
Activity of Lactobacillus brevis Alcohol Dehydrogenase on Primary and Secondary Alcohol Biofuel Precursors


Abstract
:
1. Introduction

2. Experimental Section
2.1. Chemicals
2.2. Microorganisms and Media
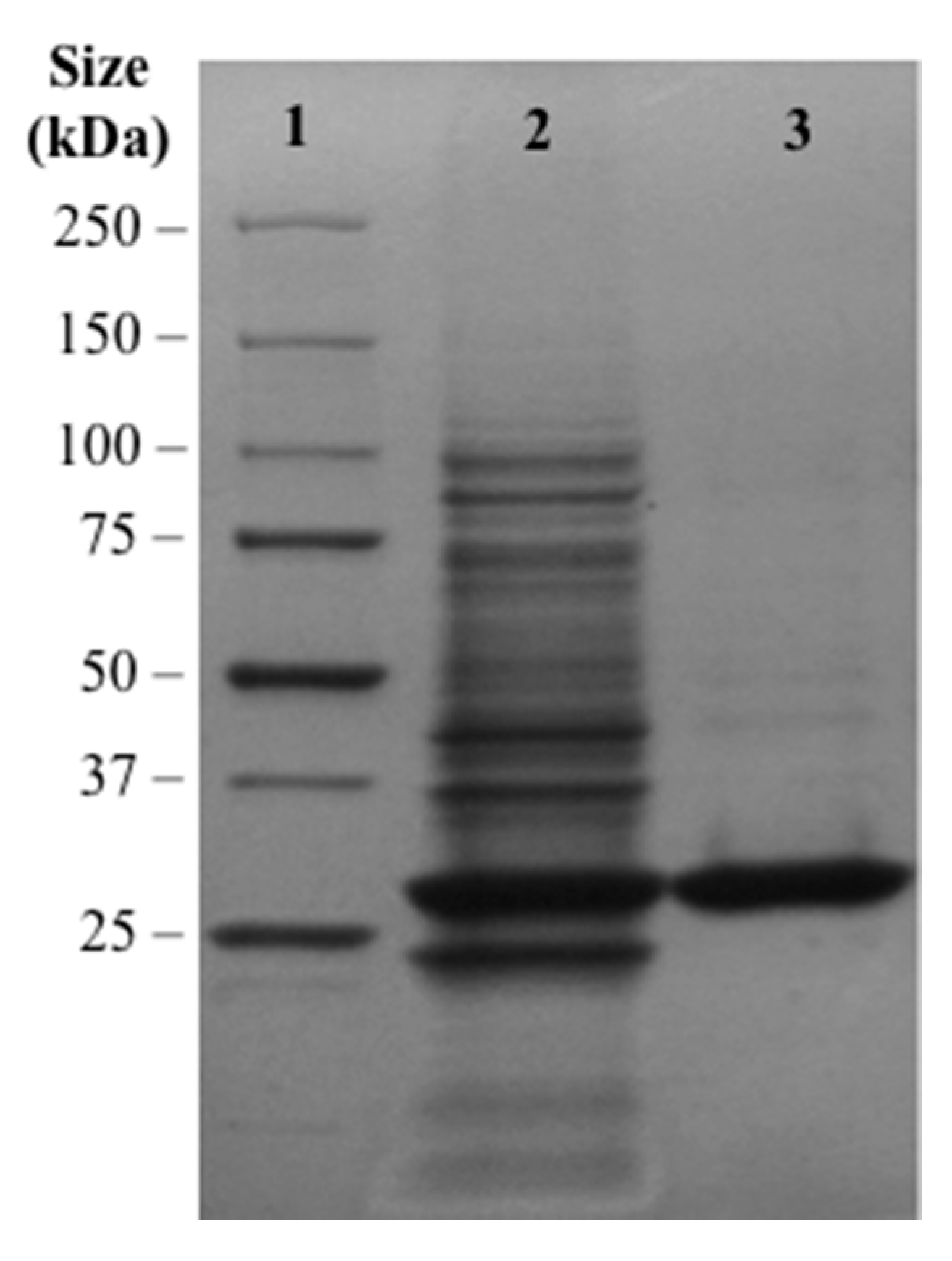
2.3. Plasmid Construction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype or Description | Source |
|---|---|---|
| E. coli NEB10-beta | araD139∆(ara-leu)7697 fhuA lacX74 galK (ϕ80 ∆(lacZ)M15) mcrA galU recA1 endA1 nupG rpsL (StrR) ∆(mrr-hsdRMS-mcrBC) | New England Biolabs |
| E. coli BW25113 | F’ λ− ∆(araD-araB)567, ∆lacZ4787(::rrnB-3), lambda−, rph-1, ∆(rhaD-rhaB)568, hsdR514 | CGSC |
| E. coli BW25113(DE3) | λDE3 lysogen of BW25113λ | This study |
| L. brevis LB19 | Genetic source of LbADH | CIRM-BIA |
| Plasmid | Features | Source |
| pACYCDuet-1 | Expression vector, Cmr, PT7, pACYC184 Ori | Novagen |
| pACYC- LbADH | LbADH from L. brevis LB19 inserted between NdeI and XhoI sites of pACYCDuet-1 | This study |
| pACYC- LbADH-His | LbADH from L. brevis LB19 inserted between BamHI and EcoRI sites of pACYCDuet-1 | This study |
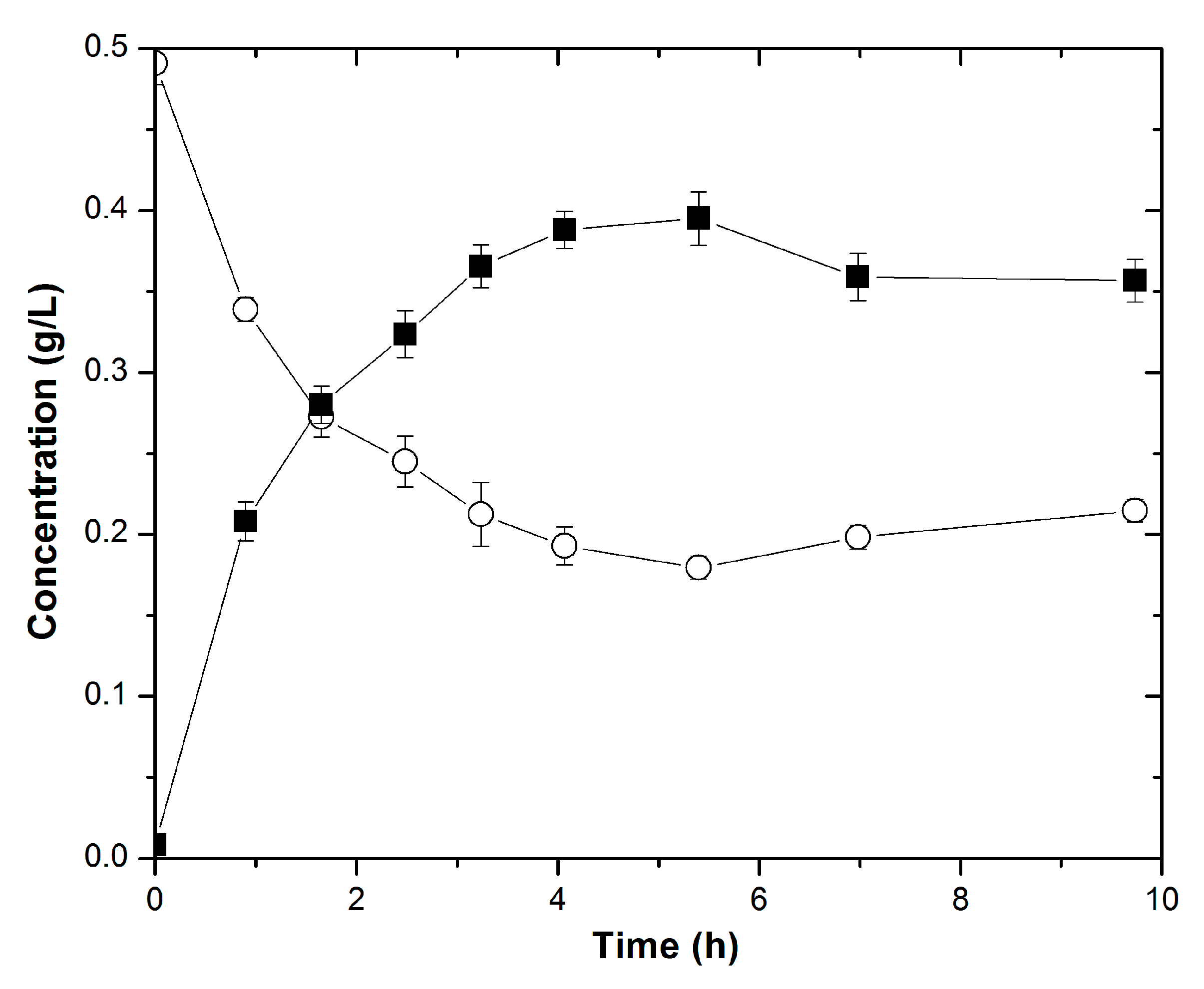
2.4. Whole Cell Conversion of 2-Butanone to 2-Butanol by E. coli Growing Cells
2.5. Whole Cell Conversion of 2-Butanone to 2-Butanol by E. coli Resting Cells
2.6. Metabolite Analyses
2.7. In Vitro Enzyme Assays
2.8. Modeling Enzyme Kinetics
3. Results and Discussion
3.1. Confirming the In Vivo Function of Recombinant LbADH for 2-Butanol Production

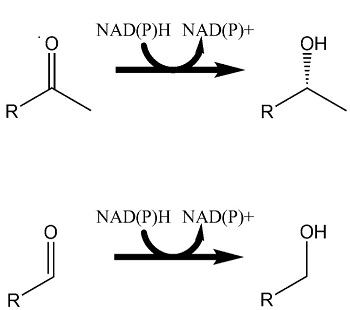
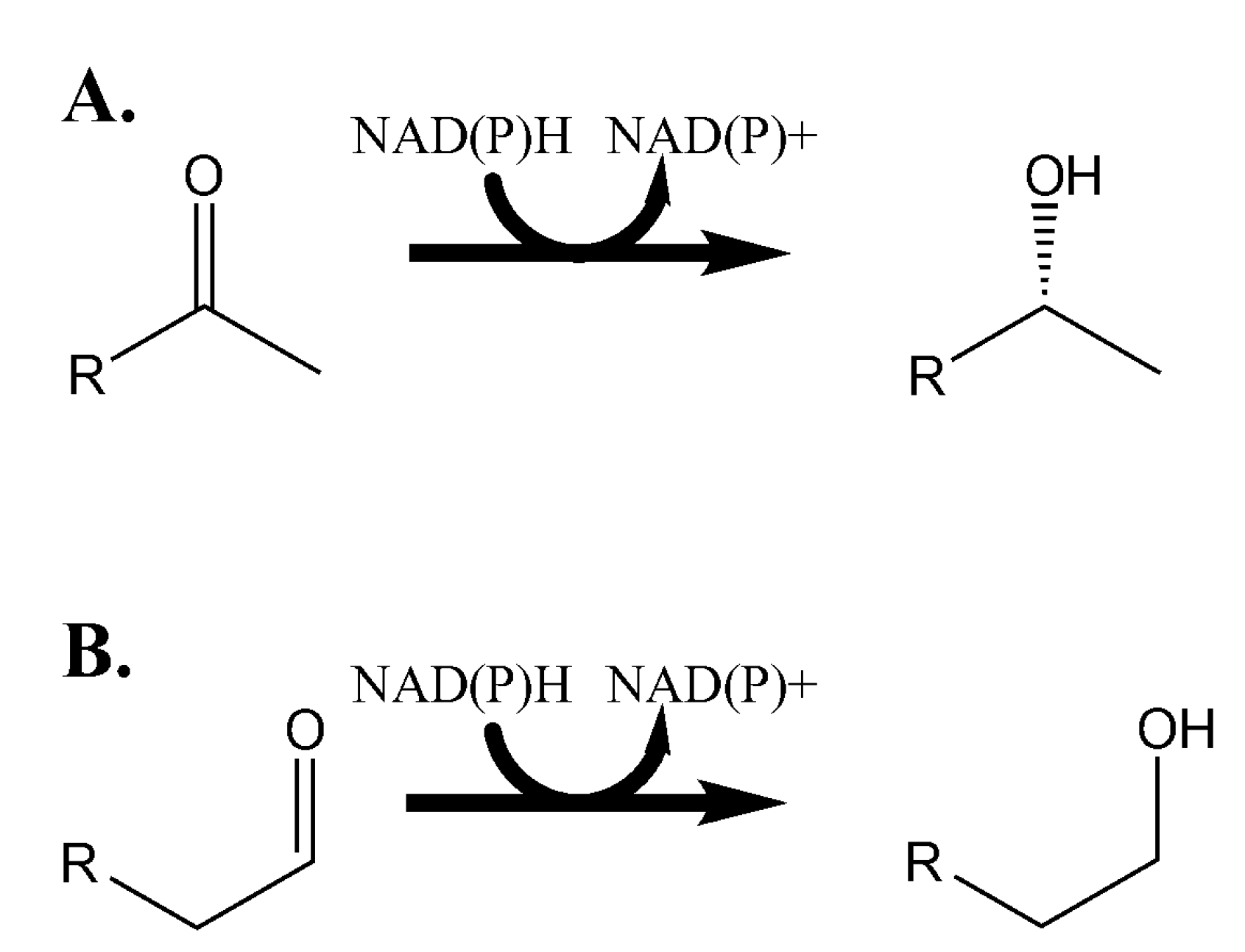
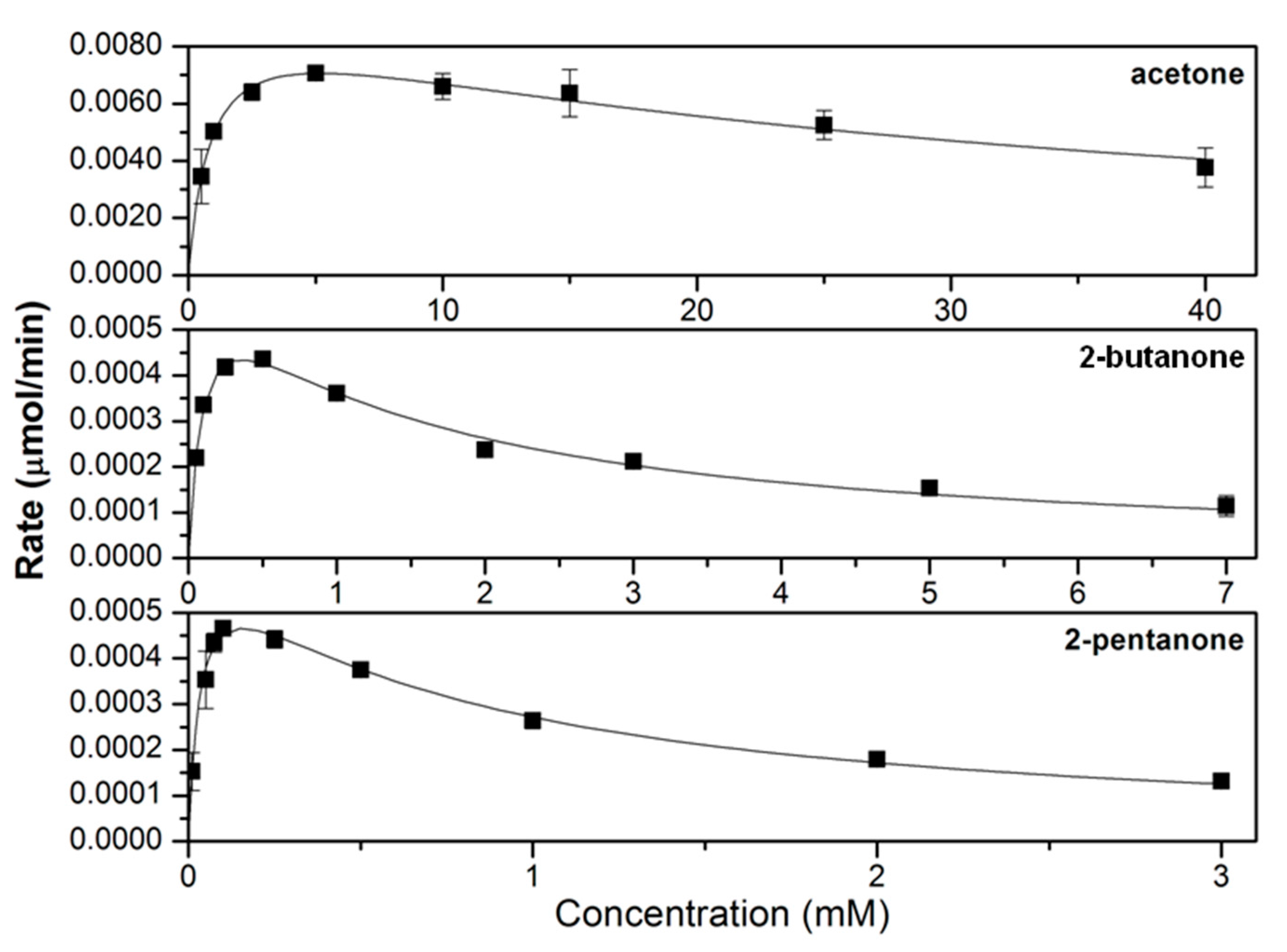
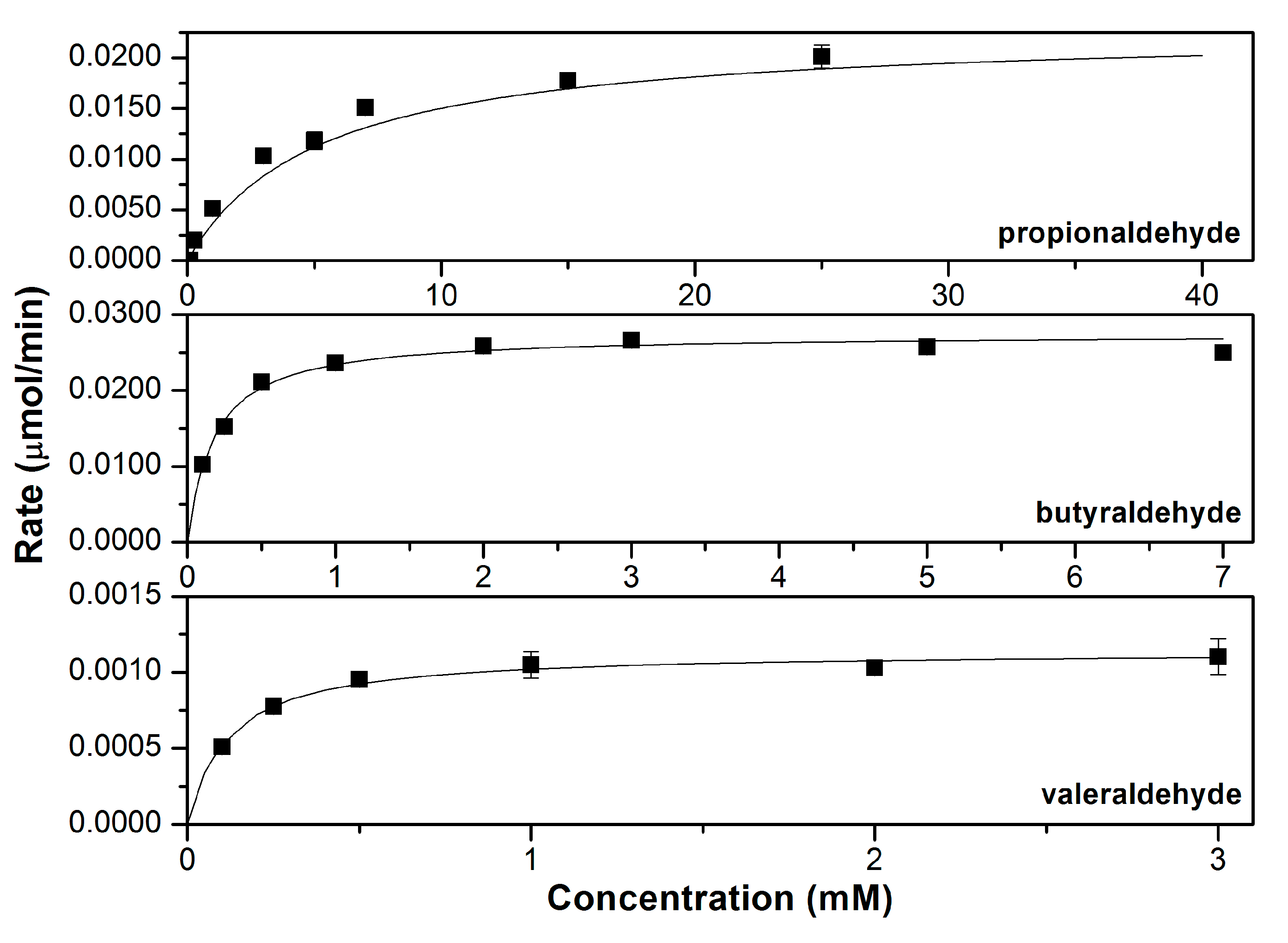
3.2. Characterizing the In Vitro Activity of Recombinant LbADH on Short-Chain 2-Alkanones and Aldehydes



| Product Alcohol | Substrate | kcat (s−1) | KM (mM) | KI (mM) | kcat/KM (mM−1s−1) |
|---|---|---|---|---|---|
| 2° | Acetone | 1.52 ± 0.01 | 0.88 ± 0.16 | 30.7 ± 6.8 | 1.73 ± 0.05 |
| 2-Butanone | 0.11 ± 0.01 | 0.10 ± 0.02 | 1.34 ± 0.26 | 1.12 ± 0.28 | |
| 2-Pentanone | 0.11 ± 0.01 | 0.04 ± 0.01 | 0.68 ± 0.17 | 3.03 ± 1.05 | |
| 1° | Propionaldehyde | 3.36 ± 0.19 | 3.1 ± 0.7 | N.D. | 1.09 ± 0.25 |
| Butyraldehyde | 4.42 ± 0.11 | 0.17 ± 0.02 | N.D. | 25.5 ± 3.5 | |
| Valeraldehyde | 0.18 ± 0.01 | 0.12 ± 0.03 | N.D. | 1.57 ± 0.41 |
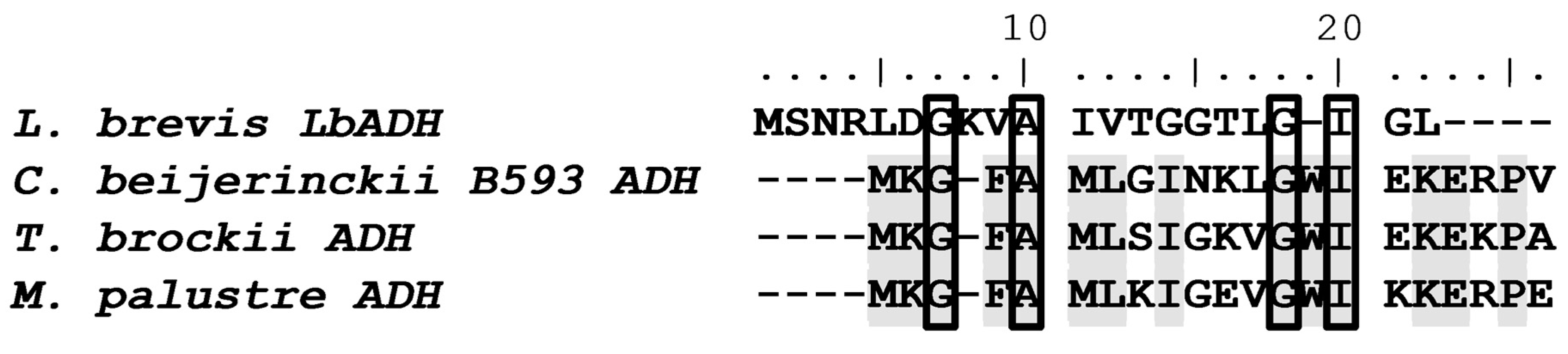
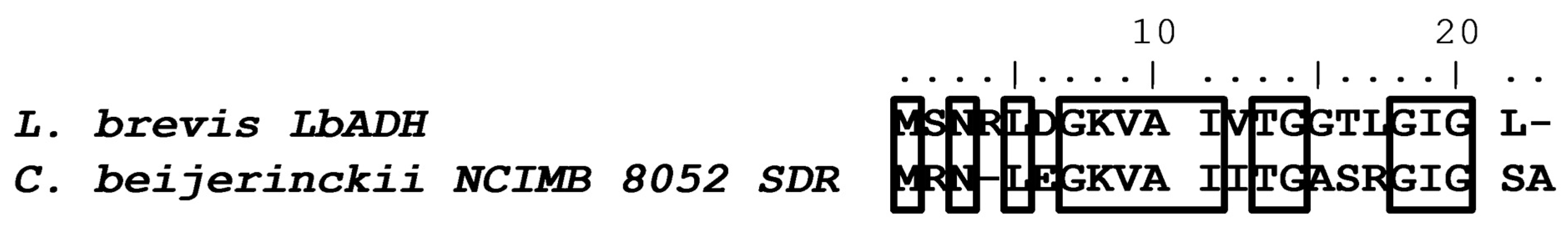
3.3. Comparing LbADH with Other Bacterial ADHs
| Organism Enzyme(s) | Substrate(s) | KM (mM) | kcat (s-1) | kcat/KM (mM−1s−1) | Relative Activity | Reference |
|---|---|---|---|---|---|---|
| L. brevis LB19 LbADH | 2-butanone | 0.096 | 0.107 | 1.12 | - | This Study |
| Clostridium beijerinckii NRRL B593 ADH | acetone 2-butanone | 0.98 1.5 | 139 64.2 | 142 43.3 | - - | [26] |
| Rhodococcus sp. GK1 SADH | acetone 2-octanone | 65 2.1 | - - | - - | - - | [21] |
| C. acetobutylicum ATCC 824 BDH I BDH II | Butyraldehyde butyraldehyde | 3.6 14 | - - | - - | - - | [25] |
| Burkholderia sp. AIU652 ADH | acetone 2-butanone 2-pentanone | 0.065 0.040 - | - - - | - - - | 100% 83% 44% | [19] |
| Pseudomonas sp. PED ADH | 2-butanone 2-pentanone | - - | - - | - - | 100% 6% | [20] |


4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hummel, W. New alcohol dehydrogenases for the synthesis of chiral compounds. Adv. Biochem. Eng./Biotechnol. 1997, 58, 145–184. [Google Scholar]
- Nakamura, K.; Yamanaka, R.; Matsuda, T.; Harada, T. Recent developments in asymmetric reduction of ketones with biocatalysts. Tetrahedron-Asymmetry 2003, 14, 2659–2681. [Google Scholar] [CrossRef]
- Leuchs, S.; Greiner, L. Alcohol dehydrogenase from Lactobacillus brevis: A versatile robust catalyst for enantioselective transformations. Chem. Biochem. Eng. Q. 2011, 25, 267–281. [Google Scholar]
- Trivedi, A.H.; Spiess, A.C.; Daussmann, T.; Buchs, J. Study on mesophilic and thermophilic alcohol dehydrogenases in gas-phase reaction. Biotechnol. Prog. 2006, 22, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Thorey, P.; Knez, Z.; Habulin, M. Alcohol dehydrogenase in non-aqueous media using high-pressure technologies: Reaction set-up and deactivation determination. J. Chem. Technol. Biotechnol. 2010, 85, 1011–1016. [Google Scholar] [CrossRef]
- Schlieben, N.H.; Niefind, K.; Muller, J.; Riebel, B.; Hummel, W.; Schomburg, D. Atomic resolution structures of R-specific alcohol dehydrogenase from Lactobacillus brevis provide the structural bases of its substrate and cosubstrate specificity. J. Mol. Biol. 2005, 349, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, M.; Villela, M.; Liese, A.; Kragl, U. Use of an ionic liquid in a two-phase system to improve an alcohol dehydrogenase catalysed reduction. Chem. Commun. 2004, 1084–1085. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, V.; Mackfeld, U.; Rother, D.; Jakoblinnert, A. Enantioselective, continuous (R)- and (S)-2-butanol synthesis: Achieving high space-time yields with recombinant E. coli cells in a micro-aqueous, solvent-free reaction system. J. Biotechnol. 2014, 191, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Eckstein, M.; Kragl, U. Influence of water-miscible organic solvents on kinetics and enantioselectivity of the (R)-specific alcohol dehydrogenase from Lactobacillus brevis. Biotechnol. J. 2006, 1, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Van den Wittenboer, A.; Schmidt, T.; Muller, P.; Ansorge-Schumacher, M.B.; Greiner, L. Biphasic mini-reactor for characterization of biocatalyst performance. Biotechnol. J. 2009, 4, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Keasling, J.D.; Chou, H. Metabolic engineering delivers next-generation biofuels. Nat. Biotechnol. 2008, 26, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Cascone, R. Biobutanol-a replacement for bioethanol? Chem. Eng. Prog. 2008, 104, S4–S9. [Google Scholar]
- Choi, Y.J.; Park, J.H.; Kim, T.Y.; Lee, S.Y. Metabolic engineering of Escherichia coli for the production of 1-propanol. Metab. Eng. 2012, 14, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T.; Atsumi, S.; Liao, J.C. Engineered synthetic pathway for isopropanol production in Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 7814–7818. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Cann, A.F.; Connor, M.R.; Shen, C.R.; Smith, K.M.; Brynildsen, M.P.; Chou, K.J.; Hanai, T.; Liao, J.C. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 2008, 10, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.R.; Leonard, E.; Yoon, S.H.; Tseng, H.C.; Yuan, C.; Prather, K.L. Engineering alternative butanol production platforms in heterologous bacteria. Metab. Eng. 2009, 11, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, S.; Wu, T.Y.; Eckl, E.M.; Hawkins, S.D.; Buelter, T.; Liao, J.C. Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl. Microbiol. Biotechnol. 2010, 85, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Prather, K.L. Controlled biosynthesis of odd-chain fuels and chemicals via engineered modular metabolic pathways. Proc. Natl. Acad. Sci. USA 2012, 109, 17925–17930. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Wakao, N. Thermostable NAD(+)-dependent (R)-specific secondary alcohol dehydrogenase from cholesterol-utilizing Burkholderia sp AIU 652. J. Biosci. Bioeng. 2003, 96, 387–393. [Google Scholar] [CrossRef]
- Bradshaw, C.W.; Fu, H.; Shen, G.J.; Wong, C.H. A Pseudomonas sp. alcohol-dehydrogenase with broad substrate-specificity and unusual stereospecificity for organic-synthesis. J. Org. Chem. 1992, 57, 1526–1532. [Google Scholar] [CrossRef]
- Kreit, J.; Elalami, A. Substrate characterization of a NAD-dependent secondary alcohol dehydrogenase from Rhodococcus sp. CKL (CIP 105335). J. Mol. Catal. B: Enzym. 2002, 19, 253–259. [Google Scholar] [CrossRef]
- Kizaki, N.; Sawa, I.; Yano, M.; Yasohara, Y.; Hasegawa, J. Purification and characterization of a yeast carbonyl reductase for synthesis of optically active (R)-styrene oxide derivatives. Biosci. Biotechnol. Biochem. 2005, 69, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Xu, Y.; Yang, M.; Mu, X.Q. A novel NADH-dependent carbonyl reductase with unusual stereoselectivity for (R)-specific reduction from an (S)-1-phenyl-1,2-ethanediol-producing microorganism: Purification and characterization. Lett. Appl. Microbiol. 2007, 44, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yamagishi, Y.; Koguchi, S.; Iwai, N.; Kitazume, T. An effective method to use ionic liquids as reaction media for asymmetric reduction by Geotrichum candidum. Tetrahedron Lett. 2006, 47, 4619–4622. [Google Scholar] [CrossRef]
- Peterson, D.J.; Welch, R.W.; Rudolph, F.B.; Bennett, G.N. Molecular cloning of an alcohol (butanol) dehydrogenase gene cluster from Clostridium acetobutylicum ATCC 824. J. Bacteriol. 1991, 173, 1831–1834. [Google Scholar]
- Ismaiel, A.A.; Zhu, C.X.; Colby, G.D.; Chen, J.S. Purification and characterization of a primary-secondary alcohol dehydrogenase from two strains of Clostridium beijerinckii. J. Bacteriol. 1993, 175, 5097–5105. [Google Scholar] [PubMed]
- Kallberg, Y.; Oppermann, U.; Jornvall, H.; Persson, B. Short-chain dehydrogenases/reductases (SDRs). Eur. J. Biochem. 2002, 269, 4409–4417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ujor, V.; Wick, M.; Ezeji, T. Identification, purification and characterization of furfural transforming enzymes from Clostridium beijerinckii NCIMB 8052. Anaerobe 2015, 33, 124–131. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halloum, I.; Thompson, B.; Pugh, S.; Nielsen, D.R. Activity of Lactobacillus brevis Alcohol Dehydrogenase on Primary and Secondary Alcohol Biofuel Precursors. Fermentation 2015, 1, 24-37. https://doi.org/10.3390/fermentation1010024
Halloum I, Thompson B, Pugh S, Nielsen DR. Activity of Lactobacillus brevis Alcohol Dehydrogenase on Primary and Secondary Alcohol Biofuel Precursors. Fermentation. 2015; 1(1):24-37. https://doi.org/10.3390/fermentation1010024
Chicago/Turabian StyleHalloum, Ibrahim, Brian Thompson, Shawn Pugh, and David R. Nielsen. 2015. "Activity of Lactobacillus brevis Alcohol Dehydrogenase on Primary and Secondary Alcohol Biofuel Precursors" Fermentation 1, no. 1: 24-37. https://doi.org/10.3390/fermentation1010024