
Unveiling the Effects of Solvent Polarity within Graphene Based Electric Double-Layer Capacitors

1
Power China Huadong Engineering Corporation Limited, Hangzhou 311122, China
2
State Key Laboratory of Clean Energy Utilization, Institute for Thermal Power Engineering, College of Energy Engineering, Zhejiang University, Hangzhou 310027, China
*
Author to whom correspondence should be addressed.
Energies 2022, 15(24), 9487; https://doi.org/10.3390/en15249487
Submission received: 7 November 2022
/
Revised: 11 December 2022
/
Accepted: 13 December 2022
/
Published: 14 December 2022
(This article belongs to the Special Issue Advances in Electrode Nanomaterials for Fast-Charging Ion Batteries)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Solvents have been considered to show a profound influence on the charge storage of electric double-layer capacitors (EDLCs). However, the corresponding mechanisms remain elusive and controversial. In this work, the influences of solvent dipole moment on the EDL structures, kinetic properties, and charging mechanisms of graphene-based EDLCs are investigated with atomistic simulations. Specifically, electrolyte structuring is conspicuously modulated by solvents, where a sharp increment of capacitance (~325.6%) and kinetics (~10-fold) is documented upon the slight descent of polarity by ~33.0%. Unusually, such an impressive enhancement is primarily attributed to the suppressed interfacial electric fields stimulated by strong-polarity solvents in the proximity of electrodes, which goes beyond the previously observed issues that stemmed from the competitive interplays between ions and solvents. Moreover, a distinctive polarity-dependent charging mechanism (i.e., from pure counterion adsorption to coion desorption) is identified, which for the first time delineates the pivotal role of solvent polarity in manipulating the charge storage evolutions. The as-obtained findings highlight that exploiting the solvent effects could be a promising avenue to further advance the performances of EDLCs.
1. Introduction
Driven by the worsening environmental pollution and diminishing fossil fuels, the development of renewable power resources and related energy storage systems are being constantly pursued. Electric double-layer capacitors (EDLCs) have garnered tremendous attention in power-needed applications owing to their superior power delivery capability (>10 kW kg−1), fast charging/discharging rate (a few seconds), and long cycle life (>105 cycles) [1,2]. Unfortunately, the energy density of EDLCs is relatively low (typically <5 Wh kg−1), which impedes them from fulfilling the ever-growing energy demands [3,4].
Graphene, a representative kind of two-dimensional carbon nanomaterial, has attracted tremendous attention as the promising electrode material for supercapacitors due to its ultrahigh specific surface area (2630 m2/g), excellent electrical conductivity, outstanding mechanical/chemical stabilities, and ultrahigh theoretical gravimetric capacitance of ~550 F/g. In the EDLCs, energy storage is completed based on the reversible adsorption/desorption of electrolytes (ions and solvents) nearby the graphene electrode, which strongly depends on the electrolyte–graphene interactions. As a result, the properties of electrolytes play a crucial role in determining the charge storage capability of EDLCs. Substantial research efforts have been primarily focused on studying the relationships between electrolyte properties and the electrochemical stability window, electrode capacitance, and the energy and power density of EDLCs. Using molecular dynamics (MD) simulation, density functional theory calculations, and experimental measurements, the effects of ion size/shape/valence, ionic conductivity, viscosity, solvent dielectric property, and polarity on the interfacial EDL structure and capacitive performance of EDLCs have been widely explored (including our previous studies) [5,6,7,8,9,10,11,12].
Recently, solvent effects on the charging mechanisms and capacitive performance of EDLCs have garnered a dramatic proliferation of theoretical studies. However, the corresponding mechanisms are still partially resolved and controversial. Shim et al. reported that the EDL capacitance was significantly decreased upon the addition of acetonitrile solvents [13]. However, employing the identical solvents, an almost invariant capacitance, comprised marginally between ~6.5 and ~7.0 μF cm−2, was recognized by Feng and colleagues [14]. Moreover, Liu et al. recently delineated a positive solvent effect on the electrode capacitance [15]. As for the kinetic properties, Chaban et al. suggested that the conductivity of solvent-free ionic liquids could be remarkably enhanced with the acetonitrile solvents (approx. by ~50 times) [16], whereas Shim et al. addressed a distinctively opposite effect [17].
These conflicting observations are presumably correlated with the solvent properties, e.g., size/shape, charge asymmetry, concentration, and dielectric constant or polarity. For instance, Uralcan et al. observed that electrode capacitance exhibited a nonmonotonic behavior with respect to the ionic or solvent concentrations, primarily ascribed to the competition between independent motions of solvated ions in the dilute regime and solvation fluctuations in the concentrated regime [18]. Furthermore, Burt et al. suggested that the specific capacitance of nanoporous carbon was a trade-off between the concentration and separability of electrolyte ions [19]. As a consequence, a molecular-level insight into the corresponding issues of the primary mechanisms/phenomena stimulated by the solvent polarity effects is highly needed.
In this work, the influences of solvent polarity on the interfacial structures, ionic kinetics, and charge storage mechanisms of graphene electrodes were delineated using MD simulations. A comprehensive examination of EDL structures on the anatomy of interfacial populations, packing locations, effective EDL thickness, and ionic separation degree was carried out. Based on the as-obtained electrolyte structuring, the responses of electrode capacitance and kinetic behaviors to the variations of dipole moments were evaluated systemically. In the end, the evolutions of charge storage were further elaborated quantitatively, in which an obvious polarity-dependent charging mechanism (transition from pure counterion adsorption to coion desorption) was for the first time proposed.
2. Simulation Methods
Consistent with our prior studies [20,21], the simulation unit cell is composed of two opposite graphene layers arranged in a parallel configuration, connecting to a confined electrolyte bath. The dimensions of the graphene electrodes along the X and Y directions were ~34.10 and ~34.45 Å, respectively, which are quantitatively comparable to or larger than those of prior theoretical studies using similar systems. The width of the electrolyte bath was sufficiently large (~50 Å), in which the density oscillations were diminished in the central region.
In this work, partial charges of ± 5 μC cm−2 were placed on the graphene surface directly contacting the electrolytes to mimic the electrode potentials, which has been widely adopted by prior simulation studies in the literature [21]. To tune the dipole moment μ (from ~1.88 to ~2.50 D), the monomeric charges δ of solvents were evolved accordingly, which has been employed by previous theoretical studies using coarse-grained models [22]. While the as-adopted algorithm presumably introduced extra artificial elements, such an all-atom MD simulation in current work is expected to characterize the solvent effects well in comparison to prior simulations employing a simple coarse-grained model.
Consistent with our prior simulation studies, electrode atoms were kept fixed and rigid during the MD simulations, described as Lennard-Jones (L–J) spheres with the parameters proposed by Cheng et al. [23]. The solvent molecules were characterized using simple point charge extended models [24], whereas the corresponding monomeric charges were tuned to produce diverse dipole moments. NaCl solutions are chosen because Na+ and Cl− are representative monovalent ions in aqueous electrolytes. Na+ and Cl− ions were modeled as charged L–J particles with the parameters proposed by Dang and collaborators [25]. In these models, the interactions between atomic sites (or ions) were expressed as a sum of coulombic and LJ 12–6 potentials as follows:
where qi, rij, εi, and σi represent the charge of ith atom or ion, the distance between the ith and jth atom or ion, the minimum energy, and the zero-energy separation distance, respectively. The LJ parameters for εij and σij are obtained with the Lorentz–Berthelot mixing rules.
All MD simulations were performed with the LAMMPS software (version: 16Feb16). The Nose–Hoover thermostat with a 100 fs damping parameter was employed to maintain the electrolyte temperature (~300 K). A cutoff of ~10.0 Å was used for the calculation of the van der Waals (VDW) and coulombic interactions in real space. The long-range coulombic interactions were treated using a particle–particle particle–mesh algorithm with a root-mean-square accuracy of 10−6. The velocity Verlet algorithm was employed to integrate Newton’s equation with a time step of 1 fs. All simulations were equilibrated for ~10 ns, followed by a ~20 ns production for data collection and analysis.
3. Results
3.1. EDL Structures
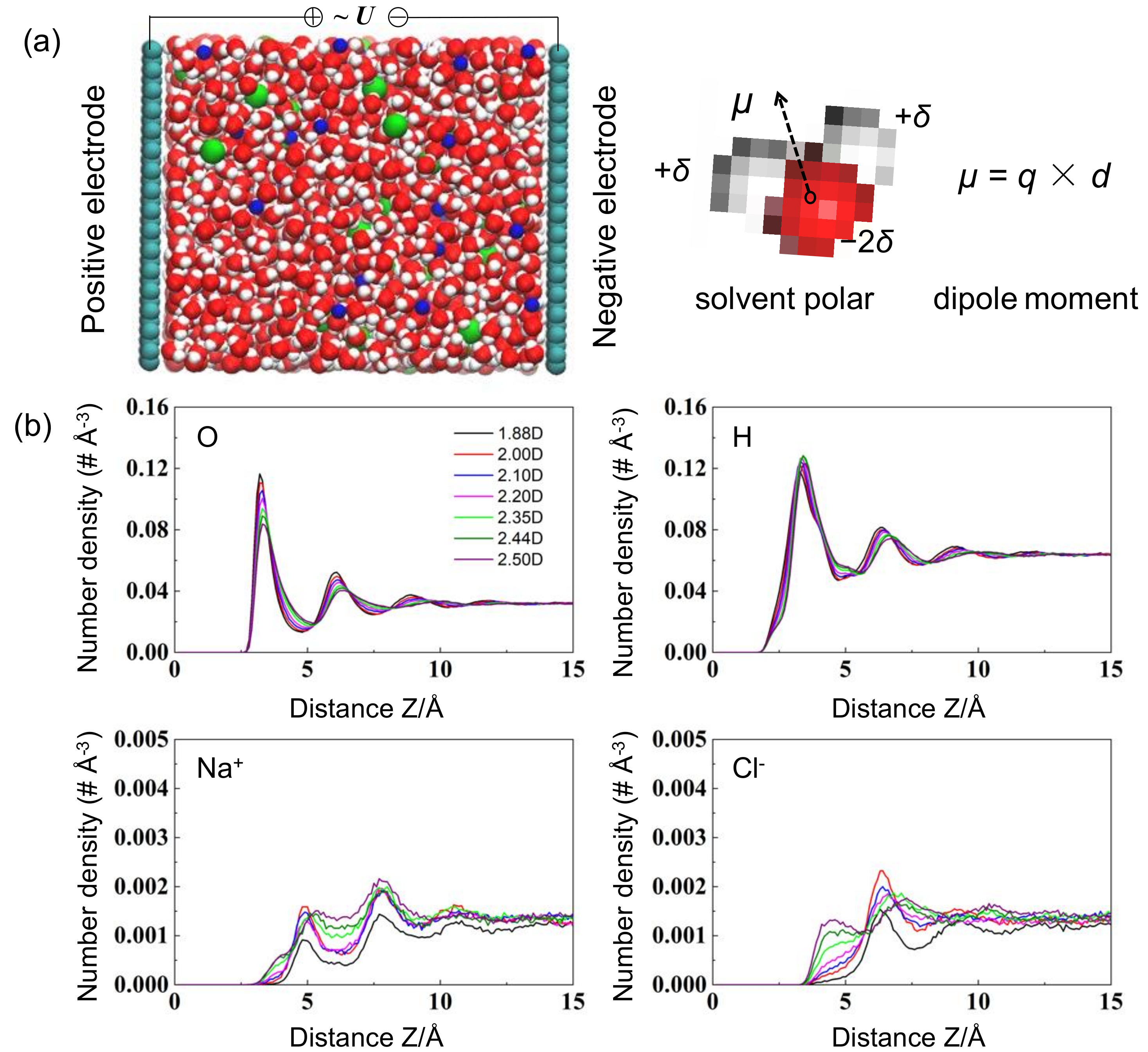
The effects of solvent polarity on the electrolyte restructuring nearby the graphene surface were first evaluated. Figure 1a delineates the schematic configurations for graphene-based EDLCs. The electrolyte number density n(z) at the position of z along the direction perpendicular to the electrified electrode was calculated as:
Figure 1b displays the density profiles n(z) of electrolytes nearby the uncharged graphene electrodes. Obviously, multilayer structures of electrolytes were observed at the interface, primarily owing to the strong interfacial VDW interactions. Specifically, the first layer of cations preferentially gathered at the valley of neighboring O atoms, in which they could maintain a complete hydration shell, whereas anions preferred to accumulate farther away from the electrified surface, probably due to the hydration effects. Nonetheless, the oscillatory patterns of the density profiles vanished about ~10–12 Å away from the electrode surface, which is in good agreement with the classic Gouy–Chapman–Stern model.
The configurations of electrolyte structuring were remarkably evolved with respect to the solvent polarities. For example, with the increment of dipole moment, the density of the first O peak was sharply suppressed by ~41.0%, approximately from ~0.117 to ~0.083 #Å−3, which was probably due to the elevated solvent–solvent repulsions stimulated by the enlarged monomeric charges. On the contrary, the corresponding magnitude of cation and anion distributions was noticeably improved with the extended dipole moment.
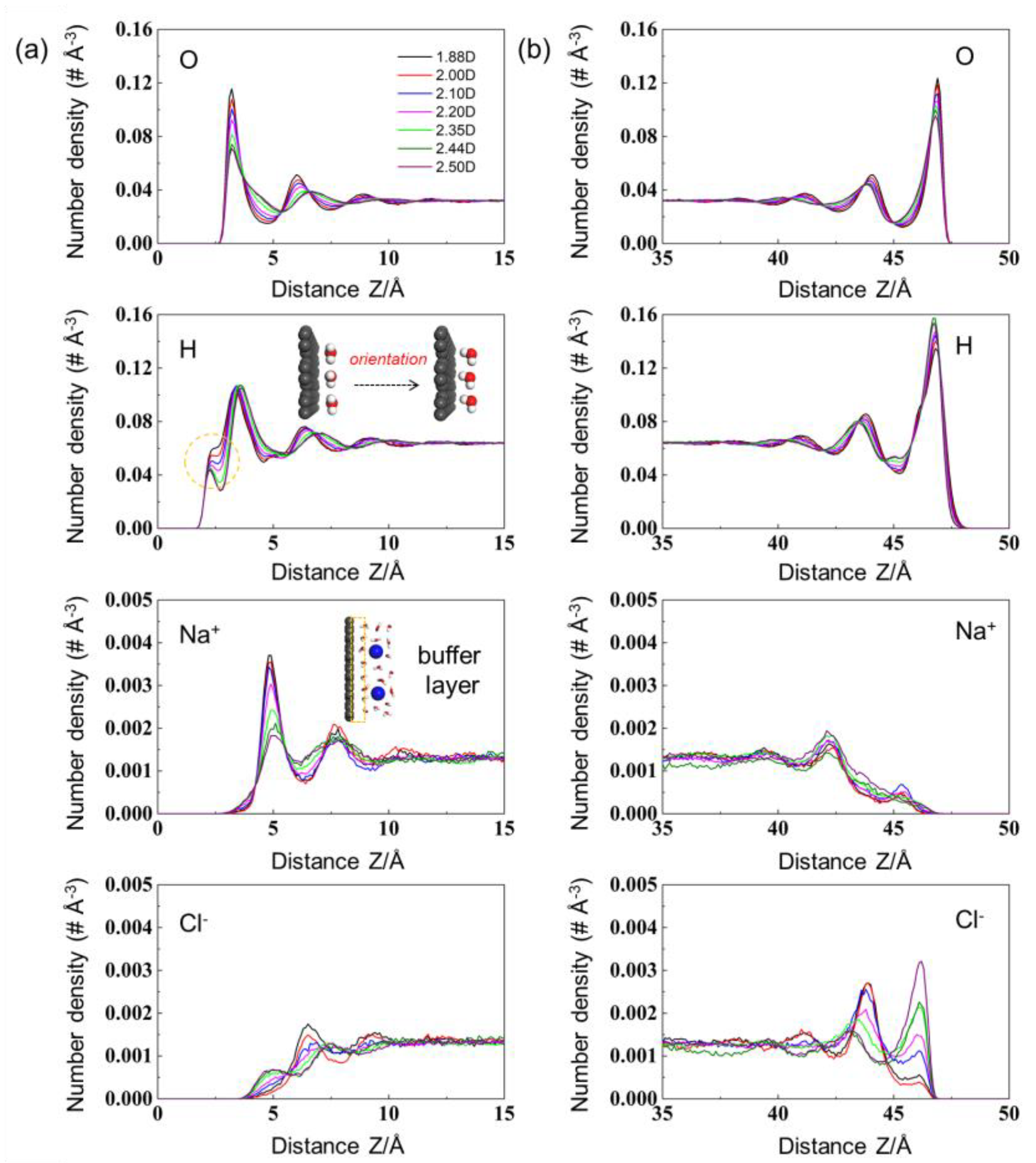
Extended to the charged cases, an analogous but more pronounced trend was recognized at the interface in Figure 2. To be specific, the intensity of the first O layer decayed more sharply (~63.4%), whereas the corresponding layer structures became smoother and broader, in which the span of the proximate peak was remarkably exaggerated from ~1.9 Å to ~2.7 Å. The decreased intensity in the first O layer can be interpreted by the elevated solvent–solvent repulsions stimulated by the enlarged monomeric charges and reduced ion populations. On the other hand, an inner peak of H atoms directly contacting the electrified surface was highly developed (see the dashed orange circle). As a result, with the increment of solvent polarity, the orientations of solvent molecules with respect to the electrified surface were thereby gradually transformed from parallel to perpendicular directions (see inset of Figure 2a). Specifically, the O and H peaks of weak-polarity solvents were almost at the same position (z = ~3.2 and ~3.3 Å for O and H atoms, respectively), gathering in a plane parallel to the electrode surface. Regarding the strong-polarity counterpart, one H atom was shifted towards the electrode surface, in which the O peak (z = ~3.2 Å) was thus sandwiched by two H layers (z = ~2.3 Å and ~3.7 Å), displaying a perpendicular direction to the electrode surface.
More specifically, a dramatic decrement of cation density (by ~205.6%) with respect to the increasing dipole moment was observed nearby the charged graphene surface. On the basis of previous studies [10,21], this phenomenon has been primarily attributed to the competitive interplays between ions and solvent molecules at the interface. The solvation strengths of organic electrolytes were considerably weak, in which the solvents around ions were easily stripped at the interface, especially under the strong external electric fields (up to ~108–109 V m−1). Therefore, ions and solvents tended to aggregate at nearly the same plane, in which the competitive interactions between ions and solvents (i.e., volume-excluded effects) played a pivotal role in determining the interfacial populations. For example, Jiang et al. suggested that strong-polarity solvents tended to intercalate inside the slit pores and the majority were aligned against the charged wall, thereby severely suppressing the ion populations due to the intensive volume-excluded effects [22].
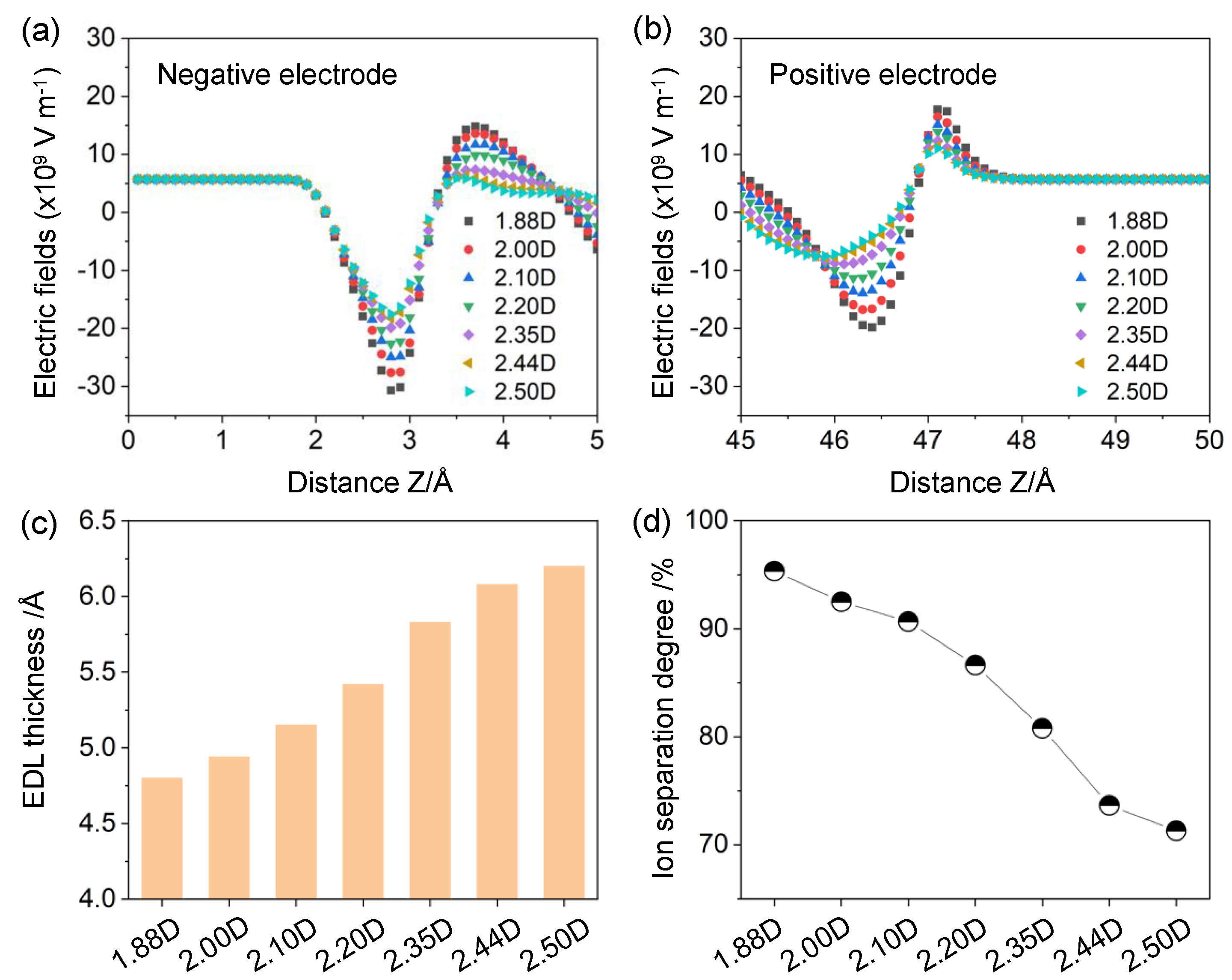
However, in this work, the as-obtained trend was principally attributed to the descended interfacial electric fields stimulated by the strong-polarity solvents in the proximity of electrodes, instead of the volume-excluded effects. Owing to the strong solvation strengths of inorganic ions, a buffer layer of solvents was usually maintained between the electrified surface and solvated ions (see the orange rectangle in Figure 2a), which effectively prohibited the electrolyte ions from approaching the electrodes directly. Considering the dielectric nature of solvents, they could noticeably screen the interfacial electric fields and thus diminish the ion–electrode interactions, which became more pronounced within the stronger-polarity solvents. Therefore, the ion populations at the interface were compressed significantly and monotonically with the increasing dipole moment. Such a phenomenon could be validated well by the interfacial electric field distributions (Figure 3a,b). Owing to the superior screening efficiency of strong-polarity solvents with higher monomeric charges, the electric field strengths in the vicinity of ion layers were decreased significantly by ~225.8% with the increment of dipole moment. Similar electrolyte distributions were also observed on the positive graphene counterpart (Figure 2b). The interfacial EDL multilayer structures almost vanish when the distance to the graphene electrode is increasing beyond ~10 Å because van der Waals interactions are short-ranged and electrostatic interactions are screened by ions and solvents.
In addition to the intensity of electrolyte populations, the capacitive behaviors of EDLCs also depend strongly on the packing positions nearby the electrode surface. To quantify the corresponding ionic locations, the effective EDL thickness dcenter was calculated as:
where z0 is the location of the electrode surface and n(z) is the ion number density at the position of z. According to the electric double-layer theory, the smaller EDL thickness benefits high capacitance values. Figure 3c depicts the effective EDL thickness for different solvent polarities. With the increment of dipole moment, the EDL thickness was monotonically improved by ~29.2%, switching from ~4.8 to ~6.2 Å. This result could be illustrated by the descended density and broader distributions of ions immersed in the strong-polarity solvents (Figure 2), indicating that solvents with higher dipole moments were presumably adverse to the formation of EDL structures.
To further probe the ion–ion interactions, the segregation degree of counterions and coions within the EDL-dominant area was evaluated. Based on a previous theoretical study [26], the ionic separation degree χ was calculated as:
Conceptually, χ = 0 or 100% indicates that the cations and anions were completely separated, whereas χ = 50% indicates that charge neutrality occurred. Figure 3d displays the separation degree as a function of solvent polarity. Different from the simulation results of the EDL thickness, the ionic separation degree declined rapidly by ~33.7% with the solvent dipole moment increasing from ~1.88 to ~2.50 D. This trend was mainly affiliated with the reduced counterion density but nearly invariant to the coion populations.
3.2. Capacitive Performances
The influences of solvent polarity were further extended to the capacitive performances of graphene-based EDLCs. Based on the as-obtained electrolyte structures, the electric potential distributions across the EDL-dominant area were examined. The electric potentials ϕ(z) within MD simulations are typically calculated by numerically solving the Poisson equation from the space charge density [27,28]:
where ε0 is the permittivity of vacuum and ρ(z) denotes the electrolyte charge density at the position of z.
Using the one-sided Green’s function method, Equation (6) could be obtained by integrating Equation (5) analytically twice via the Leibnitz integral rules:
Considering the complete screening of interfacial electric fields within the bulk region, the electrode potential Uelectrode was calculated, defined as the potential drop within electrode/electrolyte interfacial district relative to the potential of zero charge (PZC):
where UEDL is defined as the potential drop across the EDL-dominant area and PZC represents the potential difference between the electroneutral electrode and bulk electrolytes. According to electric double-layer theory, the area-specific capacitance of a single electrode could be calculated as:
where σ is the surface charge density of electrodes.
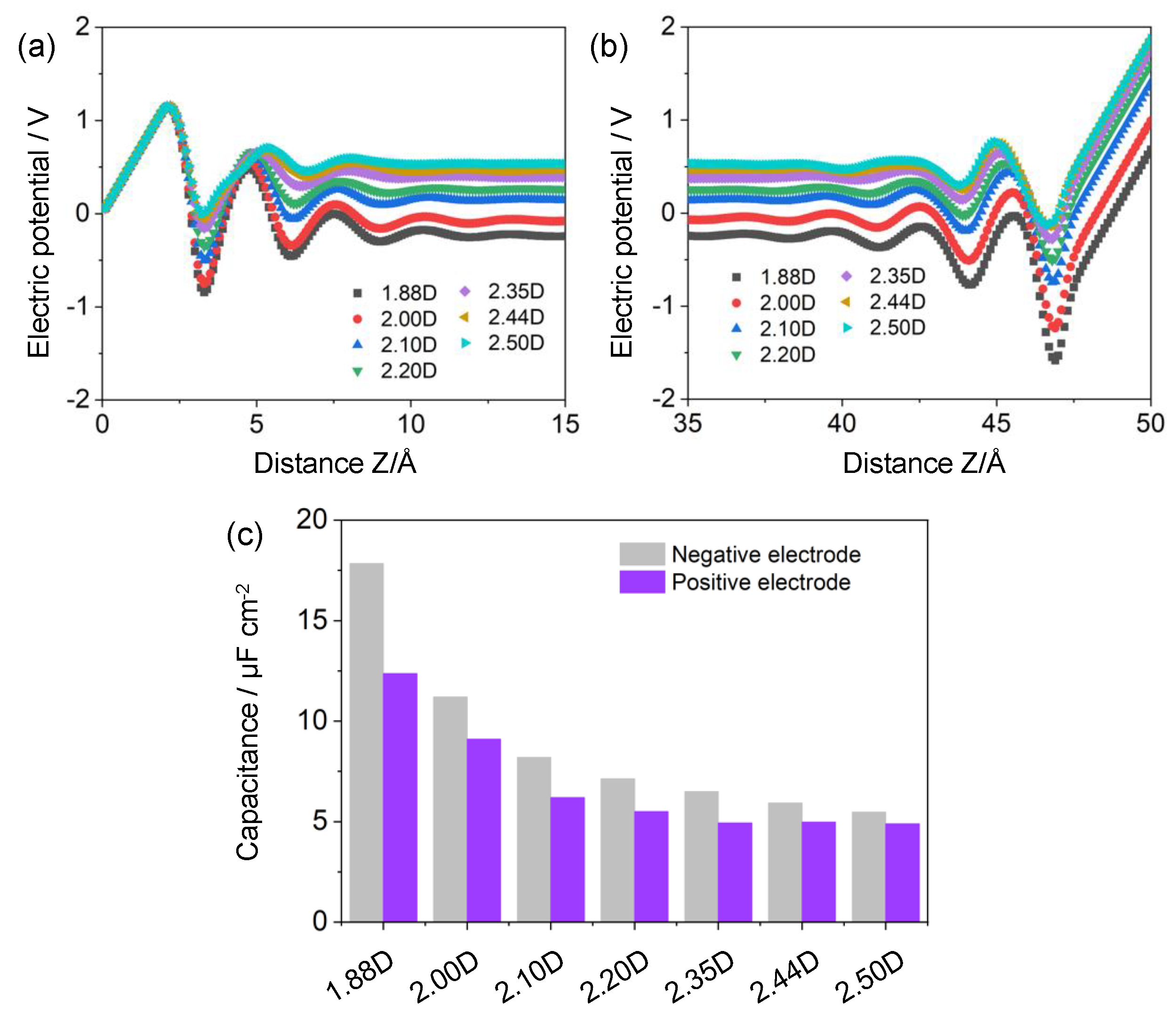
By integrating the Poisson equation, the potential of zero charge for electroneutral graphene electrodes was first calculated. With enlarging solvent polarity, the PZC values decayed monotonically, which were calculated as ~0.520, ~0.497, ~0.465, ~0.441, ~0.375, and ~0.374 V for μ = ~1.88, ~2.00, ~2.10, ~2.20, ~2.44, and ~2.50 D, respectively. This trend was mainly triggered by the reduced H density at the electrode/electrolyte interface.
Figure 4a represents the electric potentials as a function of distance z from the electrified electrode surface. The electric potential ϕ(z) displayed a remarkably oscillatory behavior in the proximity of electrodes, and progressively grew smooth with the increasing z. Specifically, the magnitude of electrode potential was monotonically extended by enlarging the solvent dipole moment from ~1.88 to ~2.50 D. This trend was intimately correlated with the aforementioned ionic distributions, where a higher ion density facilitated a more rapid screen of interfacial electric fields, and thereby resulted in a lower electrode potential drop. Similar results were also observed on the positive counterpart (Figure 4b).
Figure 4c illustrates the specific capacitances for the cathode (Ccathode) and anode (Canode). The as-calculated capacitances at the negative and positive polarizations of ~2.35 D were ~6.50 and ~4.93 μF cm−2, respectively, which are in good agreement with prior theoretical and experimental studies. For instance, using an MD simulation, Xu and colleagues pointed out that the specific capacitances were ~6.32 and ~6.04 μF cm−2 at the negative and positive polarizations of graphene electrodes, respectively [29]. In another experimental study, a representative capacitance value of ~8–10 μF cm−2 was observed for the carbon electrodes immersed in a neutral NaCl solution [30].
Interestingly, the capacitive performances of graphene electrodes could be conspicuously modulated by the solvent polarity. For example, with the slight decrement of dipole moment by ~33.0%, the specific capacitance was remarkably boosted by ~325.6% at the negative polarizations, from ~5.48 to ~17.84 μF cm−2, which was calculated as ~252.5% for the positive counterpart. Compared to prior studies concerning solvent effects [19,22], such a capacitance enhancement was rather impressive and tremendous. For instance, Jiang and colleagues found that the specific capacitance experienced a volcano-shaped trend as the dipole moment of solvents varied from ~2.5 to ~5.0 D, which was enhanced first (less than ~20%) and then descended [22]. In these theoretical studies, the influences of solvent polarity on the electrode capacitance or EDL structures have been predominantly attributed to the competitive interplays between solvents and ions accumulating at the electrified surface (i.e., volume-excluded effects). In this work, the variations of EDL capacitance or ion density were intensively correlated with the evolutions of interfacial electric fields mediated by solvents in the vicinity of electrodes, which became more pronounced within the stronger-polarity solvents. Noteworthy, we pointed out that solvents with low polarity were more preferable for further advancing the energy density of EDLCs.
Furthermore, the capacitance at the negative polarization was much higher than that at the positive counterpart. More specifically, an obvious dependence of cathode–anode asymmetry on the solvent polarity was recognized in this work, where the capacitance divergences between negative and positive polarizations were noticeably diminished by enlarging the dipole moment. For instance, within a weak-polarity solvent of ~1.88 D, the specific capacitance of the negative electrode could be approximately ~44.2% larger than that of positive polarizations, which was sharply descended to ~11.2% at a higher polarity of ~2.50 D. The polarity-dependent asymmetric cathode–anode capacitance was closely correlated with the solvent polarity and ion size effect. The increment of solvent polarity decreased the size dissymmetry between cations and anions, thus leading to similar hydration radius and capacitive performances.
3.3. Kinetic Performances
In addition to the structural arrangements (i.e., static or equilibrium characters) at the interface, the effects of solvent polarity on the kinetic characteristics were also examined. The self-diffusion coefficients D for cations, anions, and solvents were calculated based on the mean square displacement (MSD) method:
where r(t) is the position of particle mass center at the time t and n is the dimensionality of the system. For the bulk solution, n = 3 and the MSDs along three dimensions are calculated.
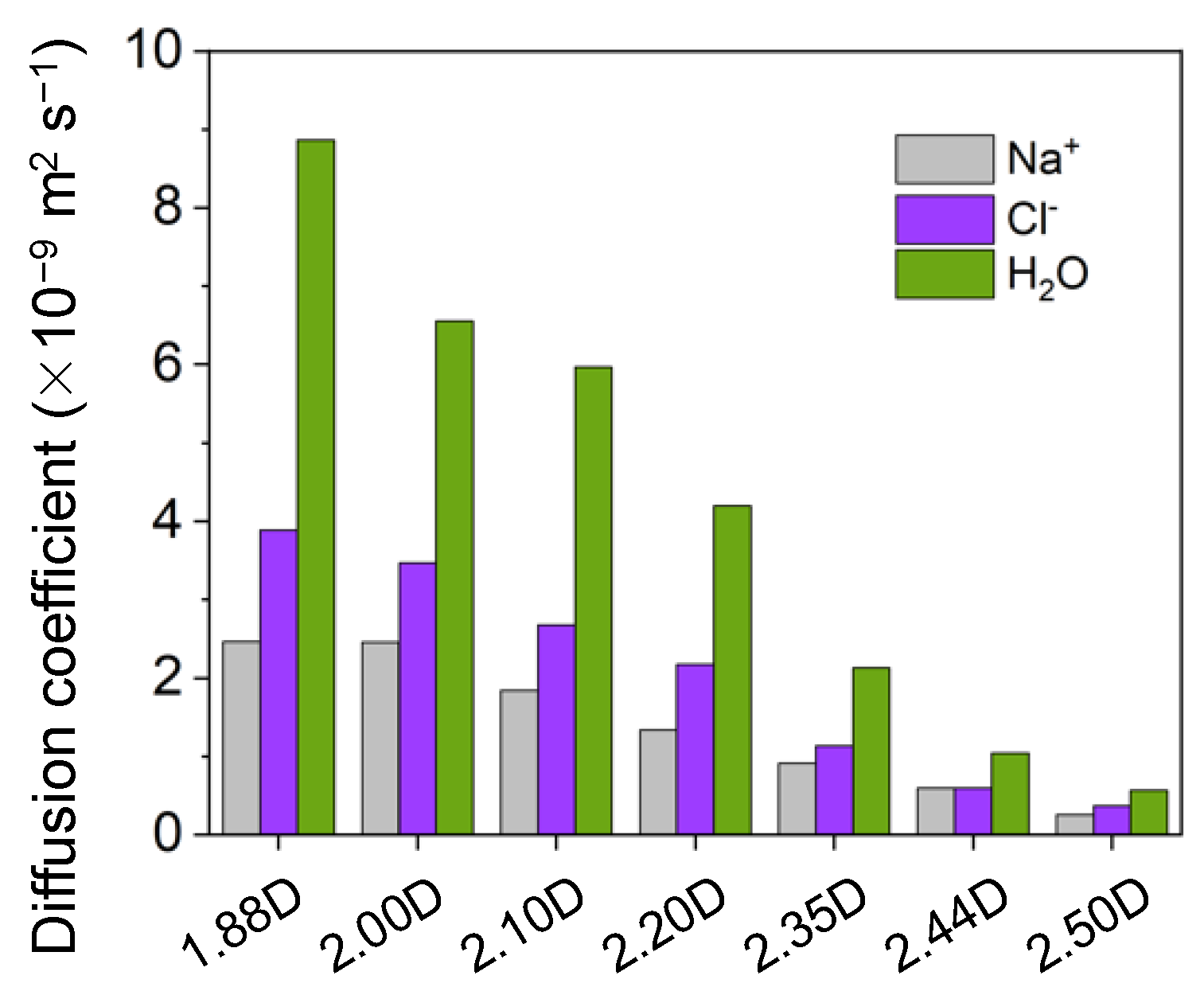
Figure 5 represents the self-diffusion coefficients D for cations, anions, and solvents as a function of dipole moment. The as-calculated self-diffusion coefficients of ~2.35 D for cations and anions were ~9.13 × 10−9 m2 s−1 and ~1.14 × 10−9 m2 s−1, respectively. With the slight decrement of dipole moment by ~33.0%, the corresponding diffusion coefficients were substantially exaggerated by ~955.2%, ~1051.6% and ~2066.8% for cations, anions, and solvents, respectively. This indicates that the dynamic behaviors of electrolytes are significantly dependent on the solvent polarity. Such an impressive enhancement of the diffusion coefficient was intensively correlated with the sharply reduced viscosity of solvents stemmed from the smaller monomeric charges. Meanwhile, the extremely high concentration of solvents could effectively decrease the ion–ion correlations to facilitate greater ion mobility. Thereby, we suggested that solvents with weak dipole moment were highly preferable for boosting the kinetic properties.
Specifically, the dynamic divergences between ions and solvents remarkably declined with respect to the increasing dipole moment. For example, with a weak-polarity solvent, the self-diffusion coefficient of solvents could be about ~260.0% higher than that of the ion counterpart, which rapidly descended to ~66.4% for strong-polarity solvents. Such a trend could be interpreted by the enhanced ion–solvent interactions stimulated by the elevated monomeric charges, which notably attenuated the kinetic divergences between ions and solvent molecules.
3.4. Changing Mechanisms
To reveal the evolving modes of graphene charge storage as a function of polarity, the ion populations nearby the charged and uncharged electrodes were examined in detail. According to previous theoretical studies [31,32], the capacitive behaviors of EDLCs were predominated by the ionic distributions in the first layer or nanoscale interfacial districts, in which the corresponding density from the second layer to the bulk region manifested a marginal influence on the electrochemical performances. For example, based on the ion number within the first layer, Jo and colleagues developed a novel measurement (i.e., differential ion capacity) to well interpret the as-obtained differential capacitance results [33]. On the other hand, Vatamanu et al. illustrated the trend of differential capacitance with respect to electrode potentials properly with the ion density in the interfacial region of z~8 Å [27]. Thus, the accumulated number density within the first layer was calculated as:
where nα(z) is the cation or anion number density at the position of z.
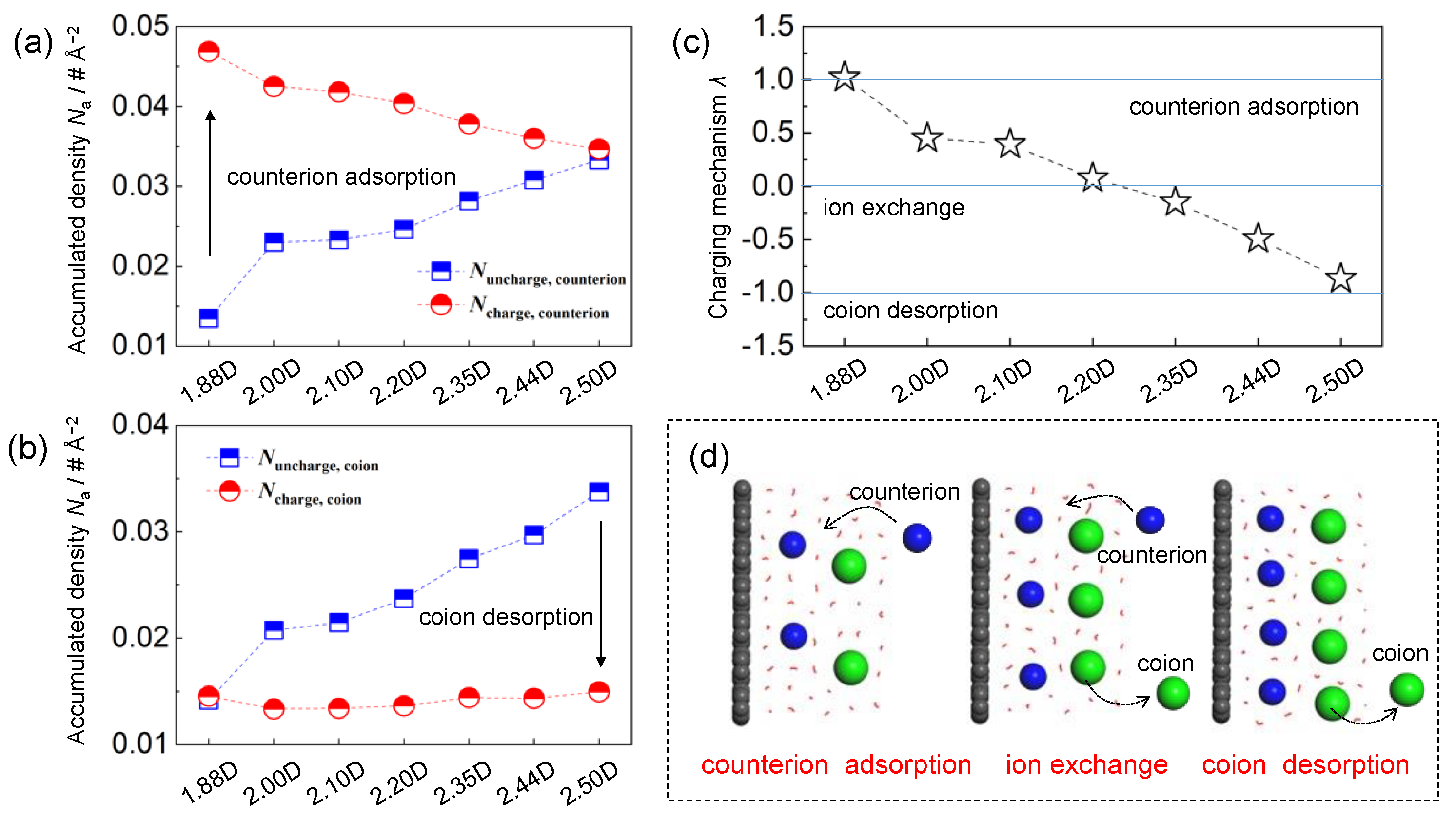
Figure 6a displays the accumulated number density of ions near the charged and uncharged electrodes. On the uncharged electrodes, both the counterion (i.e., cation) and coion (i.e., anion) populations were enlarged monotonically with the increment of solvent polarity, whereas an opposite trend was observed nearby the charged case, which is in good agreement with the as-obtained number density in Figure 2.
Based on previous experimental and theoretical studies, three distinctive charging mechanisms have been proposed, including counterion adsorption, ion exchange, and coion desorption. To quantify the charge storage mechanisms of graphene-based EDLCs, a charging mechanism parameter λ was thus calculated as [34]:
where N(Q) and N(Q0) denote the total number of ions under a surface charge density of Q (i.e., −5 μC cm−2) and Q0 (i.e., 0 μC cm−2), respectively. Nnet(Q) and Nnet(Q0) are then the net ionic charges near the charged and uncharged electrodes, respectively. Conceptually, λ = −1, 0, and 1 denote the coion desorption, ion exchange, and counterion adsorption (i.e., classic charging mechanisms), respectively. Regarding the intermediate values, for instance, λ = 0.9 meant that both the counterion adsorption and ion exchange occurred, whereas the counterion adsorption was dominated in the charging process (λ closer to ~1).
As demonstrated in Figure 6, with the increment of solvent polarity from weak to strong, a complex charging behavior transformed from pure counterion adsorption, via ion exchange, to coion desorption was recognized in this work, highlighting the pivotal role of solvent polarity in regulating the charge storage principles. Specifically, with the weak-polarity solvent of ~1.88 D, the as-calculated λ was just equal to ~1, suggesting that the charging process was achieved principally by adsorbing counterions (i.e., Na+ ions). Such a phenomenon was intensively correlated with the initial ion populations in the vicinity of electrodes. Near the electroneutral graphene surface with low-polarity solvents, the interfacial ion concentrations were much lower, primarily owing to the considerably weak ion–solvent interactions. Therefore, the system exhibited a notable preference for counterion adsorptions, which was also identified as the classic charge storage mechanism in the dilute solutions.
However, for the strong-polarity solvent of ~2.50 D, the as-obtained parameter λ approached ~−1, indicating that an unusual charging process dominated by the coion desorption was observed. Due to the reinforced ion–solvent interactions within strong-polarity solvents, the initial counterion and coion densities at the interface were drastically improved (approximately ~4 M). In such a situation, the removal of coions in the proximity of charged electrodes (with same charges) was more favorable, considering that inserting counterions would sharply exaggerate the cation–cation repulsions (ion–ion interactions increased exponentially as their separations decreased). Regarding the solvents with weak to strong polarity (~1.88 D < μ < ~2.50 D), the cation and anion concentrations were moderate, where this system did not manifest a noticeable priority between counterion insertion and coion removal. As a result, a combination of ion exchange and counterion adsorption (0 ≤ λ < 1) or coion desorption (−1 < λ ≤ 0) could be observed, highlighting the complex behaviors of charge storage mechanisms stimulated by the dielectric nature of solvents.
In fact, the critical role of solvents in regulating the charging mechanisms has been observed in recent theoretical studies [19,35]. For example, Burt et al. suggested that the solvent concentrations could obviously alter the charge storage mechanisms, switching from counterion adsorption in the diluted organic electrolytes to ion exchange in the pure solvent-free ionic liquids [19]. Using MD simulations, our work revealed that a slight decrement in dipole moment benefited an obvious enhancement of capacitance and kinetics, which agreed well with previous studies [15]. However, our work also showed conflicting observations with previously reported simulation results [13,14]. Specifically, the polarity-dependent charging mechanism (from pure counterion adsorption, via ion exchange, to coion desorption) has never been reported before. These conflicting observations were presumably correlated with the substantially diverse solvent properties in previous reports, e.g., size/shape, charge asymmetry, and concentration in addition to the dielectric constant (or solvent polarity). Therefore, a comprehensive experimental study of solvent molecules with different polarities is needed to confirm and clarify the simulation results in future work.
This work reveals the role of solvent polarity in regulating the interfacial electrolyte structure, ion solvation structure, dynamics, and charging mechanisms. The as-obtained results can provide new guidance on the design of high-performance aqueous supercapacitors and batteries based on the strategy of solvation chemistry etc. [36,37]. For example, recent studies suggested that the solvation structure of electrolytes (i.e., ion–solvent interactions) played a key role in the stability of electrodes during cell cycling [37]. In addition, this work also benefited seawater electrolysis and relevant electrochemical energy storage applications [38,39]. Moreover, it will be interesting to explore the solvent polarity effect in the nanoporous electrodes as future work, which is expected to significantly affect the ion entrance barrier, desolvation energy, and thus capacitive performance.
4. Conclusions
In summary, MD simulations were employed to characterize the evolutions of EDL capacitance, kinetic behaviors, and charge storage mechanisms in response to the variations of solvent polarities. Consistent with prior theoretical studies, the dipole moment of solvents, ranging from ~1.88 to ~2.50 D, was tuned by changing the monomeric charges. The simulation results suggested that the electrolyte structuring was significantly disturbed by the solvent polarity. To be specific, with a slight decrement of dipole moment by ~33.0%, an obvious enhancement of electrode capacitance (~325.6%) and kinetic properties (~10-fold) was recognized. In contrast to the previously reported volume-excluded effects stimulated by the competitive interplays between ions and solvents, the as-obtained impressive enhancement of capacitance was predominantly attributed to the descended interfacial electric fields issued from the strong-polarity solvents at the interface. Moreover, this work for the first time illustrated that the charge storage mechanisms of EDLCs could be effectively manipulated by the solvent dipole moment, in which a complex polarity-dependent charging mechanism (from pure counterion adsorption, via ion exchange, to coion desorption) was proposed. The as-obtained atomic insights highlight the crucial role of solvent polarity in modulating the charge storage mechanisms, and provide useful guidance for the rational design of solvents aiming for advanced performances of EDLCs.
Author Contributions
C.X.: Conceptualization, Methodology, Investigation, Formal analysis, Visualization, Writing—original draft. J.Z.: Conceptualization, Investigation, Formal analysis. D.L.: Conceptualization, Investigation, Writing—review and editing, Funding acquisition. X.Q.: Formal analysis, Investigation, Writing—review and editing. G.C.: Validation, Investigation, Writing—review and editing. H.Y.: Conceptualization, Supervision, Resources, Writing—review and editing, Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 51906211).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dai, H.; Zhang, G.; Rawach, D.; Fu, C.; Wang, C.; Liu, X.; Dubois, M.; Lai, C.; Sun, S. Polymer gel electrolytes for flexible supercapacitors: Recent progress, challenges, and perspectives. Energy Storage Mater. 2021, 34, 320–355. [Google Scholar] [CrossRef]
- Wang, J.; Malgras, V.; Sugahara, Y.; Yamauchi, Y. Electrochemical energy storage performance of 2d nanoarchitectured hybrid materials. Nat. Commun. 2021, 12, 3563. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, J.; Kim, W. Redox-active water-in-salt electrolyte for high-energy-density supercapacitors. ACS Energy Lett. 2022, 7, 1266–1273. [Google Scholar] [CrossRef]
- Xiao, J.; Han, J.; Zhang, C.; Ling, G.; Kang, F.; Yang, Q.-H. Dimensionality, function and performance of carbon materials in energy storage devices. Adv. Energy Mater. 2022, 12, 2100775. [Google Scholar] [CrossRef]
- Jalali, H.; Lotfi, E.; Boya, R.; Neek-Amal, M. Abnormal dielectric constant of nanoconfined water between graphene layers in the presence of salt. J. Phys. Chem. B 2021, 125, 1604–1610. [Google Scholar] [CrossRef]
- Zhan, C.; Cerón, M.R.; Hawks, S.A.; Otani, M.; Wood, B.C.; Pham, T.A.; Stadermann, M.; Campbell, P.G. Specific ion effects at graphitic interfaces. Nat. Commun. 2019, 10, 4858. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Koishi, A.; Bourg, I.C.; Fenter, P. Ion correlations drive charge overscreening and heterogeneous nucleation at solid–aqueous electrolyte interfaces. Proc. Natl. Acad. Sci. USA 2021, 118, e2105154118. [Google Scholar] [CrossRef]
- Le, J.-B.; Fan, Q.-Y.; Li, J.-Q.; Cheng, J. Molecular origin of negative component of helmholtz capacitance at electrified pt(111)/water interface. Sci. Adv. 2022, 6, eabb1219. [Google Scholar] [CrossRef]
- Yang, H.; Yang, J.; Li, C.; Huang, Z.; Bendavid, A.; Yan, J.; Cen, K.; Han, Z.; Bo, Z. Gel polymer dominated ion charging mechanisms within graphene nanochannels. J. Power Sources 2022, 541, 231684. [Google Scholar] [CrossRef]
- Bo, Z.; Xu, C.; Huang, Z.; Chen, P.; Yan, G.; Yang, H.; Yan, J.; Cen, K.; Ostrikov, K.K. Photo-electric capacitive deionization enabled by solar-driven nano-ionics on the edges of plasma-made vertical graphenes. Chem. Eng. J. 2021, 422, 130156. [Google Scholar] [CrossRef]
- Fleischmann, S.; Zhang, Y.; Wang, X.; Cummings, P.T.; Wu, J.; Simon, P.; Gogotsi, Y.; Presser, V.; Augustyn, V. Continuous transition from double-layer to faradaic charge storage in confined electrolytes. Nat. Energy 2022, 7, 222–228. [Google Scholar] [CrossRef]
- Boyd, S.; Ganeshan, K.; Tsai, W.-Y.; Wu, T.; Saeed, S.; Jiang, D.-e.; Balke, N.; van Duin, A.C.T.; Augustyn, V. Effects of interlayer confinement and hydration on capacitive charge storage in birnessite. Nat. Mater. 2021, 20, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.; Jung, Y.; Kim, H.J. Graphene-based supercapacitors: A computer simulation study. J. Phys. Chem. C 2011, 115, 23574–23583. [Google Scholar] [CrossRef]
- Feng, G.; Huang, J.; Sumpter, B.G.; Meunier, V.; Qiao, R. A “counter-charge layer in generalized solvents” framework for electrical double layers in neat and hybrid ionic liquid electrolytes. Phys. Chem. Chem. Phys. 2011, 13, 14723–14734. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wu, J. Boosting the performance of ionic-liquid-based supercapacitors with polar additives. J. Phys. Chem. C 2016, 120, 24041–24047. [Google Scholar] [CrossRef]
- Chaban, V.V.; Voroshylova, I.V.; Kalugin, O.N.; Prezhdo, O.V. Acetonitrile boosts conductivity of imidazolium ionic liquids. J. Phys. Chem. B 2012, 116, 7719–7727. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.; Kim, H.J.; Jung, Y. Graphene-based supercapacitors in the parallel-plate electrode configuration: Ionic liquids versus organic electrolytes. Faraday Discuss. 2012, 154, 249–263. [Google Scholar] [CrossRef]
- Uralcan, B.; Aksay, I.A.; Debenedetti, P.G.; Limmer, D.T. Concentration fluctuations and capacitive response in dense ionic solutions. J. Phys. Chem. Lett. 2016, 7, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.; Breitsprecher, K.; Daffos, B.; Taberna, P.-L.; Simon, P.; Birkett, G.; Zhao, X.S.; Holm, C.; Salanne, M. Capacitance of nanoporous carbon-based supercapacitors is a trade-off between the concentration and the separability of the ions. J. Phys. Chem. Lett. 2016, 7, 4015–4021. [Google Scholar] [CrossRef] [Green Version]
- Bo, Z.; Li, H.; Yang, H.; Li, C.; Wu, S.; Xu, C.; Xiong, G.; Mariotti, D.; Yan, J.; Cen, K.; et al. Combinatorial atomistic-to-ai prediction and experimental validation of heating effects in 350 f supercapacitor modules. Int. J. Heat Mass Transf. 2021, 171, 121075. [Google Scholar] [CrossRef]
- Yang, H.; Bo, Z.; Yan, J.; Cen, K. Influence of wettability on the electrolyte electrosorption within graphene-like nonconfined and confined space. Int. J. Heat Mass Transf. 2019, 133, 416–425. [Google Scholar] [CrossRef]
- Jiang, D.-E.; Wu, J. Unusual effects of solvent polarity on capacitance for organic electrolytes in a nanoporous electrode. Nanoscale 2014, 6, 5545–5550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Steele, W.A. Computer simulation of ammonia on graphite. I. Low temperature structure of monolayer and bilayer films. J. Chem. Phys. 1990, 92, 3858–3866. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Dang, L.X. Mechanism and thermodynamics of ion selectivity in aqueous solutions of 18-crown-6 ether: A molecular dynamics study. J. Am. Chem. Soc. 1995, 117, 6954–6960. [Google Scholar] [CrossRef]
- Pak, A.J.; Paek, E.; Hwang, G.S. Impact of graphene edges on enhancing the performance of electrochemical double layer capacitors. J. Phys. Chem. C 2014, 118, 21770–21777. [Google Scholar] [CrossRef]
- Bedrov, D.; Vatamanu, J.; Hu, Z. Ionic liquids at charged surfaces: Insight from molecular simulations. J. Non-Cryst. Solids 2015, 407, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Jenel, V.; Mihaela, V.; Oleg, B.; Dmitry, B. A comparative study of room temperature ionic liquids and their organic solvent mixtures near charged electrodes. J. Phys. Condens. Matter 2016, 28, 464002. [Google Scholar] [CrossRef]
- Xu, K.; Ji, X.; Chen, C.; Wan, H.; Miao, L.; Jiang, J. Electrochemical double layer near polar reduced graphene oxide electrode: Insights from molecular dynamic study. Electrochim. Acta 2015, 166, 142–149. [Google Scholar] [CrossRef]
- Conway, B.E. Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications; Springer Science & Business Media: Ottawa, Canada, 2013. [Google Scholar]
- Bo, Z.; Huang, Z.; Xu, C.; Chen, Y.; Wu, E.; Yan, J.; Cen, K.; Yang, H.; Ostrikov, K. Anion-kinetics-selective graphene anode and cation-energy-selective mxene cathode for high-performance capacitive deionization. Energy Storage Mater. 2022, 50, 395–406. [Google Scholar] [CrossRef]
- Bi, S.; Li, Z.; Xiao, D.; Li, Z.; Mo, T.; Feng, G.; Zhang, X. Pore-size-dependent capacitance and charging dynamics of nanoporous carbons in aqueous electrolytes. J. Phys. Chem. C 2022, 126, 6854–6862. [Google Scholar] [CrossRef]
- Jo, S.; Park, S.-W.; Shim, Y.; Jung, Y. Effects of alkyl chain length on interfacial structure and differential capacitance in graphene supercapacitors: A molecular dynamics simulation study. Electrochim. Acta 2017, 247, 634–645. [Google Scholar] [CrossRef]
- Forse, A.C.; Merlet, C.; Griffin, J.M.; Grey, C.P. New perspectives on the charging mechanisms of supercapacitors. J. Am. Chem. Soc. 2016, 138, 5731–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, C.; Liu, K.; Liu, H.L.; Wu, J.Z. Impurity effects on charging mechanism and energy storage of nanoporous supercapacitors. J. Phys. Chem. C 2017, 121, 14066–14072. [Google Scholar] [CrossRef]
- Li, C.; Wu, W.; Wang, P.; Zhou, W.; Wang, J.; Chen, Y.; Fu, L.; Zhu, Y.; Wu, Y.; Huang, W. Fabricating an aqueous symmetric supercapacitor with a stable high working voltage of 2 V by using an alkaline–acidic electrolyte. Adv. Sci. 2019, 6, 1801665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, Y. Electrolyte solvation structure as a stabilization mechanism for electrodes. Energy Mater. 2021, 1, 100004. [Google Scholar] [CrossRef]
- Jia, Z.; Cheng, C.; Chen, X.; Liu, L.; Ding, R.; Ye, J.; Wang, J.; Fu, L.; Cheng, Y.; Wu, Y. Applications of all-inorganic perovskites for energy storage. Mater. Adv. 2022, 4. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, L.; Huang, K.; Zhang, B.; Wu, J.; Huang, Y. Strained carbon steel as a highly efficient catalyst for seawater electrolysis. Energy Mater. 2022, 2, 200010. [Google Scholar] [CrossRef]
Figure 1.
(a) Representative snapshots of MD systems for graphene-based EDLCs and schematics of solvent molecules and dipole moment calculations. Gray spheres: carbon atoms; blue spheres: Na+ ions; green spheres: Cl− ions; red spheres: O ions; white spheres: H ions. (b) Number density of electrolytes nearby the uncharged graphene electrode.
Figure 1.
(a) Representative snapshots of MD systems for graphene-based EDLCs and schematics of solvent molecules and dipole moment calculations. Gray spheres: carbon atoms; blue spheres: Na+ ions; green spheres: Cl− ions; red spheres: O ions; white spheres: H ions. (b) Number density of electrolytes nearby the uncharged graphene electrode.

Figure 2.
Number density profiles of electrolytes for graphene electrodes at the surface charge densities of (a) −5 μC cm−2 and (b) +5 μC cm−2.
Figure 2.
Number density profiles of electrolytes for graphene electrodes at the surface charge densities of (a) −5 μC cm−2 and (b) +5 μC cm−2.

Figure 3.
Electric field distributions near the (a) negative and (b) positive graphene electrodes; (c) EDL thickness and (d) ionic separation degree at different solvent polarities.
Figure 3.
Electric field distributions near the (a) negative and (b) positive graphene electrodes; (c) EDL thickness and (d) ionic separation degree at different solvent polarities.

Figure 4.
Electric potential distributions ϕ(z) near the (a) negative and (b) positive graphene electrodes; (c) specific capacitance for graphene electrodes at different solvent polarities.
Figure 4.
Electric potential distributions ϕ(z) near the (a) negative and (b) positive graphene electrodes; (c) specific capacitance for graphene electrodes at different solvent polarities.

Figure 5.
Self-diffusion coefficients for cations, anions, and solvents as a function of dipole moment.
Figure 5.
Self-diffusion coefficients for cations, anions, and solvents as a function of dipole moment.

Figure 6.
Accumulated number density for (a) counterions and (b) coions near the uncharged and charged graphene electrodes; (c) charging mechanism parameter λ as a function of solvent dipole moment; (d) schematic of charging mechanisms from counterion adsorption to coion desorption.
Figure 6.
Accumulated number density for (a) counterions and (b) coions near the uncharged and charged graphene electrodes; (c) charging mechanism parameter λ as a function of solvent dipole moment; (d) schematic of charging mechanisms from counterion adsorption to coion desorption.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, C.; Zhu, J.; Li, D.; Qian, X.; Chen, G.; Yang, H. Unveiling the Effects of Solvent Polarity within Graphene Based Electric Double-Layer Capacitors. Energies 2022, 15, 9487. https://doi.org/10.3390/en15249487
AMA Style
Xu C, Zhu J, Li D, Qian X, Chen G, Yang H. Unveiling the Effects of Solvent Polarity within Graphene Based Electric Double-Layer Capacitors. Energies. 2022; 15(24):9487. https://doi.org/10.3390/en15249487
Chicago/Turabian StyleXu, Chenxuan, Jingdong Zhu, Dedi Li, Xu Qian, Gang Chen, and Huachao Yang. 2022. "Unveiling the Effects of Solvent Polarity within Graphene Based Electric Double-Layer Capacitors" Energies 15, no. 24: 9487. https://doi.org/10.3390/en15249487
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.