012. [Google Scholar] [CrossRef]
- Zou, G.; Zhang, T.; Cheng, X.; Igelman, A.D.; Wang, J.; Qian, X.; Fu, S.; Wang, K.; Koenekoop, R.K.; Fishman, G.A.; et al. Noncoding mutation in RPGRIP1 contributes to inherited retinal degenerations. Mol. Vis. 2021, 27, 95–106. [Google Scholar]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Wang, J.; Wang, M.; Igelman, A.D.; Jones, K.D.; Li, Y.; Wang, K.; Goetz, K.E.; Birch, D.G.; Yang, P.; et al. Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients with Inherited Retinal Diseases. Front. Genet. 2021, 12, 647400. [Google Scholar] [CrossRef] [PubMed]
- Branham, K.; Matsui, H.; Biswas, P.; Guru, A.A.; Hicks, M.; Suk, J.J.; Li, H.; Jakubosky, D.; Long, T.; Telenti, A.; et al. Establishing the involvement of the novel gene AGBL5 in retinitis pigmentosa by whole genome sequencing. Physiol. Genom. 2016, 48, 922–927. [Google Scholar] [CrossRef]
- Soens, Z.T.; Li, Y.; Zhao, L.; Eblimit, A.; Dharmat, R.; Li, Y.; Chen, Y.; Naqeeb, M.; Fajardo, N.; Lopez, I.; et al. Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet. Med. 2016, 18, 1044–1051. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soens, Z.T.; Branch, J.; Wu, S.; Yuan, Z.; Li, Y.; Li, H.; Wang, K.; Xu, M.; Rajan, L.; Motta, F.L.; et al. Leveraging splice-affecting variant predictors and a minigene validation system to identify Mendelian disease-causing variants among exon-captured variants of uncertain significance. Hum. Mutat. 2017, 38, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Sun, W.; Xu, Y.; Xin, W.; Guo, X.; Zhang, Q. Molecular genetics of cone-rod dystrophy in Chinese patients: New data from 61 probands and mutation overview of 163 probands. Exp. Eye Res. 2016, 146, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Paavo, M.; Lee, W.; Allikmets, R.; Tsang, S.; Sparrow, J.R. Photoreceptor cells as a source of fundus autofluorescence in recessive Stargardt disease. J. Neurosci. Res. 2019, 97, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hu, X.; Liu, F.; Soares, D.C.; Liu, X.; Yu, S.; Gao, M.; Han, S.; Qin, Y.; Li, C.; et al. Ablation of EYS in zebrafish causes mislocalisation of outer segment proteins, F-actin disruption and cone-rod dystrophy. Sci. Rep. 2017, 7, 46098. [Google Scholar] [CrossRef] [PubMed]
- Messchaert, M.; Dona, M.; Broekman, S.; Peters, T.A.; Corral-Serrano, J.C.; Slijkerman, R.W.N.; van Wijk, E.; Collin, R.W.J. Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. PloS ONE 2018, 13, e0200789. [Google Scholar] [CrossRef]
- Yu, M.; Liu, Y.; Li, J.; Natale, B.N.; Cao, S.; Wang, D.; Amack, J.D.; Hu, H. Eyes shut homolog is required for maintaining the ciliary pocket and survival of photoreceptors in zebrafish. Biol. Open 2016, 5, 1662–1673. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; van Kerkhoff, L.P.; Wieringa, B.; Ropers, H.H. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990, 347, 674–677. [Google Scholar] [CrossRef]
- Seabra, M.C.; Brown, M.S.; Goldstein, J.L. Retinal degeneration in choroideremia: Deficiency of rab geranylgeranyl transferase. Science 1993, 259, 377–381. [Google Scholar] [CrossRef]
- Van den Hurk, J.A.; Schwartz, M.; van Bokhoven, H.; van de Pol, T.J.; Bogerd, L.; Pinckers, A.J.; Bleeker-Wagemakers, E.M.; Pawlowitzki, I.H.; Rüther, K.; Ropers, H.H.; et al. Molecular basis of choroideremia (CHM): Mutations involving the Rab escort protein-1 (REP-1) gene. Hum. Mutat. 1997, 9, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.A.; Lopes, V.S.; Sanagustin, J.T.; Keady, B.T.; Williams, D.S.; Pazour, G.J. Distinct functions for IFT140 and IFT20 in opsin transport. Cytoskeleton 2014, 71, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Owen, N.; Islam, F.; Tracey-White, D.; Plagnol, V.; Holder, G.E.; Michaelides, M.; Carss, K.; Raymond, F.L.; Rozet, J.M.; et al. Nonsyndromic Retinal Dystrophy due to Bi-Allelic Mutations in the Ciliary Transport Gene IFT140. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 2015, 47, 834–841. [Google Scholar] [CrossRef]
- Bai, S.W.; Herrera-Abreu, M.T.; Rohn, J.L.; Racine, V.; Tajadura, V.; Suryavanshi, N.; Bechtel, S.; Wiemann, S.; Baum, B.; Ridley, A.J. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 2011, 9, 54. [Google Scholar] [CrossRef]
- Suga, A.; Mizota, A.; Kato, M.; Kuniyoshi, K.; Yoshitake, K.; Sultan, W.; Yamazaki, M.; Shimomura, Y.; Ikeo, K.; Tsunoda, K.; et al. Identification of Novel Mutations in the LRR-Cap Domain of C21orf2 in Japanese Patients with Retinitis Pigmentosa and Cone-Rod Dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4255–4263. [Google Scholar] [CrossRef]
- Fadaie, Z.; Whelan, L.; Ben-Yosef, T.; Dockery, A.; Corradi, Z.; Gilissen, C.; Haer-Wigman, L.; Corominas, J.; Astuti, G.D.N.; de Rooij, L.; et al. Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. NPJ Genom. Med. 2021, 6, 97. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Mullaney, B.G.; Bhaskar, S.S.; Dickerson, J.E.; Hall, G.; O’Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef]
- Khan, K.N.; Chana, R.; Ali, N.; Wright, G.; Webster, A.R.; Moore, A.T.; Michaelides, M. Advanced diagnostic genetic testing in inherited retinal disease: Experience from a single tertiary referral centre in the UK National Health Service. Clin. Genet. 2017, 91, 38–45. [Google Scholar] [CrossRef]
- Wen, S.; Wang, M.; Qian, X.; Li, Y.; Wang, K.; Choi, J.; Pennesi, M.E.; Yang, P.; Marra, M.; Koenekoop, R.K.; et al. Systematic assessment of the contribution of structural variants to inherited retinal diseases. bioRxiv 2023. [Google Scholar] [CrossRef]




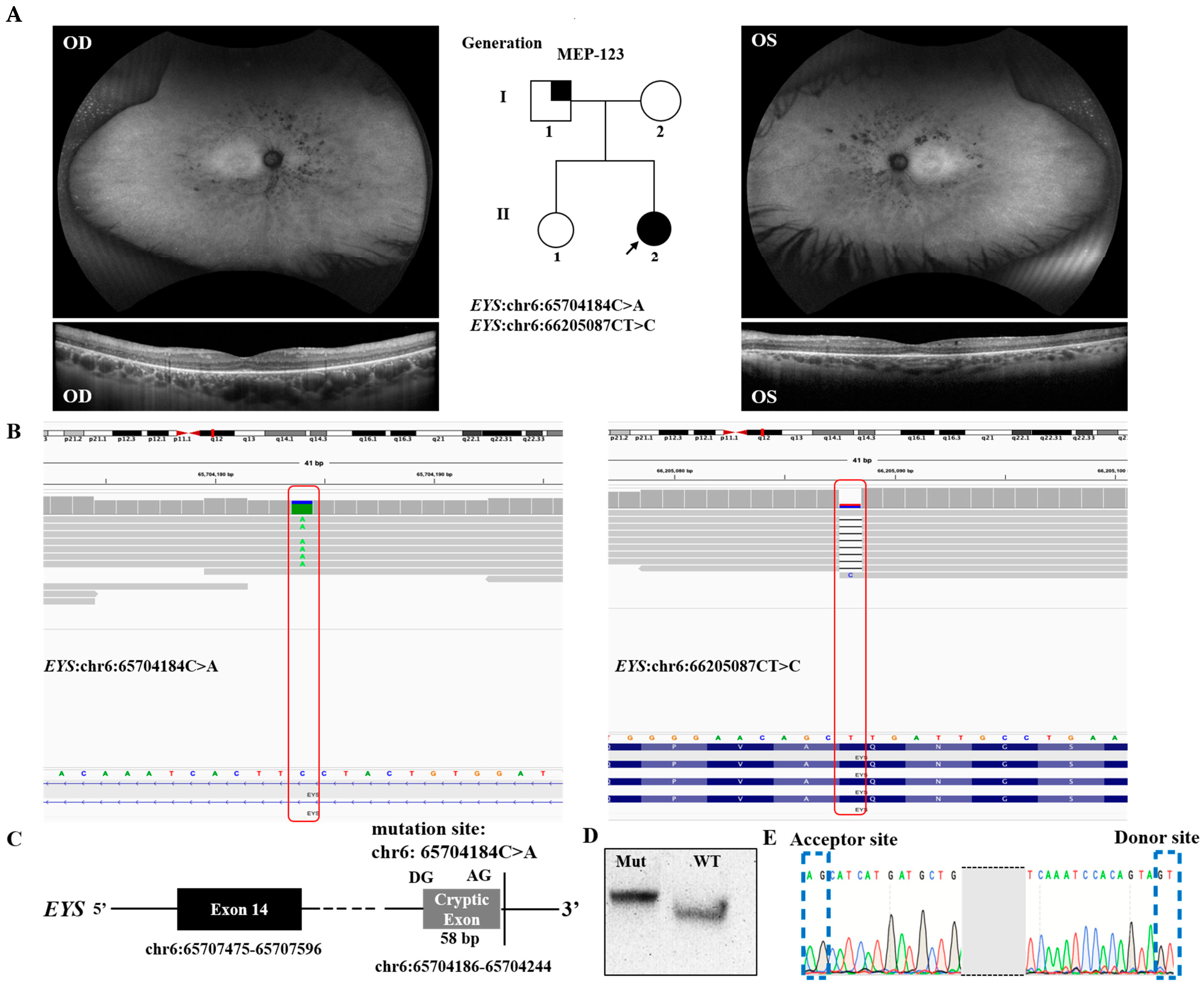{kind=link}
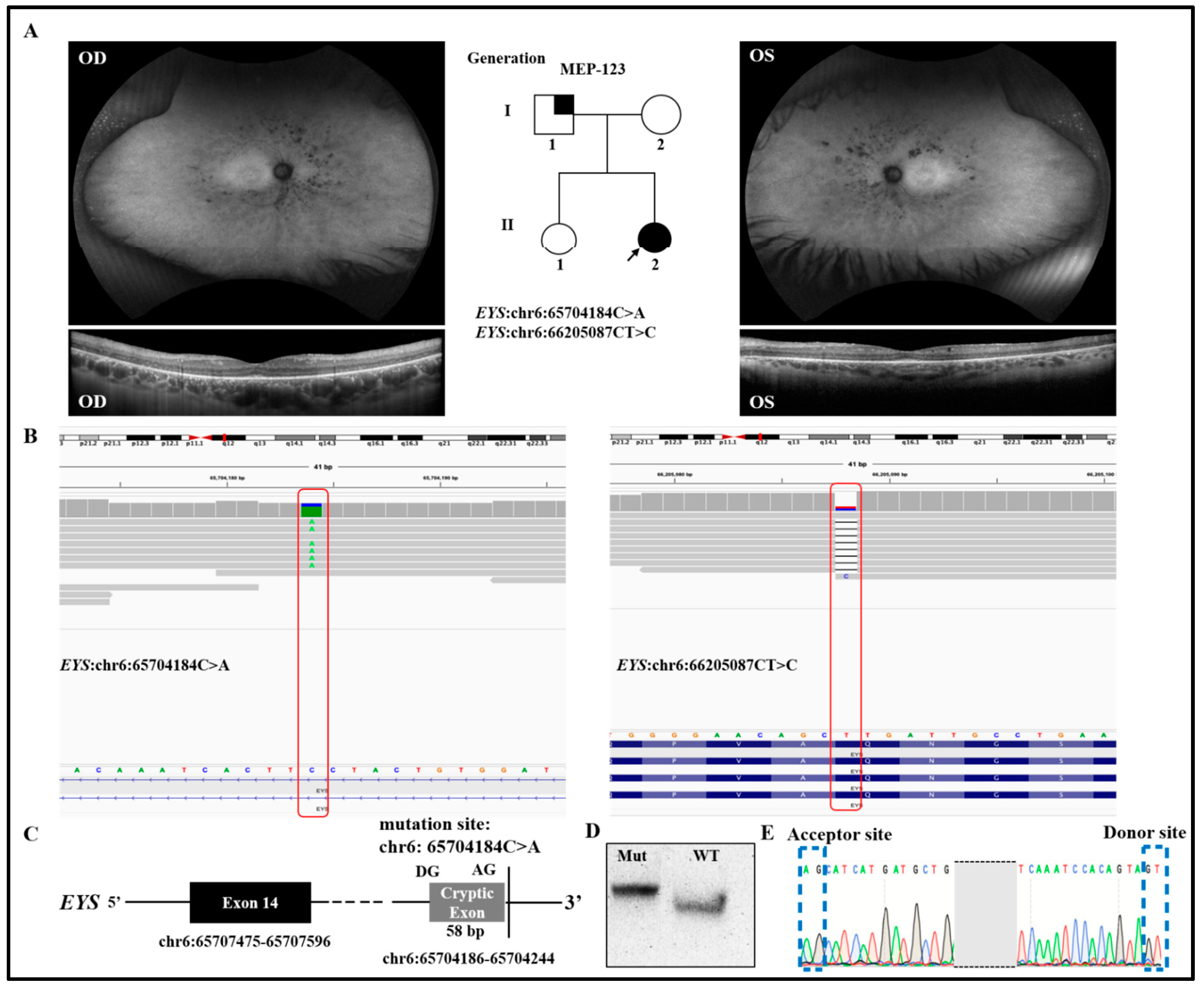{kind=link}
{kind=link}
| Patient ID | Gene | Genomic Variant | cDNA Variant | Protein Variant | Zygosity | Variant Type | Reference |
|---|---|---|---|---|---|---|---|
| MEP-123 | EYS | chr6:65704184C>A | NM_001142800.2:c.2259+3291G>T | - | Heterozygous | Splicing | Novel |
| EYS | chr6: 66205087CT>C | NM_001142800.2:c.216delA | NP_00113627.1:p. A73Lfs*12 | Heterozygous | Frameshift | Novel | |
| MEP-395 | CHM | chrX:85237775C>G | NM_000390.4:c.117-962G>C | - | Hemizygous | Splicing | Novel |
| MEP-398 | IFT140 | chr16:1630797G>A | NM_014714.4:c.1487C>T | NP_055529.2:T496M | Heterozygous | Missense | Novel |
IFT140 | chr16:1634305ins22 | NM_014714.4:c.1250_1271dup | NP_055529.2:p. S425Gfs*66 | Heterozygous | Frameshift | Novel | |
| MEP-662 | C21orf2/ CFAP410 | chr21:45750712 CCT>C | NM_004928.3:c.634_635del | NP_004919.1:p. R212Gfs | Heterozygous | Frameshift | Novel |
| C21orf2/ CFAP410 | chr21:45752937Gins12 | NM_004928.3:c.351_353dup12 | NP_004919.1:p. L118delinsTLPRL | Heterozygous | Insertion | [23] | |
| MEP-663 | C21orf2/ CFAP410 | chr21:45750712 CCT>C | NM_004928.3:c.634_635del | NP_004919.1:p. R212Gfs | Heterozygous | Frameshift | Novel |
| C21orf2/ CFAP410 | chr21:45752937Gins12 | NM_004928.3:c.352_352dup12 | NP_004919.1:p. L118delinsTLPRL | Heterozygous | Insertion | [23] | |
| MEP-082 | ABCA4 | chr1:94484001C>T | NM_000350.3:c.5196+1137G>A | - | Heterozygous | Splicing | [24] |
ABCA4 | chr1:94531618T>C | NM_000350.3:c.1555-2745A>G | - | Heterozygous | Splicing | [25] |
| Patient ID | Clinical Diagnosis | Gender | Race | Age (Years) | Age of Onset (Years) | BCVA | Progression | Other Symptoms | Family History | |
|---|---|---|---|---|---|---|---|---|---|---|
| Right | Left | |||||||||
| MEP-123 | RP | F | Caucasian | 34 | 20 | 20/50- | 20/50- | Mild progression of cystoid macular edema, peripheral vision, and night blindness | NA | Isolated case |
| MEP-395 | Choroideremia | M | Caucasian | 50 | 38 | 20/20-2 | 20/80 | Mild progression of visual field | Night blindness, legally blind | Mother with possible “slight night blindness” |
| MEP-398 | RP | M | Asian | 19 | 9 | 20/25 + 2 | 20/20-2 | Patient had minimal progression | Nyctalopia, midperipheral scotomas, midperipheral atrophy, reduced ERGs, visual fields, loss of outer retinal structures on OCT, high myopia | Some relatives with weak vision but not a similar phenotype |
| MEP-662 | CRD | M | Asian | 3 | 12 | 20/70 + 1 | 20/70+1 | Gradual tilting of RNFL over time | Primary congenital glaucoma, subnormal visual acuity, severe cone and mild rod dysfunction | Affected sister with negative family history of similar eye problems |
| MEP-663 | CRD | F | Asian | 1 | 9 | 20/150 | 20/150 + 1 | Low vision from cone–rod dystrophy | Hyperopia, nystagmus, astigmatism, intermittent exotropia OU, progressive cone–rod dystrophy | Affected brother with negative family history of similar eye problems |
| MEP-082 | ABCA4 related retinopathy | F | Caucasian | 46 | NA | CF | 20/100 | Progression of retinopathy OU with foveal sparing | Deceased in January 2022 | Family history of ALS but not vision problems |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, H.M.J.; Wang, M.; Huang, A.; Schmidt, R.; Qian, X.; Yang, P.; Marra, M.; Li, Y.; Pennesi, M.E.; Chen, R. Novel Pathogenic Mutations Identified from Whole-Genome Sequencing in Unsolved Cases of Patients Affected with Inherited Retinal Diseases. Genes 2023, 14, 447. https://doi.org/10.3390/genes14020447
Hussain HMJ, Wang M, Huang A, Schmidt R, Qian X, Yang P, Marra M, Li Y, Pennesi ME, Chen R. Novel Pathogenic Mutations Identified from Whole-Genome Sequencing in Unsolved Cases of Patients Affected with Inherited Retinal Diseases. Genes. 2023; 14(2):447. https://doi.org/10.3390/genes14020447
Chicago/Turabian StyleHussain, Hafiz Muhammad Jafar, Meng Wang, Austin Huang, Ryan Schmidt, Xinye Qian, Paul Yang, Molly Marra, Yumei Li, Mark E. Pennesi, and Rui Chen. 2023. "Novel Pathogenic Mutations Identified from Whole-Genome Sequencing in Unsolved Cases of Patients Affected with Inherited Retinal Diseases" Genes 14, no. 2: 447. https://doi.org/10.3390/genes14020447