
Unique Variant Spectrum in a Jordanian Cohort with Inherited Retinal Dystrophies

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Exome Sequencing and Data Analysis
2.3. Sanger Validation and Co-Segregation Analysis
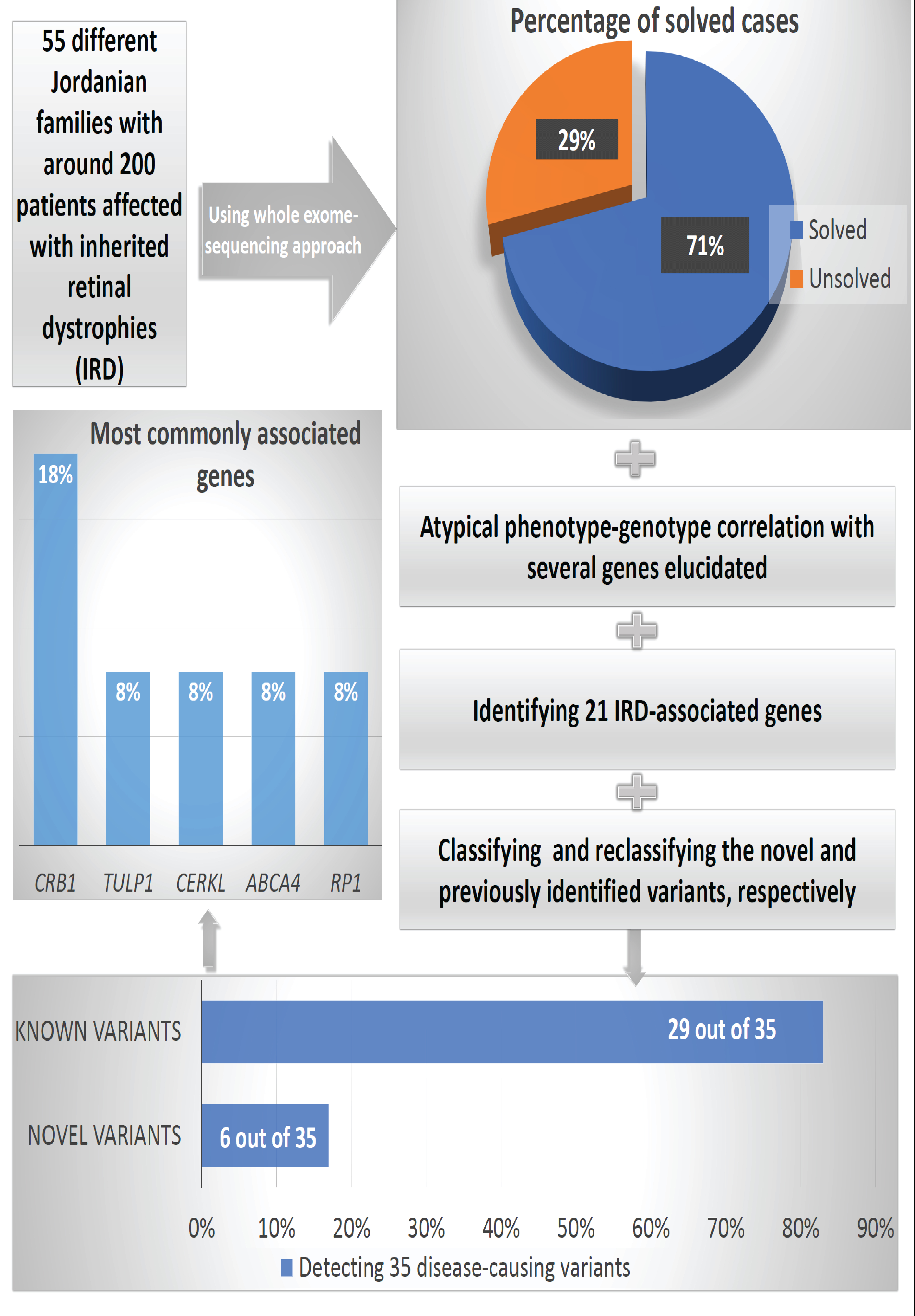
3. Results
3.1. Patients and Clinical Information
3.2. Identification of Potential Pathogenic Variants in the IRD Cohort
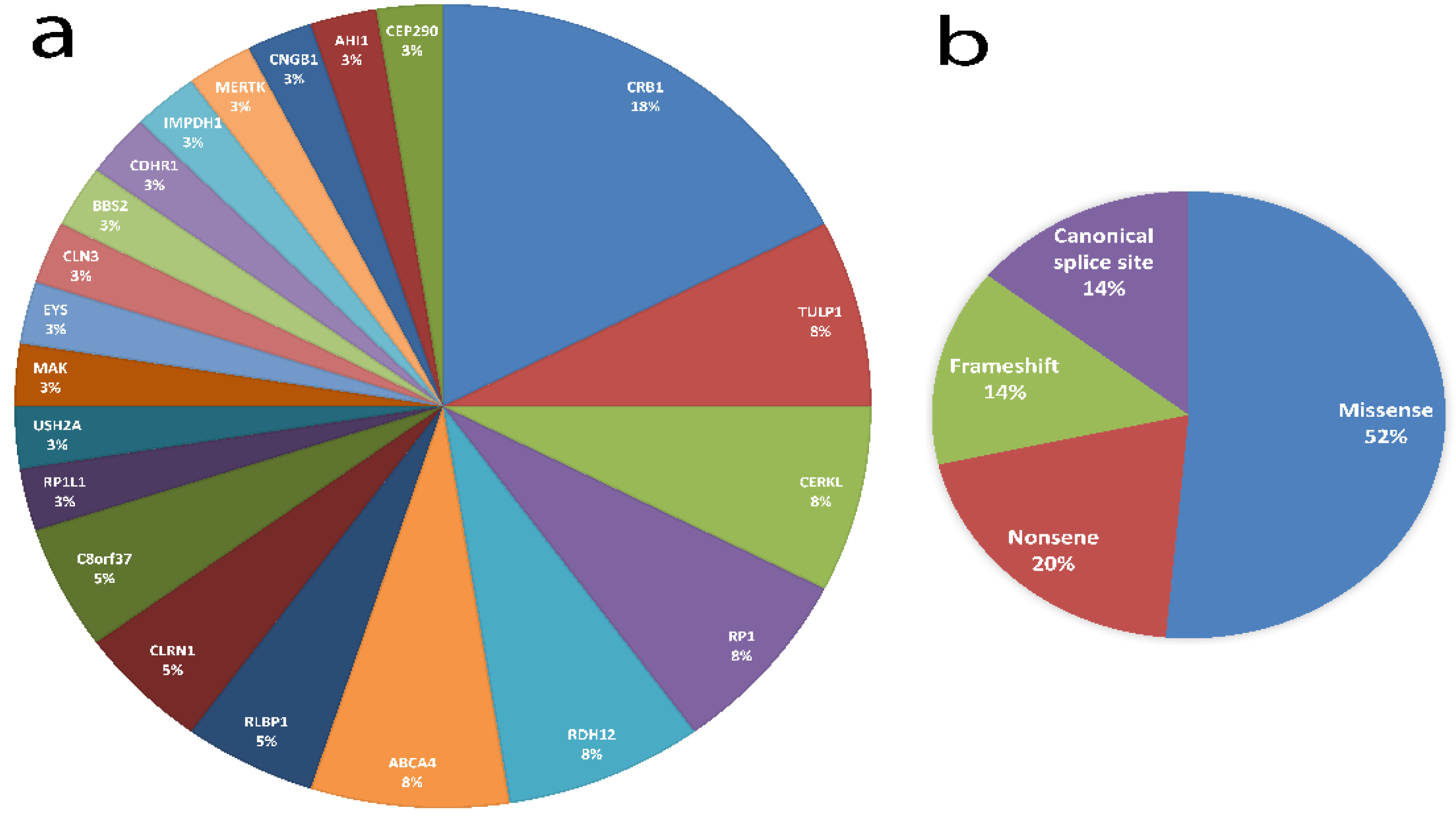
3.3. Variant Spectrum in Jordanian Patients with IRD
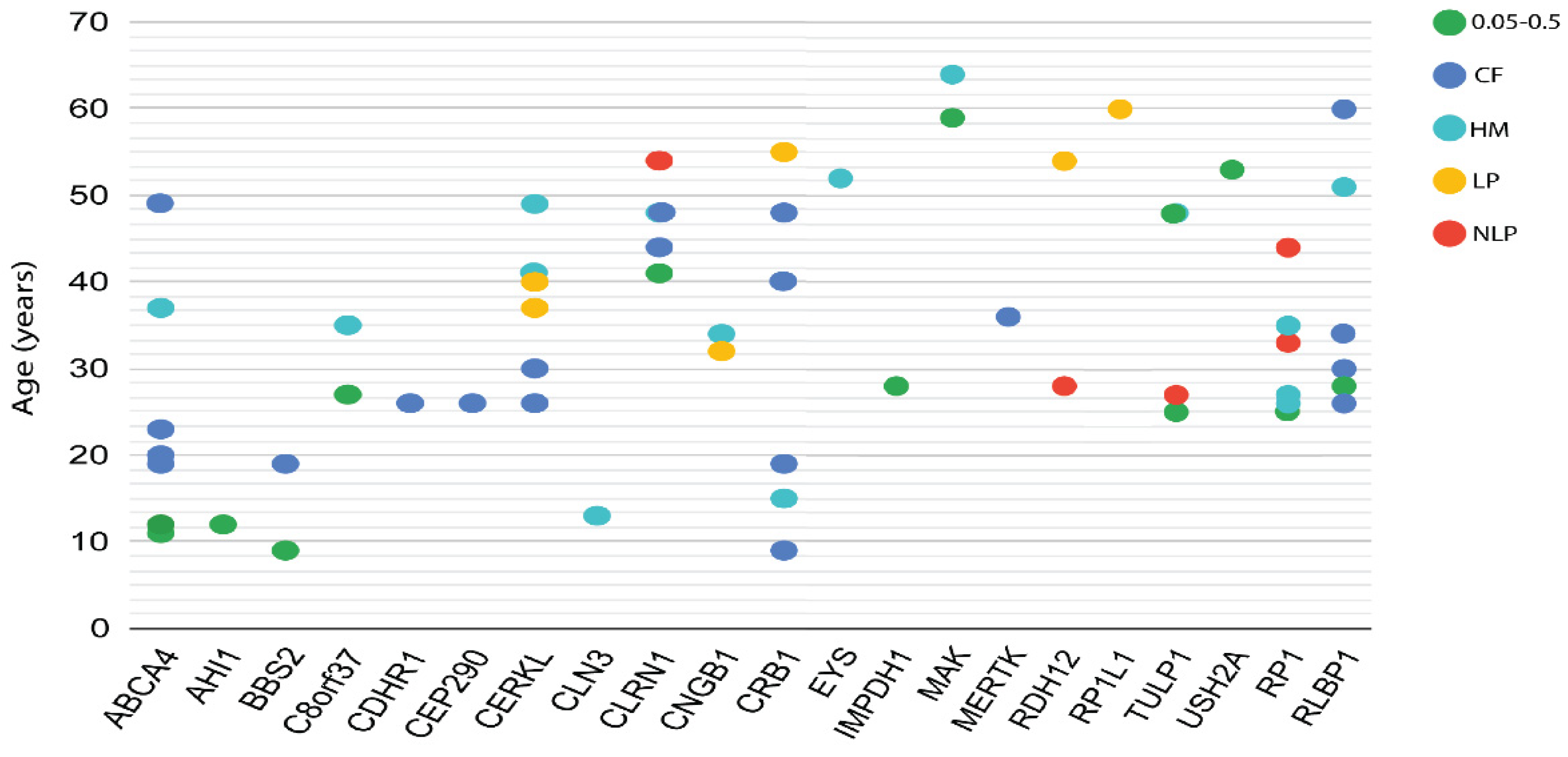
3.4. Phenotypic and Genotypic Information
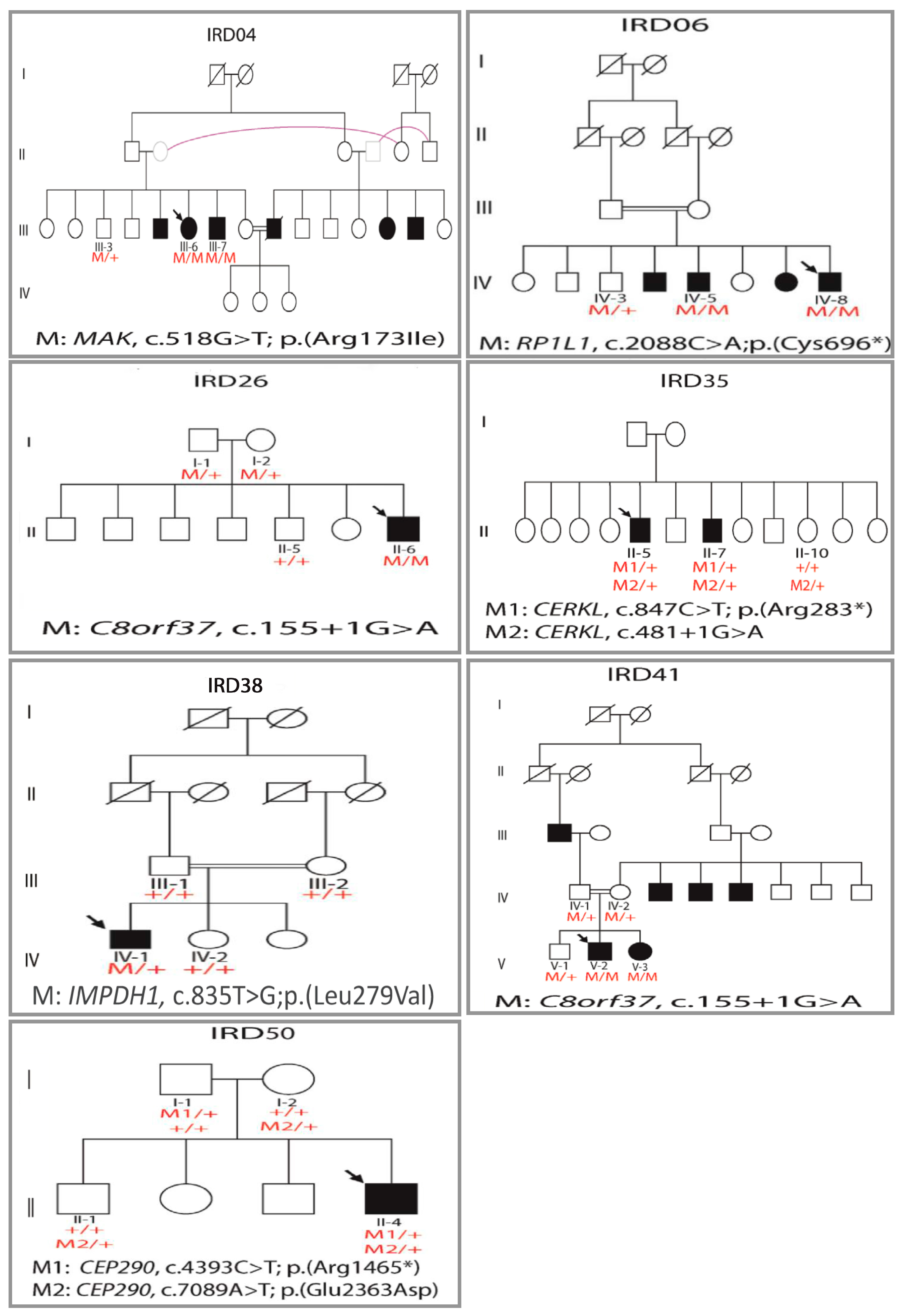
3.5. Investigating the Less Commonly Studied Genotype–Phenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sullivan, L.S.; Daiger, S.P. Inherited retinal degeneration: Exceptional genetic and clinical heterogeneity. Mol. Med. Today 1996, 2, 380–386. [Google Scholar] [CrossRef]
- Lev, S. Molecular aspects of retinal degenerative diseases. Cell. Mol. Neurobiol. 2001, 21, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef]
- Venturini, G.; Rose, A.M.; Shah, A.Z.; Bhattacharya, S.S.; Rivolta, C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012, 8, e1003040. [Google Scholar] [CrossRef] [Green Version]
- Poloschek, C.M.; Bach, M.; Lagrèze, W.A.; Glaus, E.; Lemke, J.R.; Berger, W.; Neidhardt, J. ABCA4 and ROM1: Implications for modification of the PRPH2-associated macular dystrophy phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Sharma, T. Progressive Cone Dystrophy and Cone-Rod Dystrophy (XL, AD, and AR). Adv. Exp. Med. Biol. 2018, 1085, 53–60. [Google Scholar] [CrossRef]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Garg, S. Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genet. Med. 2015, 17, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Yohe, S.; Thyagarajan, B. Review of Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Hamamy, H.; Al-Hait, S.; Alwan, A.; Ajlouni, K. Jordan: Communities and community genetics. Community Genet. 2007, 10, 52–60. [Google Scholar] [CrossRef]
- Zanetti, D.; Sadiq, M.; Carreras-Torres, R.; Khabour, O.; Alkaraki, A.; Esteban, E.; Via, M.; Moral, P. Human diversity in Jordan: Polymorphic Alu insertions in general Jordanian and Bedouin groups. Hum. Biol. 2014, 86, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Al-Eitan, L.N.; Darwish, N.N.; Hakooz, N.M.; Dajani, R.B. Investigation of the forensic GlobalFiler loci in the genetically isolated Circassian subpopulation in Jordan. Gene 2020, 733, 144269. [Google Scholar] [CrossRef]
- Azab, B.; Barham, R.; Ali, D.; Dardas, Z.; Rashdan, L.; Bijawi, M.; Maswadi, R.; Awidi, A.; Jafar, H.; Abu-Ameerh, M.; et al. Novel CERKL variant in consanguineous Jordanian pedigrees with inherited retinal dystrophies. Can. J. Ophthalmol. 2019, 54, 51–59. [Google Scholar] [CrossRef]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Almontashiri, N.A.M.; Alswaid, A.; Oza, A.; Al-Mazrou, K.A.; Elrehim, O.; Tayoun, A.A.; Rehm, H.L.; Amr, S.S. Recurrent variants in OTOF are significant contributors to prelingual nonsydromic hearing loss in Saudi patients. Genet. Med. 2018, 20, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Abu-Ameerh, M.; Mohammad, H.; Dardas, Z.; Barham, R.; Ali, D.; Bijawi, M.; Tawalbeh, M.; Amr, S.; Hatmal, M.M.; Al-Bdour, M.; et al. Extending the spectrum of CLRN1- and ABCA4-associated inherited retinal dystrophies caused by novel and recurrent variants using exome sequencing. Mol. Genet. Genom. Med. 2020, 8, e1123. [Google Scholar] [CrossRef] [PubMed]
- Al-Bdour, M.; Pauleck, S.; Dardas, Z.; Barham, R.; Ali, D.; Amr, S.; Mustafa, L.; Abu-Ameerh, M.; Maswadi, R.; Azab, B.; et al. Clinical heterogeneity in retinitis pigmentosa caused by variants in RP1 and RLBP1 in five extended consanguineous pedigrees. Mol. Vis. 2020, 26, 445–458. [Google Scholar]
- Beryozkin, A.; Zelinger, L.; Bandah-Rozenfeld, D.; Harel, A.; Strom, T.A.; Merin, S.; Chowers, I.; Banin, E.; Sharon, D. Mutations in CRB1 are a relatively common cause of autosomal recessive early-onset retinal degeneration in the Israeli and Palestinian populations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2068–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benayoun, L.; Spiegel, R.; Auslender, N.; Abbasi, A.H.; Rizel, L.; Hujeirat, Y.; Salama, I.; Garzozi, H.J.; Allon-Shalev, S.; Ben-Yosef, T. Genetic heterogeneity in two consanguineous families segregating early onset retinal degeneration: The pitfalls of homozygosity mapping. Am. J. Med. Genet. A 2009, 149a, 650–656. [Google Scholar] [CrossRef]
- Abouzeid, H.; Li, Y.; Maumenee, I.H.; Dharmaraj, S.; Sundin, O. A G1103R mutation in CRB1 is co-inherited with high hyperopia and Leber congenital amaurosis. Ophthalmic Genet. 2006, 27, 15–20. [Google Scholar] [CrossRef]
- Zernant, J.; Kulm, M.; Dharmaraj, S.; den Hollander, A.I.; Perrault, I.; Preising, M.N.; Lorenz, B.; Kaplan, J.; Cremers, F.P.; Maumenee, I.; et al. Genotyping microarray (disease chip) for Leber congenital amaurosis: Detection of modifier alleles. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3052–3059. [Google Scholar] [CrossRef]
- Hanein, S.; Perrault, I.; Gerber, S.; Tanguy, G.; Barbet, F.; Ducroq, D.; Calvas, P.; Dollfus, H.; Hamel, C.; Lopponen, T.; et al. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum. Mutat. 2004, 23, 306–317. [Google Scholar] [CrossRef] [Green Version]
- den Hollander, A.I.; Davis, J.; van der Velde-Visser, S.D.; Zonneveld, M.N.; Pierrottet, C.O.; Koenekoop, R.K.; Kellner, U.; van den Born, L.I.; Heckenlively, J.R.; Hoyng, C.B.; et al. CRB1 mutation spectrum in inherited retinal dystrophies. Hum. Mutat. 2004, 24, 355–369. [Google Scholar] [CrossRef]
- Weisschuh, N.; Feldhaus, B.; Khan, M.I.; Cremers, F.P.M.; Kohl, S.; Wissinger, B.; Zobor, D. Molecular and clinical analysis of 27 German patients with Leber congenital amaurosis. PLoS ONE 2018, 13, e0205380. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Kousal, B.; Dudakova, L.; Gaillyova, R.; Hejtmankova, M.; Diblik, P.; Michaelides, M.; Liskova, P. Phenotypic features of CRB1-associated early-onset severe retinal dystrophy and the different molecular approaches to identifying the disease-causing variants. Graefe′s Arch. Clin. Exp. Ophthalmol. 2016, 254, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; De Roach, J.N.; McLaren, T.L.; Montgomery, H.E.; Hoffmann, L.H.; Campbell, I.R.; Chen, F.K.; Mackey, D.A.; Lamey, T.M. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol. Genet. Genom. Med. 2017, 5, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Prokudin, I.; Yu, C.; Liang, J.; Xie, Y.; Flaherty, M.; Tian, L.; Crofts, S.; Wang, F.; Snyder, J.; et al. Advantage of Whole Exome Sequencing over Allele-Specific and Targeted Segment Sequencing in Detection of Novel TULP1 Mutation in Leber Congenital Amaurosis. Ophthalmic Genet. 2015, 36, 333–338. [Google Scholar] [CrossRef]
- Glockle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.H.; Garzozi, H.J.; Ben-Yosef, T. A novel splice-site mutation of TULP1 underlies severe early-onset retinitis pigmentosa in a consanguineous Israeli Muslim Arab family. Mol. Vis. 2008, 14, 675–682. [Google Scholar]
- Boulanger-Scemama, E.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.P.; Letexier, M.; Souied, E.; et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: Mutation spectrum and new genotype-phenotype correlation. Orphanet J. Rare Dis. 2015, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Aleman, T.S.; Soumittra, N.; Cideciyan, A.V.; Sumaroka, A.M.; Ramprasad, V.L.; Herrera, W.; Windsor, E.A.; Schwartz, S.B.; Russell, R.C.; Roman, A.J.; et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5944–5954. [Google Scholar] [CrossRef] [Green Version]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1219. [Google Scholar] [CrossRef] [Green Version]
- Bolinches-Amoros, A.; Leon, M.; Del Buey Furio, V.; Marfany, G.; Gonzalez-Duarte, R.; Erceg, S.; Lukovic, D. Generation of an iPSC line from a retinitis pigmentosa patient carrying a homozygous mutation in CERKL and a healthy sibling. Stem Cell Res. 2019, 38, 101455. [Google Scholar] [CrossRef] [PubMed]
- Ezquerra-Inchausti, M.; Anasagasti, A.; Barandika, O.; Garay-Aramburu, G.; Galdos, M.; Lopez de Munain, A.; Irigoyen, C.; Ruiz-Ederra, J. A new approach based on targeted pooled DNA sequencing identifies novel mutations in patients with Inherited Retinal Dystrophies. Sci. Rep. 2018, 8, 15457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Avela, K.; Sankila, E.M.; Seitsonen, S.; Kuuluvainen, L.; Barton, S.; Gillies, S.; Aittomaki, K. A founder mutation in CERKL is a major cause of retinal dystrophy in Finland. Acta Ophthalmol. 2018, 96, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Gil, N.; Gonzalez-Del Pozo, M.; Martin-Sanchez, M.; Mendez-Vidal, C.; Rodriguez-de la Rua, E.; Borrego, S.; Antinolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, 41937. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, V.W.; Feng, Y.; Tian, X.; Li, F.Y.; Truong, C.; Wang, G.; Chiang, P.W.; Lewis, R.A.; Wong, L.J. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6213–6223. [Google Scholar] [CrossRef]
- Rodriguez-Flores, J.L.; Fakhro, K.; Hackett, N.R.; Salit, J.; Fuller, J.; Agosto-Perez, F.; Gharbiah, M.; Malek, J.A.; Zirie, M.; Jayyousi, A.; et al. Exome sequencing identifies potential risk variants for Mendelian disorders at high prevalence in Qatar. Hum. Mutat. 2014, 35, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Nishiguchi, K.M.; Tearle, R.G.; Liu, Y.P.; Oh, E.C.; Miyake, N.; Benaglio, P.; Harper, S.; Koskiniemi-Kuendig, H.; Venturini, G.; Sharon, D.; et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc. Natl. Acad. Sci. USA 2013, 110, 16139–16144. [Google Scholar] [CrossRef] [Green Version]
- Bornancin, F.; Mechtcheriakova, D.; Stora, S.; Graf, C.; Wlachos, A.; Devay, P.; Urtz, N.; Baumruker, T.; Billich, A. Characterization of a ceramide kinase-like protein. Biochim. Biophys. Acta 2005, 1687, 31–43. [Google Scholar] [CrossRef]
- Tuson, M.; Marfany, G.; Gonzalez-Duarte, R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am. J. Hum. Genet. 2004, 74, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; Huang, F.; Wu, K.C.; Wu, J.; Chen, J.; Pang, C.P.; Lu, F.; Qu, J.; Jin, Z.B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Pierrache, L.H.M.; Messchaert, M.; Thiadens, A.; Haer-Wigman, L.; de Jong-Hesse, Y.; van Zelst-Stams, W.A.G.; Collin, R.W.J.; Klaver, C.C.W.; van den Born, L.I. Extending the Spectrum of EYS-Associated Retinal Disease to Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Messchaert, M.; Haer-Wigman, L.; Khan, M.I.; Cremers, F.P.M.; Collin, R.W.J. EYS mutation update: In silico assessment of 271 reported and 26 novel variants in patients with retinitis pigmentosa. Hum. Mutat. 2018, 39, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Haines, R.L.; Codlin, S.; Mole, S.E. The fission yeast model for the lysosomal storage disorder Batten disease predicts disease severity caused by mutations in CLN3. Dis. Model. Mech. 2009, 2, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Munroe, P.B.; Mitchison, H.M.; O’Rawe, A.M.; Anderson, J.W.; Boustany, R.M.; Lerner, T.J.; Taschner, P.E.; de Vos, N.; Breuning, M.H.; Gardiner, R.M.; et al. Spectrum of mutations in the Batten disease gene, CLN3. Am. J. Hum. Genet. 1997, 61, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Zaghloul, N.A.; Liu, Y.; Gerdes, J.M.; Gascue, C.; Oh, E.C.; Leitch, C.C.; Bromberg, Y.; Binkley, J.; Leibel, R.L.; Sidow, A.; et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10602–10607. [Google Scholar] [CrossRef] [Green Version]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, R.H.; Li, Z.; Abd El Aziz, M.M.; Mackay, D.S.; Eljinini, M.A.; Zeidan, M.; Moore, A.T.; Bhattacharya, S.S.; Webster, A.R. Biallelic mutation of protocadherin-21 (PCDH21) causes retinal degeneration in humans. Mol. Vis. 2010, 16, 46–52. [Google Scholar]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Perrault, I.; Hanein, S.; Gerber, S.; Barbet, F.; Ducroq, D.; Dollfus, H.; Hamel, C.; Dufier, J.L.; Munnich, A.; Kaplan, J.; et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am. J. Hum. Genet. 2004, 75, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Beryozkin, A.; Zelinger, L.; Bandah-Rozenfeld, D.; Shevach, E.; Harel, A.; Storm, T.; Sagi, M.; Eli, D.; Merin, S.; Banin, E.; et al. Identification of mutations causing inherited retinal degenerations in the israeli and palestinian populations using homozygosity mapping. Investig. Ophthalmol Vis. Sci. 2014, 55, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.A.; Janecke, A.R.; Lange, J.; Feathers, K.L.; Hubner, C.A.; McHenry, C.L.; Stockton, D.W.; Rammesmayer, G.; Lupski, J.R.; Antinolo, G.; et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum. Mol. Genet. 2005, 14, 3865–3875. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.S.; Adams, M.; Nassar, N.; Bizon, C.; Lee, K.; Schmitt, C.P.; Wilhelmsen, K.C.; Evans, J.P. An informatics approach to analyzing the incidentalome. Genet. Med. 2013, 15, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Nishiguchi, K.M.; Rivolta, C. Genes associated with retinitis pigmentosa and allied diseases are frequently mutated in the general population. PLoS ONE 2012, 7, e41902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancati, F.; Barrano, G.; Silhavy, J.L.; Marsh, S.E.; Travaglini, L.; Bielas, S.L.; Amorini, M.; Zablocka, D.; Kayserili, H.; Al-Gazali, L.; et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am. J. Hum. Genet. 2007, 81, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita, D.A.; Mole, S.E.; Minassian, B.A. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016, 18, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, H.; Tuan, H.F.; Nguyen, D.H.; Sun, V.; Keser, V.; Bowne, S.J.; Sullivan, L.S.; Luo, H.; Zhao, L.; et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 2014, 133, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.A.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.W.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, V.M.; Nassisi, M.; Solis Hernandez, C.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients With Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021. [Google Scholar] [CrossRef]
- Mizobuchi, K.; Hayashi, T.; Yoshitake, K.; Fujinami, K.; Tachibana, T.; Tsunoda, K.; Iwata, T.; Nakano, T. Novel homozygous CLN3 missense variant in isolated retinal dystrophy: A case report and electron microscopic findings. Mol. Genet. Genom. Med. 2020, 8, e1308. [Google Scholar] [CrossRef]
- Chen, F.K.; Zhang, X.; Eintracht, J.; Zhang, D.; Arunachalam, S.; Thompson, J.A.; Chelva, E.; Mallon, D.; Chen, S.C.; McLaren, T.; et al. Clinical and molecular characterization of non-syndromic retinal dystrophy due to c.175G>A mutation in ceroid lipofuscinosis neuronal 3 (CLN3). Doc. Ophthalmol. 2019, 138, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Hull, S.; Roepman, R.; van den Born, L.I.; Oud, M.M.; de Vrieze, E.; Hetterschijt, L.; Letteboer, S.J.F.; van Beersum, S.E.C.; Blokland, E.A.; et al. Missense mutations in the WD40 domain of AHI1 cause non-syndromic retinitis pigmentosa. J. Med. Genet. 2017, 54, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.; Glass, I. Joubert Syndrome. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beryozkin, A.; Shevah, E.; Kimchi, A.; Mizrahi-Meissonnier, L.; Khateb, S.; Ratnapriya, R.; Lazar, C.H.; Blumenfeld, A.; Ben-Yosef, T.; Hemo, Y.; et al. Whole Exome Sequencing Reveals Mutations in Known Retinal Disease Genes in 33 out of 68 Israeli Families with Inherited Retinopathies. Sci. Rep. 2015, 5, 13187. [Google Scholar] [CrossRef]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [Green Version]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, Y.; Jiao, X.; Jin, C.; Jiang, D.; Tanwar, M.; Ma, Z.; Huang, L.; Ma, X.; Sun, W.; et al. Homozygosity Mapping and Genetic Analysis of Autosomal Recessive Retinal Dystrophies in 144 Consanguineous Pakistani Families. Investig. Ophthalmol Vis. Sci. 2017, 58, 2218–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmaninejad, A.; Bedoni, N.; Ravesh, Z.; Quinodoz, M.; Shoeibi, N.; Mojarrad, M.; Pasdar, A.; Rivolta, C. Whole exome sequencing and homozygosity mapping reveals genetic defects in consanguineous Iranian families with inherited retinal dystrophies. Sci. Rep. 2020, 10, 19413. [Google Scholar] [CrossRef]
- Hosono, K.; Nishina, S.; Yokoi, T.; Katagiri, S.; Saitsu, H.; Kurata, K.; Miyamichi, D.; Hikoya, A.; Mizobuchi, K.; Nakano, T.; et al. Molecular Diagnosis of 34 Japanese Families with Leber Congenital Amaurosis Using Targeted Next Generation Sequencing. Sci. Rep. 2018, 8, 8279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibi, I.; Chebil, A.; Falfoul, Y.; Allaman-Pillet, N.; Kort, F.; Schorderet, D.F.; Matri, L.E. Corrigendum: Identifying mutations in Tunisian families with retinal dystrophy. Sci. Rep. 2017, 7, 46776. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Families | AR | Sporadic | Total | Consanguineous |
|---|---|---|---|---|
| Solved (n) | 26 | 13 | 39 | 31 |
| Unsolved (n) | 9 | 7 | 16 | 8 |
| Total, n (%) | 35 (64%) | 20 (36%) | 55 (100%) | 39 (71%) |
| Detection rate of potential DCVs (%) | 74.3% | 65% | 71% | 79.5% |
| Family ID | Gene | Variant Coordinate hg19 | HGVS Variant Nomenclature | dbSNP ID | gnomAD v3.1.1 Frequency | Zygo. | Segregation | ClinVar * | In silico Predictions SIFT, PP, MT | ACMG Classification | References | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Highest | SAS | ME $ | |||||||||||
| IRD03 | CRB1 | Chr1:197404300 | NM_201253.2:c.3307G > A; p.(Gly1103Arg) | rs62636275 | 2.1 × 10−4 | 2.1 × 10−4 | NA | Hom | Not done | P | D, D, A | LP | [25,26,27,28,29] |
| IRD47 | CRB1 | Chr1:197404300 | NM_201253.2:c.3307G > A; p.(Gly1103Arg) | rs62636275 | 2.1 × 10−4 | 2.1 × 10−4 | NA | Hom | Not done | P | D, D, A | LP | [25,26,27,28,29] |
| IRD14 | CRB1 | Chr1:197390691 | NM_201253.2:c.1733T > A; p.(Val578Glu) | rs1266363944 | NA | NA | NA | Hom | Not done | LP | D, D, DC | VUS | [25,30] |
| IRD19 | CRB1 | Chr1:197390691 | NM_201253.2:c.1733T > A; p.(Val578Glu) | rs1266363944 | NA | NA | NA | Hom | Not done | LP | D, D, DC | VUS | [25,30] |
| IRD28 | CRB1 | Chr1:197390691 | NM_201253.2:c.1733T > A; p.(Val578Glu) | rs1266363944 | NA | NA | NA | Hom | Yes | LP | D, D, DC | VUS | [25,30] |
| IRD33 | CRB1 | Chr1:197396763 | NM_201253.2:c.2308G > A; p.(Gly770Ser) | rs767648174 | 6.6 × 10−5 | NA | NA | Hom | Not done | D, D, DC | LP | [31,32,33] | |
| IRD39 | CRB1 | Chr1:197390802 | NM_201253.2:c.1844G > T; p.(Gly615Val) | 1.5 × 10−5 | NA | NA | Hom | Yes | D, D, DC | LP | [25] | ||
| IRD09 | TULP1 | Chr6:35473549 | NM_003322.3:c.1081C > T; p.(Arg361 *) | 2.4 × 10−5 | NA | NA | Hom | Yes | P | [34,35,36] | |||
| IRD12 | TULP1 | Chr6:35467755 | NM_003322.3: c.1495 + 2dupT | rs1581735836 | NA | NA | NA | Hom | Yes | P | [37] | ||
| IRD31 | TULP1 | Chr6:35473543 | NM_003322.3:c.1087G > A; p.(Gly363Arg) | 4.8 × 10−4 | NA | NA | Hom | Not done | D, D, DC | VUS | [38] | ||
| IRD11 | CERKL | Chr2:182468594 | NM_001030311.2: c.450_451delAT; p.(Ile150Metfs * 3) | NA | NA | NA | Hom | Yes | P | [19] | |||
| IRD18 | CERKL | Chr2:182413318 | NM_001030311.2: c.1164_1165delTG; p.(Cys388 *) | rs776727320 | 1.1 × 10−3 | NA | NA | Hom | Yes | P | [19,39] | ||
| IRD35 | CERKL | Chr2:182423344 | NM_001030311.2: c.847C > T; p.(Arg283 *) | rs121909398 | 9.6 × 10−4 | NA | NA | Com. het | Yes | P | P | [32,40,41,42,43,44,45,46,47,48,49,50,51,52] | |
| IRD35 | CERKL | Chr2:182468563 | NM_001030311.2: c.481 + 1G > A | NA | NA | NA | Com.het | Yes | P | Novel | |||
| IRD02 | CLRN1 | Chr3:150659368 | NM_001195794.1: c.433 + 1G > A | rs201205811 | NA | NA | NA | Hom | Yes | P | [23] | ||
| IRD36 | CLRN1 | Chr3:150659479 | NM_001195794.1:c.323T > C; p.(Leu108Pro) | 4.6 × 10−4 | NA | NA | Hom | Yes | D, D, DC | VUS | [23] | ||
| IRD05 | RP1 | Chr8:55537568 | NM_006269.1: c.1126C > T; p.(Arg376*) | rs760689800 | NA | NA | NA | Hom | Yes | P | [24] | ||
| IRD08 | RP1 | Chr8:55534133 | NM_006269.1: c.607G > A; p.(Gly203Arg) | rs786205589 | NA | NA | NA | Hom | Yes | LP | D, D, DC | LP | [24] |
| IRD22 | RP1 | Chr8:55534133 | NM_006269.1: c.607G > A; p.(Gly203Arg) | rs786205589 | NA | NA | NA | Hom | Yes | LP | D, D, DC | LP | [24] |
| IRD10 | RLBP1 | Chr15:89761858 | NM_000326.4: c.79delA; p.(Thr27Profs * 26) | rs1567124404 | NA | NA | NA | Hom | Yes | P | P | [24] | |
| IRD17 | RLBP1 | Chr15:89758418 | NM_000326.4: c.398delC; p.(Pro133Glnfs * 126) | NA | NA | NA | NA | Hom | Yes | P | [24] | ||
| IRD26 | C8orf37 | Chr8:96281262 | NM_177965.3: c.155 + 1G > A | 6.5 × 10−5 | NA | NA | Hom | Yes | P | Novel | |||
| IRD41 | C8orf37 | Chr8:96281262 | NM_177965.3: c.155 + 1G > A | 6.5 × 10−5 | NA | NA | Hom | Yes | P | Novel | |||
| IRD02 | ABCA4 | Chr1:94480098 | NM_000350.2: c.5460 + 1G > A | rs61753030 | 2.4 × 10−5 | NA | NA | Hom | Yes | P | [23] | ||
| IRD24 | ABCA4 | Chr1:94528780 | NM_000350.2: c.1648G > A; p.(Gly550Arg) | rs61748558 | 1.4 × 10−5 | NA | NA | Hom | Yes | LP | D, D, DC | LP | [23] |
| IRD48 | ABCA4 | Chr1:94480098 | NM_000350.2: c.5460 + 1G > A | rs61753030 | 2.4 × 10−5 | NA | NA | Hom | Yes | P | [23] | ||
| IRD37 | USH2A | Chr1:216019303 | NM_206933.2: c.8917_8918del; p.(Leu2973Lysfs * 79) | NA | NA | NA | Hom | Not done | P | [53] | |||
| IRD04 | MAK | Chr6:10804098 | NM_001242957.1: c.518G > T; p.(Arg173Ile) | NA | NA | NA | Hom | Yes | D, D, DC | VUS | Novel | ||
| IRD07 | EYS | Chr6:65655759 | NM_001142800.1: c.2308C > T; p.(Gln770 *) | NA | NA | NA | Hom | Not done | P | [54,55] | |||
| IRD16 | CLN3 | Chr16:28493482 | NM_001042432.1: c.1000C > T; p.(Arg334Cys) | rs386833694 | NA | NA | NA | Hom | Not done | LP | D, D, DC | VUS | [56,57] |
| IRD20 | BBS2 | Chr16:56536365 | NM_031885.3:c.944G > A; p.(Arg315Gln) | rs544773389 | NA | NA | NA | Hom | Not done | VUS | D, D, DC | VUS | [58,59] |
| IRD27 | CDHR1 | Chr10:85957581 | NM_033100.3:c.338delG; p.(Gly113Alafs * 2) | rs747425652 | NA | NA | NA | Hom | Yes | P | [60] | ||
| IRD38 | IMPDH1 | Chr7:128040188 | NM_000883.3:c.835T > G; p.(Leu279Val) | NA | NA | NA | Het | Yes | D, D, DC | LP | Novel | ||
| IRD46 | MERTK | Chr2:112779847 | NM_006343.2:c.2362G > A; p.(Val788Met) | rs769691218 | 6.5 × 10−5 | NA | NA | Hom | Not done | D, D, DC | VUS | ClinVar | |
| IRD56 | CNGB1 | Chr16:57937858 | NM_001297.4:c.2662G > A; p.(Ala888Thr) | rs368328328 | 8.3 x10−4 | 8.3 × 10−4 | NA | Hom | Yes | D, D, DC | VUS | ClinVar | |
| IRD06 | RP1L1 | Chr8:10469520 | NM_178857.5:c.2088C > A; p.(Cys696 *) | NA | NA | NA | Hom | Yes | P | Novel | |||
| IRD21 | RDH12 | Chr14:68192803 | NM_152443.2:c.379G > T; p.(Gly127 *) | rs104894474 | NA | NA | NA | Hom | Not done | P | P | [47,61,62] | |
| IRD55 | RDH12 | Chr14:68196070 | NM_152443.2:c.821T > C; p.(Leu274Pro) | NA | NA | NA | Hom | Yes | D, D, DC | VUS | [63,64] | ||
| IRD25 | RDH12 | Chr14:68196070 | NM_152443.2:c.821T > C; p.(Leu274Pro) | NA | NA | NA | Hom | Yes | D, D, DC | VUS | [63,64] | ||
| IRD49 | AHI1 | Chr6:135752384 | NM_017651.4:c.2335G > A; p.(Asp779Asn) | 3.2 × 10−3 | NA | 3.2 × 10−3 | Hom | Yes | VUS | T, D, N | VUS | ClinVar | |
| IRD50 | CEP290 | Chr12:88479860 | NM_025114.3:c.4393C > T; p.(Arg1465 *) | rs539400286 | 2.1 × 10−4 | 2.1 × 10−4 | NA | Com. het | Yes | P | P | [46,47,65,66,67,68] | |
| IRD50 | CEP290 | Chr12:88447469 | NM_025114.3:c.7089A > T; p.(Glu2363Asp) | NA | NA | NA | Com. het | Yes | T, B, N | VUS | Novel | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azab, B.; Dardas, Z.; Aburizeg, D.; Al-Bdour, M.; Abu-Ameerh, M.; Saleh, T.; Barham, R.; Maswadi, R.; Ababneh, N.A.; Alsalem, M.; et al. Unique Variant Spectrum in a Jordanian Cohort with Inherited Retinal Dystrophies. Genes 2021, 12, 593. https://doi.org/10.3390/genes12040593
Azab B, Dardas Z, Aburizeg D, Al-Bdour M, Abu-Ameerh M, Saleh T, Barham R, Maswadi R, Ababneh NA, Alsalem M, et al. Unique Variant Spectrum in a Jordanian Cohort with Inherited Retinal Dystrophies. Genes. 2021; 12(4):593. https://doi.org/10.3390/genes12040593
Chicago/Turabian StyleAzab, Bilal, Zain Dardas, Dunia Aburizeg, Muawyah Al-Bdour, Mohammed Abu-Ameerh, Tareq Saleh, Raghda Barham, Ranad Maswadi, Nidaa A Ababneh, Mohammad Alsalem, and et al. 2021. "Unique Variant Spectrum in a Jordanian Cohort with Inherited Retinal Dystrophies" Genes 12, no. 4: 593. https://doi.org/10.3390/genes12040593