1. Introduction
With the completion of the human genome project and the rapid development of high-throughput sequencing technologies, tremendous amounts of genetic data, such as single nucleotide polymorphism (SNP) data, have been generated. There is a wealth of information in these data, and how to mine information in a large amount of data has become the key to many studies. Genome-wide association studies (GWAS) have emerged as a powerful way for discovering genetic variants associated with complex diseases, and GWAS often uses SNPs as genetic markers for case-control association studies.
The interaction between different genes when expressing a specific phenotype is called epistasis. The importance of epistasis in phenotypic-genotype associations has been established. Epistasis can be defined in different ways. Here we focus on and significantly expand upon statistical epistasis which is the deviation from additivity in mapping multi-locus genotypes to phenotypic variation [
1]. To analyze epistatic interactions between SNPs, several epistasis detection methods have been proposed [
2,
3,
4]. Although using a real biological SNP dataset is necessary, the real process behind it is usually unknown. Therefore, it is very complicated to evaluate the accuracy of these epistatic detection methods by using real biological SNP datasets [
5]. The use of simulated data provides a new way to test the accuracy of the SNP epistasis method because the expected result of simulated data is known. Running the detection methods on the simulated data and analyzing the comparison between the running results and the expected results provide a good way to evaluate these detection methods. In addition, simulated data has very little data generation cost. Therefore, simulated data has turned out to be an important tool for data analysis. To better evaluate the performance of epistasis detection methods to detect SNP interactions in real biological SNP data, simulated datasets are critical.
In simulated data, a penetrance function, or a penetrance table, is commonly used to describe the epistatic relationship between SNPs. The penetrance table contains the probability of a particular combination of alleles and their expression phenotype. This probability is usually expressed as a penetrance value which is the probability of being affected given the genotype of the sample. We calculate the epistasis model in the simulated data, that is, we calculate the values of a penetrance table.
According to whether there is a marginal effect of a single SNP in the epistatic model, the models are divided into two types: the epistatic model with marginal effect (eME model) and the epistatic model without marginal effect (eNME model). The SNP epistatic model with marginal effect means that one or more SNPs in this is model have marginal effects, but the combined epistatic effect of all SNPs is stronger. To facilitate the calculation of this type of model, the penetrance value for each genotype combination is defined as a function of one or more variables, and each variable typically represents a statistical parameter of the interaction. Typically, the penetrance of a marginal effect epistatic model can be constrained to specific expressions for the baseline penetrance
and relative penetrance
[
1,
6,
7]:
In this type of model, given the prevalence
(the probability of a population affected by the epistasis model) and the heritability
(the variation in phenotypes affected by epistatic models of SNP interactions) of the model where their formulas are as follows:
then both the values of
and
can be determined. Thus, the penetrance value of the combined genotype can be determined, and a specific epistatic model to be solved can be obtained. Under the constraints of prevalence and heritability, EpiSIM [
1] solves
and
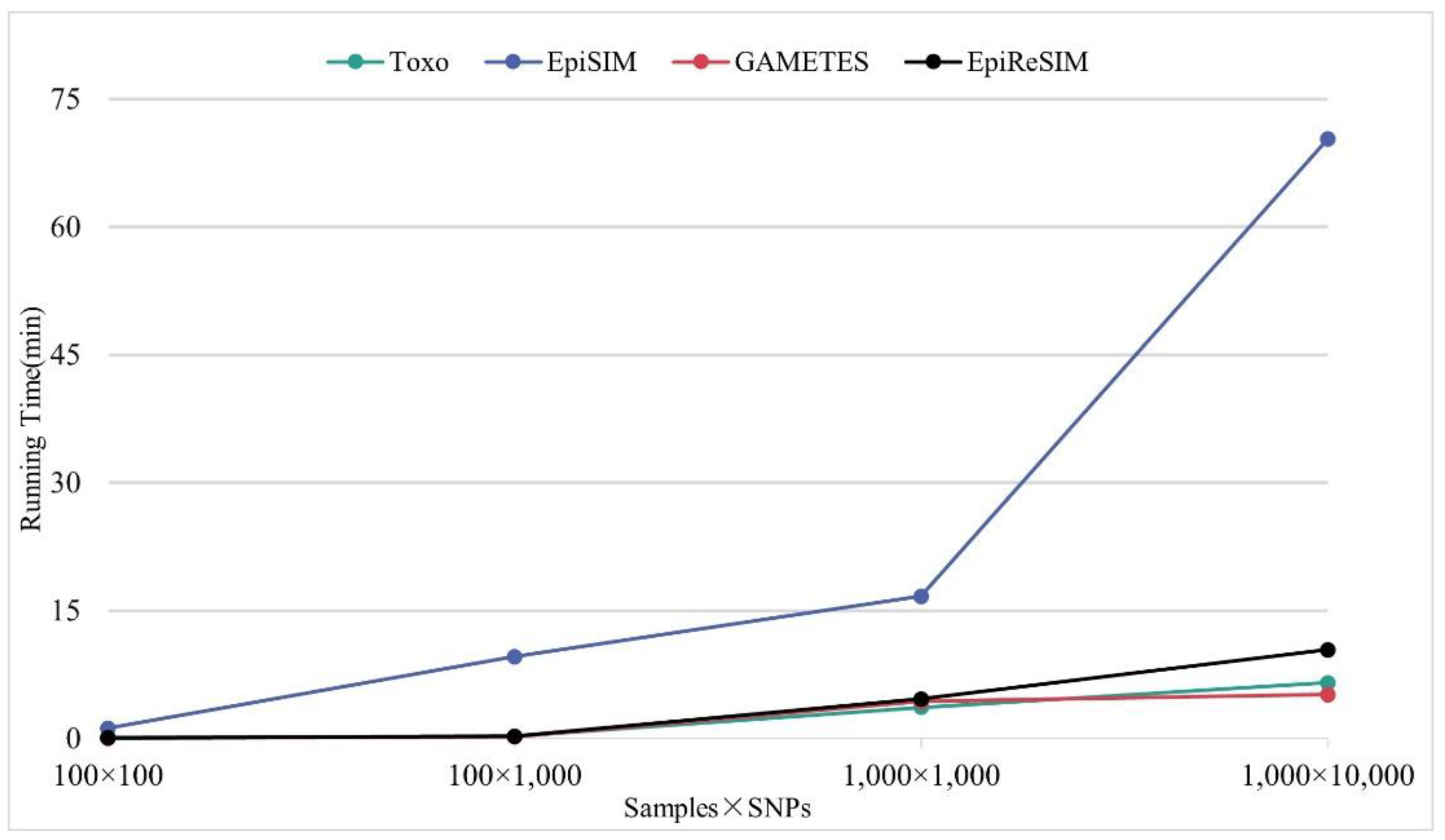
to obtain the eME model by solving equations, and at the same time, EpiSIM generates simulated data with linkage disequilibrium patterns and haplotype blocks using the Markov Chain process. This calculation method brings a lot of computational burdens when calculating high-order models. Toxo [
8] calculates an eME model by specifying either prevalence or heritability and maximizing the other. In this way, it reduces the computational burden and can calculate high-order eME models, but Toxo does not have its own data generation method and needs to use other simulation software to generate simulated data. EpiGEN [
9] also uses a similar parameter baseline risk to generate the model and it can generate both categorical and quantitative phenotypes, but it generates simulated data also with the help of other simulators.
The epistatic model without marginal effects means that a single SNP has no main effect, but the combination of the several specific SNPs has a strong epistatic effect [
10,
11]. The penetrance of the epistatic model without marginal effect has no obvious mathematical law, so it is impossible to constrain the epistatic models without marginal effects with functional expressions. Therefore, an epistatic model without marginal effect needs to calculate the penetrance value corresponding to the genotype separately, and make it satisfy the situation that the individual marginal effect of each SNP is equal to 0, which brings a heavy computational burden to the calculation of the model. GAMETES [
12] is an epistasis simulator, which uses a random architecture to generate a pure and strict penetrance table of epistasis models without marginal effects, and the prevalence and heritability of the models to be generated are specified by users. EpiSIM [
1] searches the value of penetrance with fixed steps in the range of 0 to 1 under the constraints of prevalence and heritability to calculate the penetrance table. Although EpiSIM uses an exhaustive search method to calculate the 2-order eNME model, it also brings a heavy computational burden.
In this study, we focus on the calculation of the epistatic model without marginal effects and propose a new simulation method called EpiReSIM. When calculating an eNME model, the simulator innovatively converts the calculation of the SNP epistasis model into the problem of solving the underdetermined equation by using the feature that the prevalence of the model is equal to the marginal penetrance. EpiReSIM divides the calculation of epistasis models displaying no marginal effects into two cases. In the first case, the calculation of the epistasis model displaying no marginal effects is a linear under-determined system of the equations-solving problem under only the prevalence constraint. In this case, we use the method of Complete Orthogonal Decomposition method to calculate this equation set. In the second case, when both prevalence and heritability are constrained simultaneously, we transform the computation of the epistasis model displaying no marginal effects into a nonlinear under-determined system of equations-solving problem, which is solved by the simulator using Newton’s method to calculate the penetrance value of the epistasis models displaying no marginal effects. Through the calculation of these two cases, EpiReSIM was able to compute penetrance tables for high-order models. Then, the simulation method also provides its own data generation, which uses a resampling method to generate samples of the simulated data and generates a label for each sample of simulated data based on the calculated penetrance table.
4. Conclusions
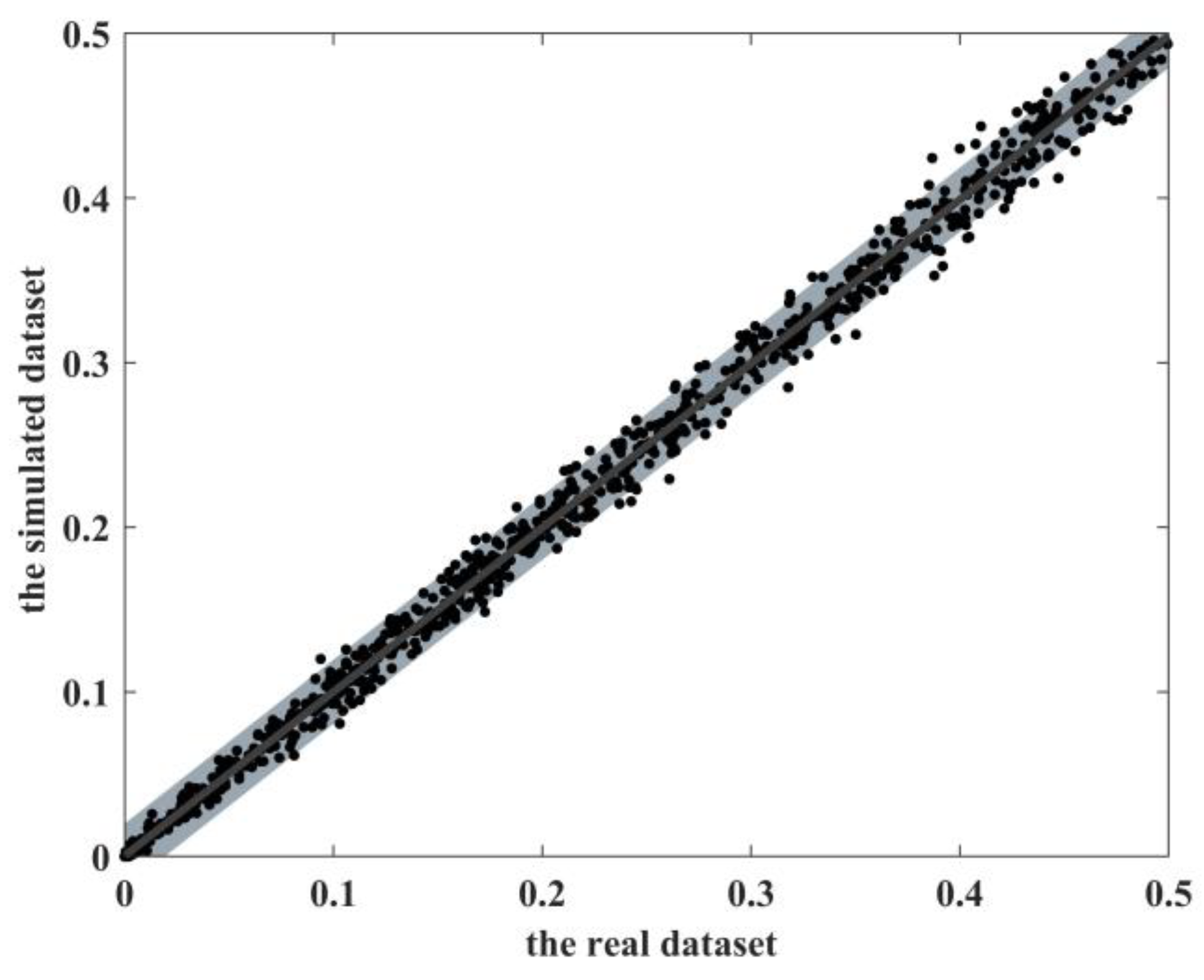
The main contribution of this work is to propose a simulator, EpiReSIM, which can calculate the penetrance table of high-order eNME models and generate simulated data by resampling method. EpiReSIM provides two calculation methods, that is users can calculate the model by using the constraints on prevalence or using the constraints of prevalence and heritability. The validity of the simulated data generated by EpiReSIM is verified by different SNP interaction recognition methods. At the same time, the simulator is open source, which facilitates the study of SNP interaction methods.
This simulation method, however, also has its own limitations and there are several areas that need to be enhanced. First, although EpiReSIM can calculate high-order models without marginal effects, there are very few cases where the heritability of the calculated models is relatively low, which puts forward higher requirements for SNP interaction identification methods. Second, the current version of EpiReSIM does not consider interactions between environmental and genetic factors. We will focus on improving the usability of EpiReSIM in the following two areas: including the influence of environmental factors when calculating the eNME model, and optimizing the data generation method of resampling.


 ,
, 



{kind=link}
{kind=link}
{kind=link}