
Treacher Collins Syndrome: Genetics, Clinical Features and Management



1
Department of Genetics, Wroclaw University of Environmental and Life Sciences, 51-631 Wroclaw, Poland
2
Department of Plastic Surgery, Wroclaw Medical University, 50-367 Wroclaw, Poland
3
Head and Neck Surgery Clinic for Children and Young Adults, Department of Clinical Pediatrics, University of Warmia and Mazury, 10-561 Olsztyn, Poland
4
Department of Pediatrics, Division Pediatric Propedeutics and Rare Disorders, Wroclaw Medical University, 51-618 Wroclaw, Poland
*
Author to whom correspondence should be addressed.
Genes 2021, 12(9), 1392; https://doi.org/10.3390/genes12091392
Submission received: 5 August 2021
/
Revised: 4 September 2021
/
Accepted: 5 September 2021
/
Published: 9 September 2021
(This article belongs to the Section Genetic Diagnosis)
Abstract
:Treacher Collins syndrome (TCS) is associated with abnormal differentiation of the first and second pharyngeal arches, occurring during fetal development. Features of TCS include microtia with conductive hearing loss, slanting palpebral fissures with possibly coloboma of the lateral part of lower eyelids, midface hypoplasia, micrognathia as well as sporadically cleft palate and choanal atresia or stenosis. TCS occurs in the general population at a frequency of 1 in 50,000 live births. Four subtypes of Treacher Collins syndrome exist. TCS can be caused by pathogenic variants in the TCOF1, POLR1D, POLR1C and POLR1B genes. Genetically, the TCOF1 gene contains 27 exons which encodes the Treacle protein. In TCOF1, over 200 pathogenic variants have been identified, of which most are deletions leading to a frame-shift, that result in the formation of a termination codon. In the presented article, we review the genetics and phenotype of TCS as well as the management and surgical procedures utilized for treatment.
1. Genetics
Treacher Collins syndrome (TCS, OMIM 154500), also referred to as Franceschetti syndrome or mandibulofacial dysostosis (MFD1), is a rare developmental disorder. The disorder has been described by Berry [1], Treacher Collins [2] and Franceschetti and Klein [3]. The incidence is about 1 in 50,000 live births [4] with 40% of cases involving a family history, while the remaining 60% occur as a result of de novo mutations.
TCS syndrome is genetically heterogeneous [5]. The literature describes four clinical subtypes: Treacher Collins syndromes 1: TCS1 is caused by pathogenic variants of the TCOF1 gene; Treacher Collins syndromes 2: TCS2 is caused by pathogenic variants in the POLR1D gene; and Treacher Collins syndromes 3: TCS3 caused by pathogenic variants in the POLR1C gene and lately identified POLR1B gene as a new gene responsible for a novel Treacher Collins syndromes 4: TCS4 [6] (Table 1).
No association has been identified between the clinical features of patients and the gene in which the alterations occur.
The TCOF1 gene is encoded by 27 exons. Two exons discovered latest are: exon 6A of length 231bp localized between exons 6 and 7 and exon 16A, 108bp in length, localized between exons 16 and 17 [11]. TCOF1 encodes a 144 kDa nuclear phosphoprotein called Treacle which consists of 1411aa [12], while the transcript containing the additional exon 6A encodes a protein longer by 77aa [11].
In the central region of treacle are the multiple casein kinase 2 (CK2)/protein kinase C (PKC) phosphorylation sites [13]. Treacle contains a putative nuclear export signal at the N-terminus and nuclear import signals at the C-terminus, suggesting dynamic localization of treacle [14]. A LisH motif is located at the N-terminus of treacle [15].
Treacle is involved in the transport of proteins and ribosomal subunits between the nucleolus and cytoplasm [16]. This protein supports ribosomal DNA gene transcription by interacting with the UBF (upstream binding factor) [17]. Treacle is involved in ribosome biogenesis by binding and requiring Pol I, UBF, Nopp140 to the rDNA promoter. UBF is usually tightly bound by treacle. A decrease in treacle could cause UBF rapid displacement from rDNA and then to the inhibition of rRNA synthesis. Increased expression of treacle may interfere with cisplatin-induced apoptosis [18]. Thus, both silencing and ectopic expression of TCOF1 coding treacle can cause apoptosis of neural crest cells during embryogenesis by influence on crucial apoptotic regulators [14].
Loss of treacle protein function in neuroblastoma cells affects the expression of genes that are involved in proliferation, apoptosis and cell cycle.
Ciccia et al. [19] identified treacle as a potential component of the NBS1 protein complex. Subunits of RNA polymerase I (POLR1A, B and E) were found to be components of TCOF1 complexes. They have also discovered TCOF1 as a DDR (DNA damage response) factor, that recruits NBS1 to nucleoli after DNA damage. DNA damage due to oxidative stress occurs during embryonic development [15].
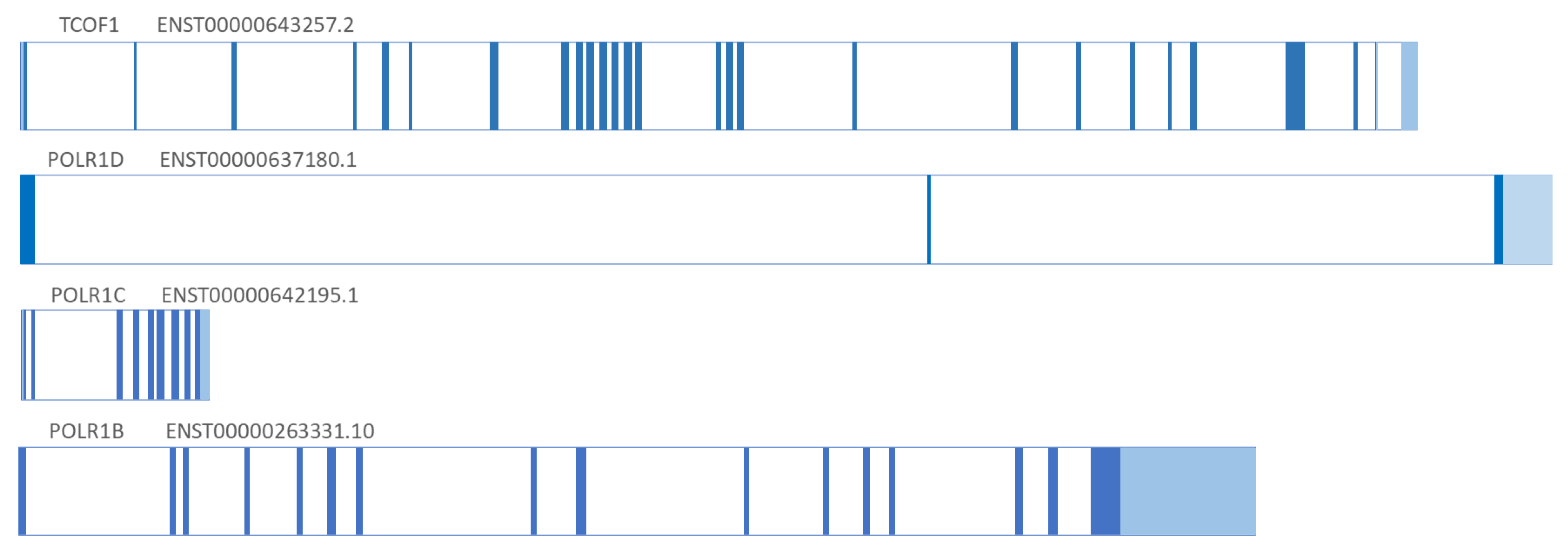
Dauwerse et al. discovered that POLR1C and POLR1D genes are involved in TCS syndrome by detecting deletion and pathogenic variants in the POLR1D and POLR1C genes in patients with TCS [7]. The POLR1C and POLR1D genes were discovered more than 10 years after the discovery of the TCOF1 gene. The POLR1D gene is encoded by 3 exons, POLR1C by 9 exons, while the POLR1B gene consists of 15 exons. The structure of TCS-associated genes, their exons, and differences in length are presented (Figure 1).
Pathogenic variants in POLR1D are autosomal dominant and autosomal recessive, in POLR1C are autosomal recessive, while in TCOF1 and POLR1B genes are autosomal dominant. Variants in POLR1D lead to haploinsufficiency of gene, whereas in POLR1C lead to functional depletion of the gene [7].
POLR1C and POLR1D are subunits that are shared between RNA polymerases I and III. RNA polymerase I and III are involved in ribosomal RNA transcription. Insufficient amounts of Pol I or Pol III can lead to a reduced number of mature ribosomes in neural crest cells during embryonic development, which could affect apoptosis of proper initially formed first and second branchial arches causing craniofacial abnormalities [7].
The POLR1B gene is a unique subunit of RNA polymerase I. Pathogenic variants of the POLR1B gene can lead to p53-dependent apoptosis and consequent cranioskeletal defects [6].
Over 200 mutations have been reported in the TCOF1 gene including deletions, insertions, substitutions [15,22,23,24,25,26,27]. Exons 10, 15, 16, 23 and 24 are hotspots in TCOF1. Conte et al. [15] detected 15 different TCOF1 pathogenic variants in multiple exons: 3, 5, 6, 10, 12, 13, 15, 16, 18, 20, 22, 23, 24. The effect of most TCOF1 pathogenic variants is haploinsufficiency.
In most TCS causes, the pathogenic variants are small deletions from 1 to 40 nucleotides, that result in premature termination of codons, producing a truncated protein [8]. Chen et al. [28] detected about 63% deletions ranging in size from 1–38 nucleotides in Chinese patients with TCS.
Gross deletions of TCOF1 were identified with a 5% frequency in those with TCS [29]. Beygo et al. [30] identified a 3.367kb deletion in one patient with a clinical diagnosis of TCS. This deletion concerned exon 3 and is the first described single exon deletion within the TCOF1 gene. Liu et al. [31] identified a novel 2–6 exon deletion of TCOF1. This was the first report of a fetal for TCS in a Chinese population.
Kantaputra et al. [32] discovered a novel TCOF1 pathogenic variant, c.4138_4142del, p.Lys1380GlufsTer12 in patients with TCS, who did not have the typical TCS phenotype. In these cases were observed short cranial bases, hypoplastic maxilla and mandible. The next most common pathogenic variants of the TCOF1 gene are insertions. The longest insertion, c.484_668ins185bp, was localized to exon 5 in twin sisters [33]. Zhang et al. [27] identified five novel variants (two nonsense, one missense and one splicing) in TCOF1 in Chinese patients. Zeng et al. [26] reported a nonsense pathogenic variant (c.1622G > A) in exon 11 of TCOF1.
Dauwerse et al. [7] detected pathogenic variants in genes POLR1C and in POLR1D (20 heterozygous variants) in TCS patients, while a report by Sanchez et al. [6] identified three novel pathogenic variants in POLR1B.
In the case of Treacher Collins syndrome patients, no correlation is observed between the type of the pathogenic variants and the phenotype.
2. Clinical Features–Phenotype
Symptoms of TCS are clinically very variable and include following mean malformations/features:
- external and middle ear abnormalities including microtia with conductive hearing loss attributed most commonly to malformation of the ossicles,
- bilateral and symmetric downslanting palpebral fissures,
- coloboma or notching of the lateral part of lower eyelids with medial absence or sparse of the eyelashes and tear ducts defect,
- hypoplasia of the facial bones with micrognathia and retrognathia as well as zygomatic bones. Hypoplasia of the zygomatic bones and mandible can cause significant feeding and respiratory difficulties.
Other common manifestations include cleft palate, unilateral or bilateral choanal stenosis or atresia and pharyngeal hypoplasia. Most patients with TCS have unaffected intellect, and TCS occurs in both genders equally (Table 2).
The phenotypic features of TCS are observed bilaterally, and disorder intensity is determined by skeletal deformities. The lateral and lower orbital walls are shorter due to hypoplasia of the zygomatic bone, there is narrowing and flattening of the bony face, as well as displacement of the outer corner of the eye and symmetric downslanting palpebral fissures [36]. A forward protruding nose is often observed, as well as abnormal occlusion, and in more severe cases the falling back of the tongue resulting in breathing problems. The severity of the deformities does not increase with age (Figure 2). Patients with TCS have various degrees of deformed auricles, leading to partial or complete deafness. Most TCS patients have unilateral or bilateral conductive type hearing loss. Conductive hearing impairment can be attributed mainly to malformations of the middle ear, which are similar in patients with hearing defects or lack of auditory ossicles.
In 60% of children is noticed a poor quality of speech. Speech difficulties have resulted from hearing loss problems and speech acquisition and are associated with structural defects of the ears. Researchers noted that speech therapy may be more effective after structural normalization; however, it is also recommended before such normalization. Patients with TCS may use speech through expressions such as gestures, facial expressions, and vocalizations, which the speech pathologist should consider to contribute to social interactions. Children who have tracheostomies require special attention from speech pathologists.
Clinical diagnosis of patients with TCS may be difficult as they possess features similar to other disorders, such as oculoauriculovertebral spectrum (known as Goldenhar syndrome), Miller and Nager syndrome, as well as mandibulofacial dysostosis with microcephaly. These disorders are caused by abnormal development of the first and second branchial arches of embryonic development [25]. In the case of oculoauriculovertebral spectrum disorders, the genes responsible for the disease have not been described in the literature. Nager syndrome is an autosomal dominant disorder. It is caused by variants in the SF3B4 gene, which is located on chromosome 1q21.2. Patients with Nager syndrome are characterized by the following phenotype: downslanting palpebral fissures, hypoplasia of zygomatic bone, micrognathia, cleft lower eyelid, dysplastic ears associated with hearing loss, combined with predominantly radial limb defects such as lateral absence or hypoplastic of thumbs and hypoplasia of the radius with radioulnar synostosis. Miller syndrome is an autosomal recessive disorder caused by mutations in the DHODH gene, which is located on chromosome 16q22.2. Patients with Miller syndrome are characterized by malar hypoplasia, micrognathia, microstomia and a cleft lip and/or palate. The limb defects in Miller syndrome are distinctive, consisting of an absence or incomplete development of the 5th digital ray of all four limbs, and, frequently, forearm abnormalities (the ulna and radius can be short with occasional radio-ulnar synostosis). Pathogenic variants in the EFTUD2 gene are responsible for mandibulofacial dysostosis with microcephaly (MFDM), characterized by the following phenotypic features: microcephaly, asymmetry of facial features with unusual ears with preauricular tags, sensorineural deafness, a small jaw and major defects including choanal atresia, cleft palate, cardiac defects and even esophageal atresia as well as significant developmental delay and speech delay.
3. Management and Surgical Treatment
Patients with TCS need specialized, multidisciplinary treatment involving pediatrics, clinical genetics, otolaryngologist, orthodontist, audiologist, psychologist and having major impact variable types of surgeons. Of significant importance is (computed tomography) CT scanning, used to document the anatomy of the head in order to prepare a plan for surgery, to recognize defects of hearing organs as well as to document progress in different stages of treatment (Figure 3). Neuro-ophthalmologic evaluation of children is necessary to determine extraocular muscle function and to assess corneal exposure and visual acuity due to orbital defects. An echocardiogram is used to identify heart defects and also ophthalmologic and dental anomalies assessments are made.
Treatment can be divided into three main phases depending on the age of the patient.
- Birth to age 2 years—facilitation of elementary living functions: breathing, e.g., tracheostomy (Figure 4), feeding and hearing as well as vision and heart.
- Age 3 to 12 years—speech therapy, integration into society, bone reconstructions including mandibular (Figure 5), which can prevent progression of defects as well as ophthalmological and orthodontic support. Pre-school children usually receive the same treatment.
- Age 13 to 18—orthognathic therapies, maxillomandibular and nasal reconstruction as well as integration into society. Multistage reconstructive treatment forming face is possible in this age and makes noticeable improvements (Figure 6). However, if possible, it is recommended to start therapy at an earlier age. The sooner therapy begins gives the higher possibility of achieving optimal results (Figure 7).
Treatment must be started as early as the first year of life in cases where there is a life-threatening risk, e.g., nocturnal apnea in children with underdevelopment of the mandible and hypertrophy of the tongue. In the most severe cases, this can lead to suffocation (apnea). In these cases, a mandibular distraction is most effective. The correction of macrostomia or cleft lip surgery is generally performed in infancy. Similarly, due to the need to protect the eyeball, coloboma surgery is generally carried out in the first year of life (even in the first months) if indicated.
To determine the severity of mandibular underdevelopment, the Kaban classification is used (types I–III). For patients with TCS, surgical treatment is critical and often begins in the first year of life and continues until growth is complete. The shortening of the mandible and falling back of the tongue results in upper airway obstruction. If macrostomia is present, surgery is needed before the child is one year of age. The lip is sewn at the age of 6 months and the cleft palate between the first and second years of life [37].
The cleft upper eyelid (coloboma) should be operated on before the end of the first year of life. This is necessary to create mechanical eye protection and to ensure proper corneal hydration. Misalignment of the eyelid crevices is corrected by lateral canthopexy, and eyelid defects and coloboma are corrected with local plasty eyelid flaps.
Another phenotypic feature seen in children with TCS is symmetric downslanting palpebral fissures, surgery for which begins at preschool age. The procedure involves displacement of a long dermal-musculocutaneous flap from the upper eyelid to the lower eyelid and canthopexy [38]. In cases of complete auricular microtia, reconstruction is performed with autogenous cartilage. Underdevelopment of the mandible is treated by conducting bone distractions, with more severe cases requiring treatment between 6 and 10 years of age [39].
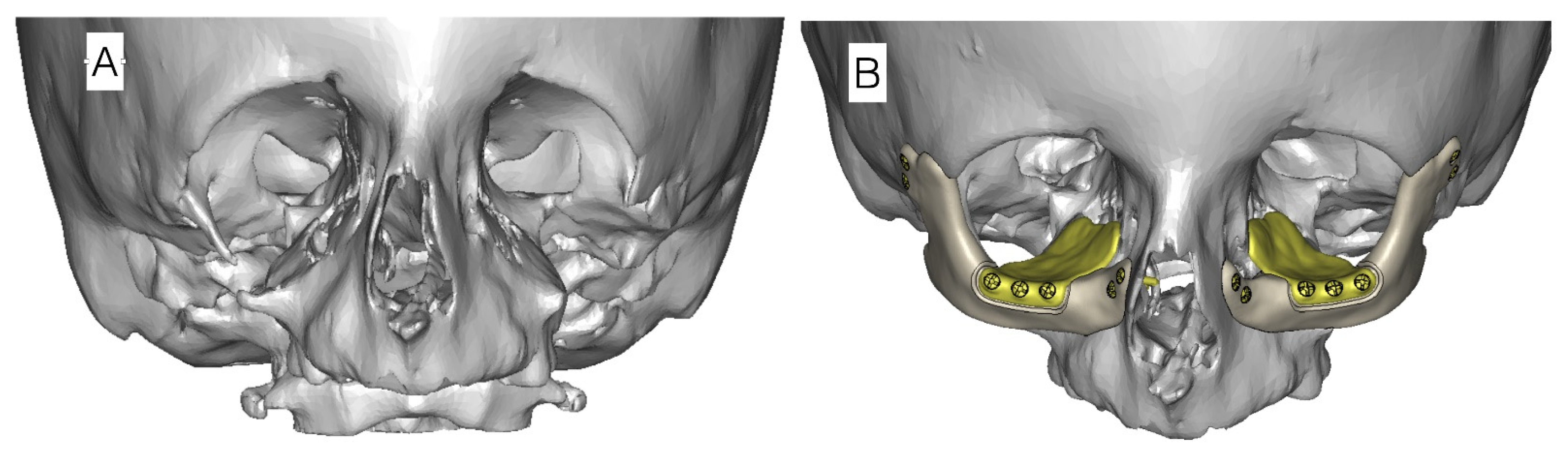
Orbital cavity wall deformations are operated simultaneously with proper eyelid positioning. To improve facial appearance due to wide and long nose structure, horizontal chin osteotomy and rhinoplasty can be performed [40]. Orbital floor defects are reconstructed with allogenic or individual orbital floor implants (Figure 8). Reconstruction of zygomatic and orbital bone deficiencies should not be performed before the age of 5 years unless there are severe problems with corneal exposure.
4. Conclusions
Molecular diagnosis plays a very significant role for patients with Treacher Collins syndrome in the prenatal and postnatal periods. Genetic counselling is very important for every family with a TCS child. Each patient with TCS is unique and treatment should be developed by a multidisciplinary team. As there are no clinical differences between the four subtypes of Treacher Collins syndrome, the surgical treatment also does not differ according to the type of pathogenic variants in the genes described. Apart from age, it is the appearance, location and severity of the deformity that determines treatment priorities.
The complete etiology of TCS is not yet fully understood. In about 10% of cases, it remains unknown what implies the disorder. This may indicate a role of other genes or other genetic mechanisms in the pathogenesis of TCS [28].
Author Contributions
B.A.M.-K. Defined the article structure, data collection and interpretation, research and study of the literature, description of genetics, final review of article; P.W. description of medical treatment; K.D. description of clinical features, detail treatment of described patients, photo documentation of the treatment including patient’s consent; R.Ś. description of clinical features, final review of article. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
This is a review article. The study did not report any data.
Acknowledgments
We want to thank to patients and their parents for agreement for publication of the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Berry, G.A. Note on a congenital defect colobomata of the lower lid. R. Lond. Ophthalmic. Hosp. Rep. 1889, 12, 255. [Google Scholar]
- Collins, E.T. Cases with symmetrical congenital notches in the outer part of each lower lid and defective development of malar bones. Trans. Ophthalmol. Soc. UK 1900, 20, 190. [Google Scholar]
- Franceschetti, A.; Klein, D. The mandibulofacial dysostosis; a new hereditary syndrome. Acta Ophthalmol. 1949, 27, 143–224. [Google Scholar]
- Gorlin, R.J.; Cohen, M.M.; Levin, L.S. Syndromes of the Head and Neck; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Dixon, J.; Trainor, P. Treacher Collins syndrome. Orthod. Craniofacial Res. 2007, 10, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Laplace-Builhé, B.; Mau-Them, F.T.; Richard, E.; Goldenberg, A.; Toler, T.; Guignard, T.; Gatinois, V.; Vincent, M.; Blanchet, C.; et al. POLR1B and neural crest cell anomalies in Treacher Collins syndrome type 4. Genet. Med. 2020, 22, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauwerse, J.G.; Dixon, J.; Seland, S.; Ruivenkamp, C.A.L.; Van Haeringen, A.; Hoefsloot, L.H.; Peters, D.J.M.; Boers, A.C.-D.; Daumer-Haas, C.; Maiwald, R.; et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2010, 43, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Geneviève, P.; Ostertag, A.; Marlin, S.; Lacombe, D.; Martin-Coignard, D.; Coubes, C.; David, G.; Lyonnet, S.; Vilain, C.; et al. Treacher Collins syndrome: A clinical and molecular study based on a large series of patients. Genet. Med. 2016, 18, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.orpha.net (accessed on 5 August 2021).
- Katsanis, S.H.; Jabs, E.W. Treacher Collins Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2004. [Google Scholar]
- So, R.B.; Gonzales, B.; Henning, D.; Dixon, J.; Dixon, M.J.; Valdez, B.C. Another face of the Treacher Collins syndrome (TCOF1) gene: Identification of additional exons. Gene 2004, 328, 49–57. [Google Scholar] [CrossRef]
- Valdez, B.C.; Henning, D.; So, R.B.; Dixon, J.; Dixon, M.J. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl. Acad. Sci. USA 2004, 101, 10709–10714. [Google Scholar] [CrossRef] [Green Version]
- Isaac, C.; Marsh, K.L.; Paznekas, W.A.; Dixon, J.; Dixon, M.J.; Jabs, E.; Meier, U.T. Characterization of the Nucleolar Gene Product, Treacle, in Treacher Collins Syndrome. Mol. Biol. Cell 2000, 11, 3061–3071. [Google Scholar] [CrossRef] [Green Version]
- Winokur, S.T.; Shiang, R. The Treacher Collins syndrome (TCOF1) gene product, treacle, is targeted to the nucleolus by signals in its C-terminus. Hum. Mol. Genet. 1998, 7, 1947–1952. [Google Scholar] [CrossRef] [Green Version]
- Sakai, D.; Trainor, P. Face off against ROS: Tcof1 /Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev. Growth Differ. 2016, 58, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Conte, C.; D’Apice, M.R.; Rinaldi, F.; Gambardella, S.; Sangiuolo, F.; Novelli, G. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome. BMC Med. Genet. 2011, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodfellow, S.J.; Zomerdijk, J.C.B.M. Basic Mechanisms in RNA Polymerase I Transcription of the Ribosomal RNA Genes. Subcell. Biochem. 2013, 61, 211–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzanka, M.; Piekiełko-Witkowska, A. The Role of TCOF1 Gene in Health and Disease: Beyond Treacher Collins Syndrome. Int. J. Mol. Sci. 2021, 22, 2482. [Google Scholar] [CrossRef]
- Ciccia, A.; Huang, J.-W.; Izhar, L.; Sowa, M.E.; Harper, J.; Elledge, S.J. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, 18631–18636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: www.ncbi.nlm.nih.gov/gene (accessed on 5 August 2021).
- Available online: http://www.ensembl.org (accessed on 5 August 2021).
- Splendore, A.; Fanganiello, R.D.; Masotti, C.; Morganti, L.S.; Passos-Bueno, M.R. TCOF1 mutation database: Novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature. Hum. Mutat. 2005, 25, 429–434. [Google Scholar] [CrossRef]
- Marszałek-Kruk, B.A.; Śmigiel, R.; Sasiadek, M. Novel mutation in the TCOF1 gene in a patient with Treacher Collins syndrome. Pediatr. Polska 2014, 89, 462–465. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Lu, Y.; Wang, Y.; Zhang, X.; Duan, H.; Cheng, J.; Yuan, H.; Han, D. Identification of a novel TCOF1 mutation in a Chinese family with Treacher Collins syndrome. Exp. Ther. Med. 2018, 16, 2645–2650. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Wang, Y.; Fan, Y.; Du, H.; Luo, N.; Zhang, S.; Chen, X. TCOF1 pathogenic variants identified by Whole-exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet J. Rare Dis. 2019, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Xie, M.; Li, J.; Xie, H.; Lu, X. A novel nonsense mutation in the TCOF1 gene in one Chinese newborn with Treacher Collins syndrome. Int. J. Pediatr. Otorhinolaryngol. 2021, 141, 110561. [Google Scholar] [CrossRef]
- Zhang, C.; An, L.; Xue, H.; Hao, S.; Yan, Y.; Zhang, Q.; Jin, X.; Li, Q.; Zhou, B.; Feng, X.; et al. Mutation analysis of TCOF1 gene in Chinese Treacher Collins syndrome patients. J. Clin. Lab. Anal. 2021, 35, e23567. [Google Scholar] [CrossRef] [PubMed]
- Chen-Long, L.; Guo, L.; Li, C.-L.; Shan, J.; Xu, H.-S.; Li, J.-Y.; Sun, S.; Hao, S.-J.; Jing, S.; Chai, G.; et al. Mutation screening of Chinese Treacher Collins syndrome patients identified novel TCOF1 mutations. Mol. Genet. Genom. 2018, 293, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.; Oldridge, M.; Archer, C.; O’Rourke, A.; McParland, J.; Brekelmans, R.; Seller, A.; Lester, T. Gross deletions in TCOF1 are a cause of Treacher–Collins–Franceschetti syndrome. Eur. J. Hum. Genet. 2012, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Beygo, J.; Buiting, K.; Seland, S.; Lüdecke, H.-J.; Hehr, U.; Lich, C.; Prager, B.; Lohmann, D.; Wieczorek, D. First Report of a Single Exon Deletion in TCOF1 Causing Treacher Collins Syndrome. Mol. Syndr. 2012, 2, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lin, P.; Pang, J.; Jia, Z.; Peng, Y.; Xi, H.; Wu, L.; Li, Z.; Wang, H. Identification of a novel gross deletion of TCOF1 in a Chinese prenatal case with Treacher Collins syndrome. Mol. Genet. Genom. Med. 2020, 8, e1313. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Tripuwabhrut, K.; Intachai, W.; Carlson, B.M.; Quarto, N.; Ngamphiw, C.; Tongsima, S.; Sonsuwan, N. Treacher Collins syndrome: A novel TCOF1 mutation and monopodial stapes. Clin. Otolaryngol. 2020, 45, 695–702. [Google Scholar] [CrossRef]
- Marszałek-Kruk, B.A.; Wojcicki, P.; Śmigiel, R.; Trzeciak, W.H. Novel insertion in exon 5 of the TCOF1 gene in twin sisters with Treacher Collins syndrome. J. Appl. Genet. 2012, 53, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Teber, A.; Gillessen-Kaesbach, G.; Fischer, S.; Böhringer, S.; Albrecht, B.; Albert, A.; Arslan-Kirchner, M.; Haan, E.; Hagedorn-Greiwe, M.; Hammans, C.; et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur. J. Hum. Genet. 2004, 12, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Splendore, A.; Silva, E.O.; Alonso, L.G.; Richieri-Costa, A.; Alonso, N.; Rosa, A.; Carakushanky, G.; Cavalcanti, D.P.; Brunoni, D.; Passos-Bueno, M.R. High mutation detection rate in TCOF1 among Treacher Collins syndrome patients reveals clustering of mutations and 16 novel pathogenic changes. Hum. Mutat. 2000, 16, 315–322. [Google Scholar] [CrossRef]
- Posnick, J.C.; Ruiz, R.L. Treacher Collins syndrome: Current evaluation, treatment, and future directions. Cleft Palate Craniofac. J. 2000, 37, 434. [Google Scholar] [CrossRef]
- Wójcicki, P.; Marszałek-Kruk, B. Genetic Factors and Treatment Principles of Treacher Collins Syndrome. Dent. Med. Probl. 2005, 42, 619–626. [Google Scholar]
- Jackson, I.T. Reconstruction of the Lower Eyelid Defect in Treacher Collins Syndrome. Plast. Reconstr. Surg. 1981, 67, 365–367. [Google Scholar] [CrossRef]
- Posnick, J.C. Treacher Collins syndrome: Perspectives in evaluation and treatment. J. Oral Maxillofac. Surg. 1997, 55, 1120–1133. [Google Scholar] [CrossRef]
- Farkas, L.G.; Posnick, J.C. Detailed Morphometry of the Nose in Patients with Treacher Collins Syndrome. Ann. Plast. Surg. 1989, 22, 211–219. [Google Scholar] [CrossRef] [PubMed]

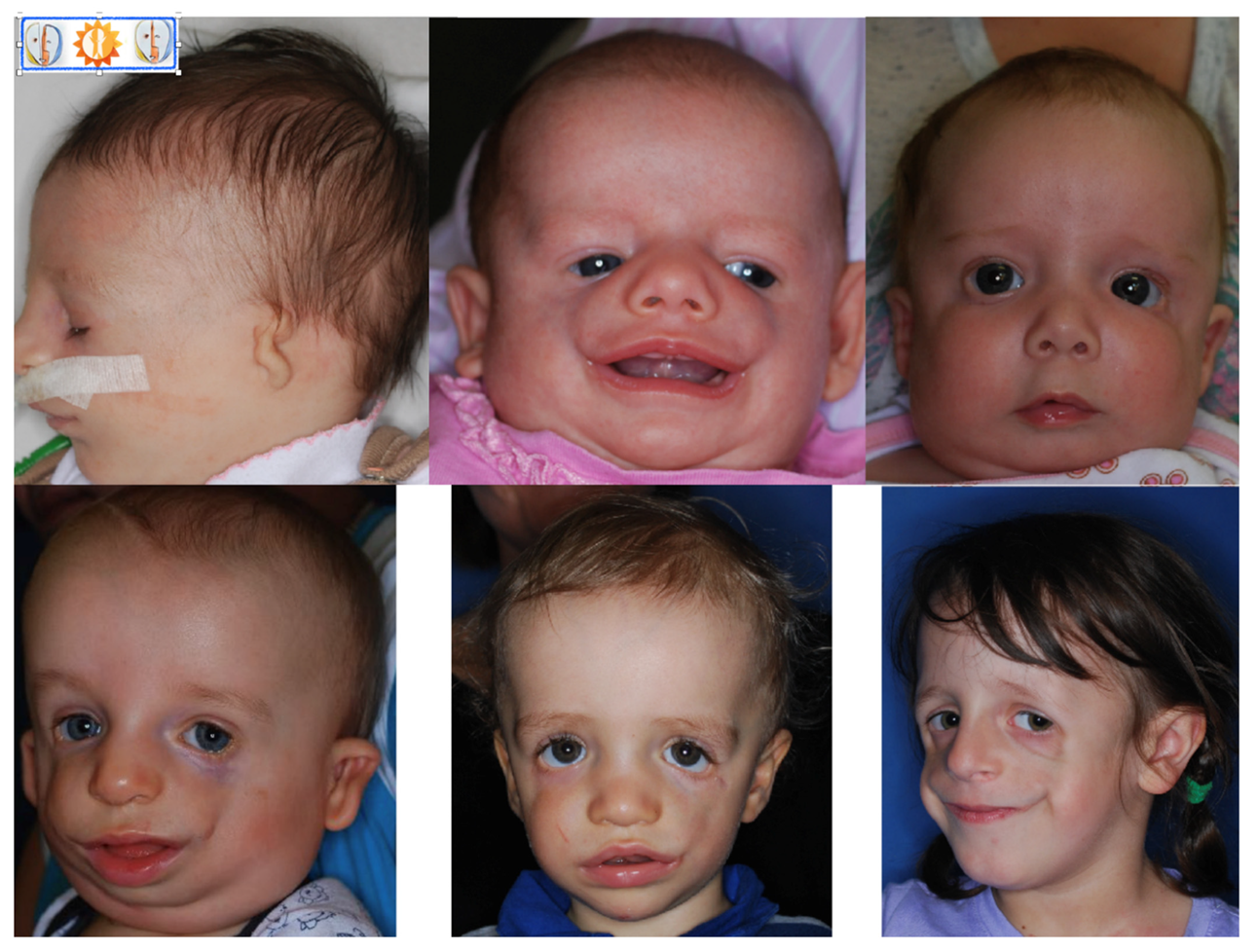
Figure 2.
Patients with TCS at different ages, visible variations in defect severity.

Figure 3.
Cephalometric radiographs showing the basic steps of treatment related to the process of mandibular and maxillary osteodistraction aimed at improving facial contour and opening the upper airway.
Figure 3.
Cephalometric radiographs showing the basic steps of treatment related to the process of mandibular and maxillary osteodistraction aimed at improving facial contour and opening the upper airway.

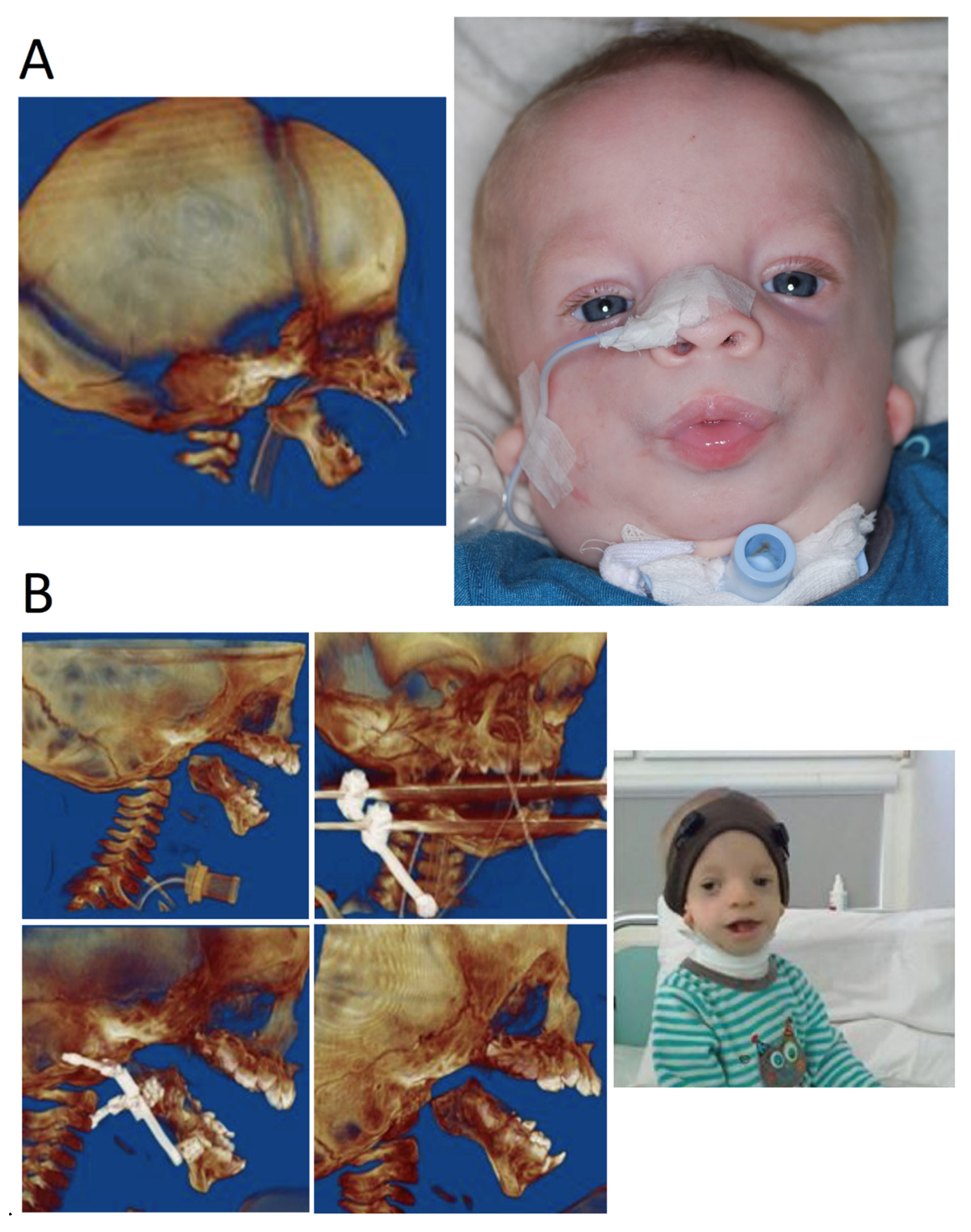
Figure 4.
The same patient at different ages during different stages of treatment. (A) Infant with TCS, CT image of 3D reconstruction, patient with tracheostomy. (B) Few years-old patient. 3D images of reconstruction during treatment—multistage mandibular osteodistraction; patient after completion of osteodistraction process, removed tracheostomy tube.
Figure 4.
The same patient at different ages during different stages of treatment. (A) Infant with TCS, CT image of 3D reconstruction, patient with tracheostomy. (B) Few years-old patient. 3D images of reconstruction during treatment—multistage mandibular osteodistraction; patient after completion of osteodistraction process, removed tracheostomy tube.

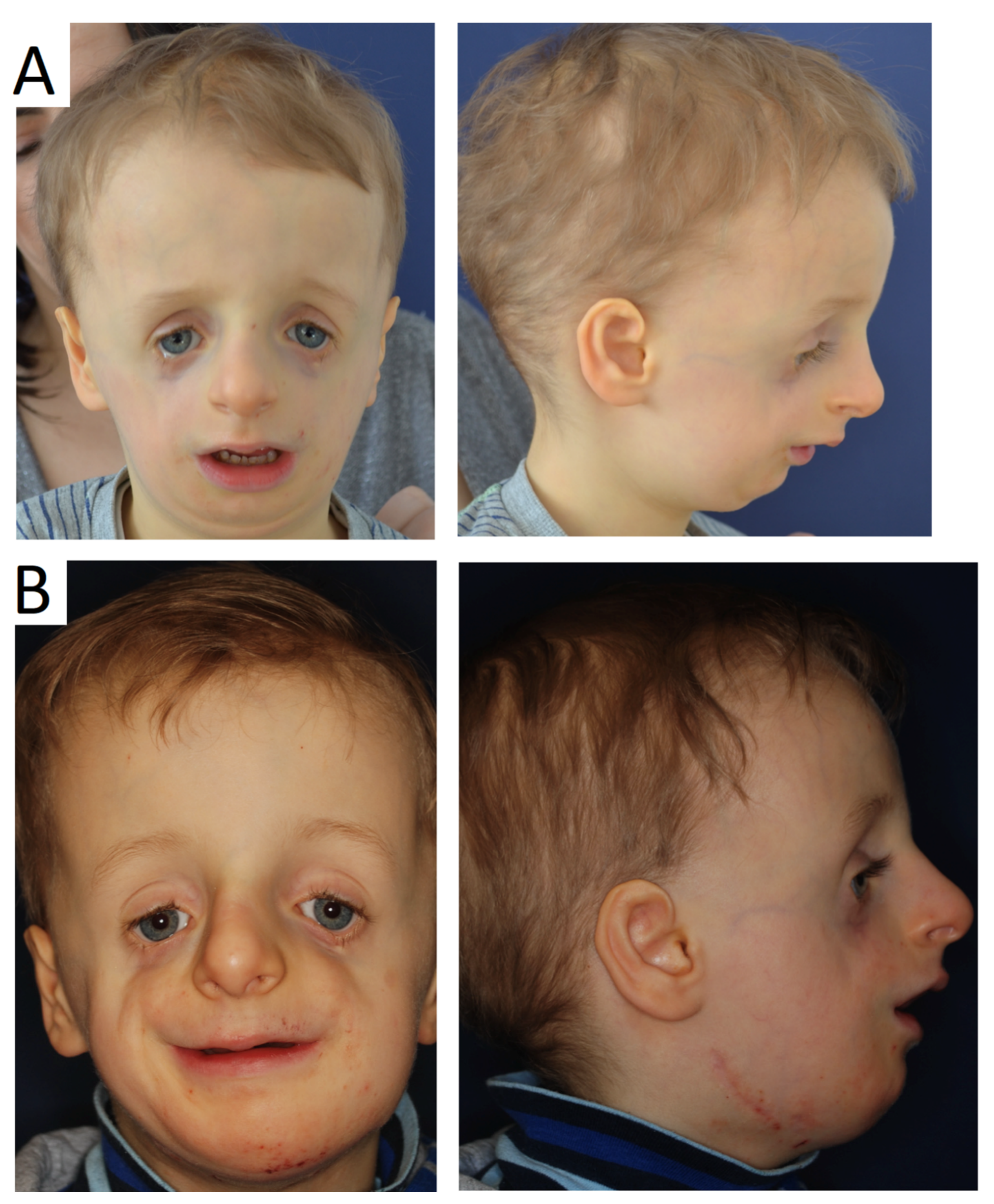
Figure 5.
Patient at the age of 4 before (A) and after (B) mandibular distraction prior to further stages of treatment.
Figure 5.
Patient at the age of 4 before (A) and after (B) mandibular distraction prior to further stages of treatment.

Figure 6.
Teenage female patient with TCS. (A) Stereolithographic model with visible bone deformations. (B) Side and frontal view before major treatment, large deformation of the middle face and mandible resulting in closure of the upper airway, absence of zygomatic bone, coloboma of the lower eyelids. (C) Patient after multistage reconstructive treatment forming the shape of face, transplant taken from the cranial vault mounted in place of the zygomatic bone; despite late notification of the therapy, noticeable improvements in facial features.
Figure 6.
Teenage female patient with TCS. (A) Stereolithographic model with visible bone deformations. (B) Side and frontal view before major treatment, large deformation of the middle face and mandible resulting in closure of the upper airway, absence of zygomatic bone, coloboma of the lower eyelids. (C) Patient after multistage reconstructive treatment forming the shape of face, transplant taken from the cranial vault mounted in place of the zygomatic bone; despite late notification of the therapy, noticeable improvements in facial features.

Figure 7.
Adult patient with TCS after correction treatment.

Figure 8.
Example of virtual planning of zygomatic bone reconstruction with alloplastic implants. (A) Computed tomography image prior to transplantation in a patient with missing zygomatic bone. (B) CT scan image with planned alloplastic zygomatic bone reconstruction and individual orbital floor recontouring plates.
Figure 8.
Example of virtual planning of zygomatic bone reconstruction with alloplastic implants. (A) Computed tomography image prior to transplantation in a patient with missing zygomatic bone. (B) CT scan image with planned alloplastic zygomatic bone reconstruction and individual orbital floor recontouring plates.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Classification of genes and subtypes of Treacher Collins syndrome (TCS) (ORPHA: 861).
| Phenotype | Gene | |||||||
|---|---|---|---|---|---|---|---|---|
| TCS Subtype | OMIM | Name | OMIM | Inheritance | ID | Chromosome Locus | Frequency of TCS Pathogenic Variant [6,7,8] | Product of Gene |
| TCS1 | 154500 | TCOF1 | 606847 | AD | 6949 | 5q32-q33 | 86% | Treacle protein |
| TCS2 | 613717 | POLR1D | 613715 | AD, AR | 51082 | 13q12.2 | 6% | DNA-directed RNA polymerases I and III subunit RPAC2 |
| TCS3 | 248390 | POLR1C | 610060 | AR | 9533 | 6p21.1 | 1.2% | DNA-directed RNA polymerases I and III subunit RPAC1 |
| TCS4 | 618939 | POLR1B | 602000 | AD | 84172 | 2q14.1 | 1.3% | DNA-directed RNA polymerase I subunit RPA2 |
Table 2.
Classic features of Treacher Collins syndrome.
| Classic Feature | Symptom, Feature | Occurrence in Affected Individuals |
|---|---|---|
| Very frequent | Downslanting palpebral fissures | 89–100% |
| Malar hypoplasia/hypoplasia of zygomatic complex | 81–97% | |
| Conductive hearing loss | 83–92% | |
| Mandibular hypoplasia/micrognathia | 78–91% | |
| Frequent | Atresia of external ear canal | 68–71% |
| Microtia | 10–77% | |
| Coloboma (notching) of the lower lid | 54–69% | |
| Delayed speech development | 57–63% | |
| Asymmetry | 52% | |
| Preauricular hair displacement | 24–49% | |
| Rare | Nasogastric tube or gastrostomy in neonates | 28% |
| Cleft palate | 21–33% | |
| Intubation or tracheostomy in neonates | 12–18% | |
| Choanal stenosis/atresia | 13–25% | |
| Cardiac malformation | 11% | |
| Very rare | Rachis malformation | 7% |
| Renal malformation | 4% | |
| Microcephaly | 3% | |
| Intellectual disability Delayed motor development | 1.7–10% | |
| Limb anomaly | 1.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marszałek-Kruk, B.A.; Wójcicki, P.; Dowgierd, K.; Śmigiel, R. Treacher Collins Syndrome: Genetics, Clinical Features and Management. Genes 2021, 12, 1392. https://doi.org/10.3390/genes12091392
AMA Style
Marszałek-Kruk BA, Wójcicki P, Dowgierd K, Śmigiel R. Treacher Collins Syndrome: Genetics, Clinical Features and Management. Genes. 2021; 12(9):1392. https://doi.org/10.3390/genes12091392
Chicago/Turabian StyleMarszałek-Kruk, Bożena Anna, Piotr Wójcicki, Krzysztof Dowgierd, and Robert Śmigiel. 2021. "Treacher Collins Syndrome: Genetics, Clinical Features and Management" Genes 12, no. 9: 1392. https://doi.org/10.3390/genes12091392
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.