
Reactivities of Hydroxycinnamic Acid Derivatives Involving Caffeic Acid toward Electrogenerated Superoxide in N,N-Dimethylformamide



1
Department of Pharmacy, Gifu Pharmaceutical University, Daigaku-Nishi, Gifu 501-1196, Japan
2
Faculty of Pharmacy, Gifu University of Medical Science, 4-3-3 Nijigaoka, Kani, Gifu 509-0923, Japan
*
Author to whom correspondence should be addressed.
Electrochem 2022, 3(3), 347-360; https://doi.org/10.3390/electrochem3030024
Submission received: 31 May 2022
/
Revised: 27 June 2022
/
Accepted: 3 July 2022
/
Published: 5 July 2022
(This article belongs to the Collection Feature Papers in Electrochemistry)
Abstract
:Reactivity of (2E)-3-(3,4-dihydroxyphenyl)prop-2-enoic acid (caffeic acid), classified as a hydroxycinnamic acid (HCA) derivative, toward electrogenerated superoxide radical anion (O2•−) was investigated through cyclic voltammetry, in situ electrolytic electron spin resonance spectrometry, and in situ electrolytic ultraviolet–visible spectrometry in N,N-dimethylformamide (DMF), aided by density functional theory (DFT) calculations. The quasi-reversible redox of dioxygen/O2•− is modified in the presence of caffeic acid, suggesting that O2•− is scavenged by caffeic acid through proton-coupled electron transfer. The reactivities of caffeic acid toward O2•− are mediated by the ortho-diphenol (catechol) moiety rather than by the acryloyl group, as experimentally confirmed in comparative analyses with other HCAs. The electrochemical and DFT results in DMF suggested that a concerted two-proton-coupled electron transfer mechanism proceeds via the catechol moiety. This mechanism embodies the superior kinetics of O2•− scavenging by caffeic acid.
1. Introduction
Caffeic acid ((2E)-3-(3,4-dihydroxyphenyl)prop-2-enoic acid, CafH2(COOH)) is a natural phenolic compound classified as a hydroxycinnamic acid (HCA) derivative. All plants produce caffeic acid as an intermediate in the biosynthesis of lignin, a principal component of woody plant biomass [1]. CafH2(COOH) delivers a surprising number of health benefits; for example, it mitigates carcinogenesis, neurodegeneration, and other age-related diseases, and induces pharmacological activities such as immunomodulatory and anti-inflammatory effects [2,3,4,5]. The carcinogenicities and anticancer activities of CafH2(COOH) have been enthusiastically researched but with mixed results [2,6]. CafH2(COOH) can target several diseases by scavenging the reactive oxygen species (ROS) generated in biochemical processes and pathologies. Therefore, ROS scavenging by HCAs involving CafH2(COOH) as an antioxidant might play a therapeutic role. However, whether CafH2(COOH) exerts a substantial effect on any human disease or delivers health benefits to humans remains unclear because solid evidence is lacking.
To confirm the medicinal effect of HCAs, especially of CafH2(COOH) that possesses antioxidant activity, we must elucidate the chemical reaction mechanism, including the electron transfer (ET) reaction that scavenges ROS such as superoxide radical anions (O2•−, the precursor of the hydroperoxyl radical, HO2•) and the hydroxyl radical (HO•). Clarifying the mechanism of ROS scavenging by HCAs is necessary for understanding the related medicinal effect, as the ROS generated around lesions and inflammatory organs are direct causes of several pathologies [4,6]. Previous studies in the field of electrochemistry have reported how HCA is oxidized by electrodes in aqueous or non-aqueous (aprotic) solvents [7,8,9], where the reactivity of HCAs toward the ROS was imitated by the heterogeneous electrochemical process. In these studies, antioxidant activity of HCA was assessed using cyclicvoltammetry, although the electrooxidation behavior varies depending on the presence or absence of protons. In the pioneering work by Hotta et al. [9], a well-developed two-electron reversible redox of CafH2(COOH) in acidic-aqueous media was observed, demonstrating that protons from the aqueous solvent relate to the oxidation. Conversely, cyclic voltammograms (CVs) of CafH2(COOH) in an aprotic acetonitrile solvent demonstrated an irreversible oxidation, as shown in the pioneering work by Masek et al. [8]. These electrochemical results imply that the chemical reaction mechanism involving proton transfer (PT) and ET between HCAs and the ROS cannot be observed in aqueous solvents, because solvent-derived protons interfere with the PT from hydroxyl groups of HCA. As is widely known, effective antioxidant activity usually requires the presence of ortho- or para-diphenolic hydroxyl groups (OH) for quinone–hydroquinone π-conjugation. CafH2(COOH) belongs to the phenylpropanoids, possessing both an ortho-diphenol (catechol, CatH2) moiety and an acryloyl group linked via a C6–C3 skeleton. Antioxidant studies of CafH2(COOH) employing numerous assays and methodologies have been published, such as 1,1-diphenyl-2-picryl-hydrazyl free radical scavenging, 2-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) radical scavenging, total antioxidant activity by the ferric thiocyanate method, and O2•− scavenging with metal chelating activities [2,3]. Meanwhile, computational studies have provided mechanistic insights into the structure–antioxidant activity relationships of HCAs [10,11,12,13]. These studies evaluated the thermochemical properties of HCAs, such as their bond dissociation energies of OH and ionization potentials, thereby clarifying the energetics of the ROS-scavenging reaction. A pioneering work by Nsangou et al. [14] investigated the antioxidant activity of CafH2(COOH) toward HO2• and HO•. Being more reactive than O2•−, these radicals are substantial cellular toxins in the living body. From the structure–property relationships of ROS scavenging by CafH2(COOH), the activity was mainly contributed by the CatH2 moiety, which has a high degree of conjugation and can delocalize π-electrons with a resonance structure [11,13]. The characteristic resonance effect of CafH2(COOH) is considered to thermodynamically stabilize its radical product. However, isolated HO2• and HO• are highly reactive and their reactions are difficult to observe experimentally; consequently, conclusive evidence of the ROS-scavenging mechanism of CafH2(COOH) is lacking in the literature.
There are several possible mechanisms of ROS (O2•−, HO•, and HO2•) scavenging by HCAs: single ET, hydrogen atom transfer (HAT) involving proton-coupled electron transfer (PCET) [15,16,17,18,19], sequential proton-loss electron transfer [20], and superoxide-facilitated oxidation (SFO) [21,22,23]. Along the SFO mechanism, the initial PT from the substrate to O2•− generates HO2•, and a rapid dismutation into hydroperoxide (H2O2) and dioxygen (O2) follows. Then, the O2 formed in the dismutation process oxidizes the substrate anion [24]. On the other side, the other mechanism involves direct ET. In an aprotic solution, various phenolic antioxidants, polyphenols [18], 1,2- and 1,4-benzendiols (catechol [16] and hydroquinone [17]), and monophenols including aminophenols [19,25,26], scavenge electrogenerated O2•− through the PCET characterized by quinone–hydroquinone π-conjugation. Although O2•− is not so electrophilic but a Brønsted base, HO2• formed through the protonation of O2•− is a strong oxidant with a short lifetime. Therefore, the initial PT and subsequent oxidation (ET) are closely related to each other through the PCET between CafH2(COOH)/anion and O2•−/HO2• that embodies the actual mechanism of O2•−/HO2• scavenging.
In this study, we investigate the homogeneous chemical reactivities between electrogenerated O2•− and HCAs involving CafH2(COOH) using cyclicvoltammetry, in situ electrolytic electron spin resonance (ESR) spectral measurements, and in situ ultraviolet–visible (UV–vis) spectral measurements in N,N-dimethylformamide (DMF) solution. Avoiding the interference of solvent-derived protons, the ROS scavenging reaction by HCAs is inferred from the electrochemical measurements by applying density functional theory (DFT). In addition, this study provides mechanistic insights into the structural features of CafH2(COOH), namely, the CatH2 moiety and the acryloyl group for the PCET mechanism characterized by π-conjugated redox reaction. Accordingly, we present valuable information regarding O2•− scavenging by HCAs including CafH2(COOH), which is assumed to provide substantial health benefits as a phytoalexin.
2. Materials and Methods
2.1. Chemicals
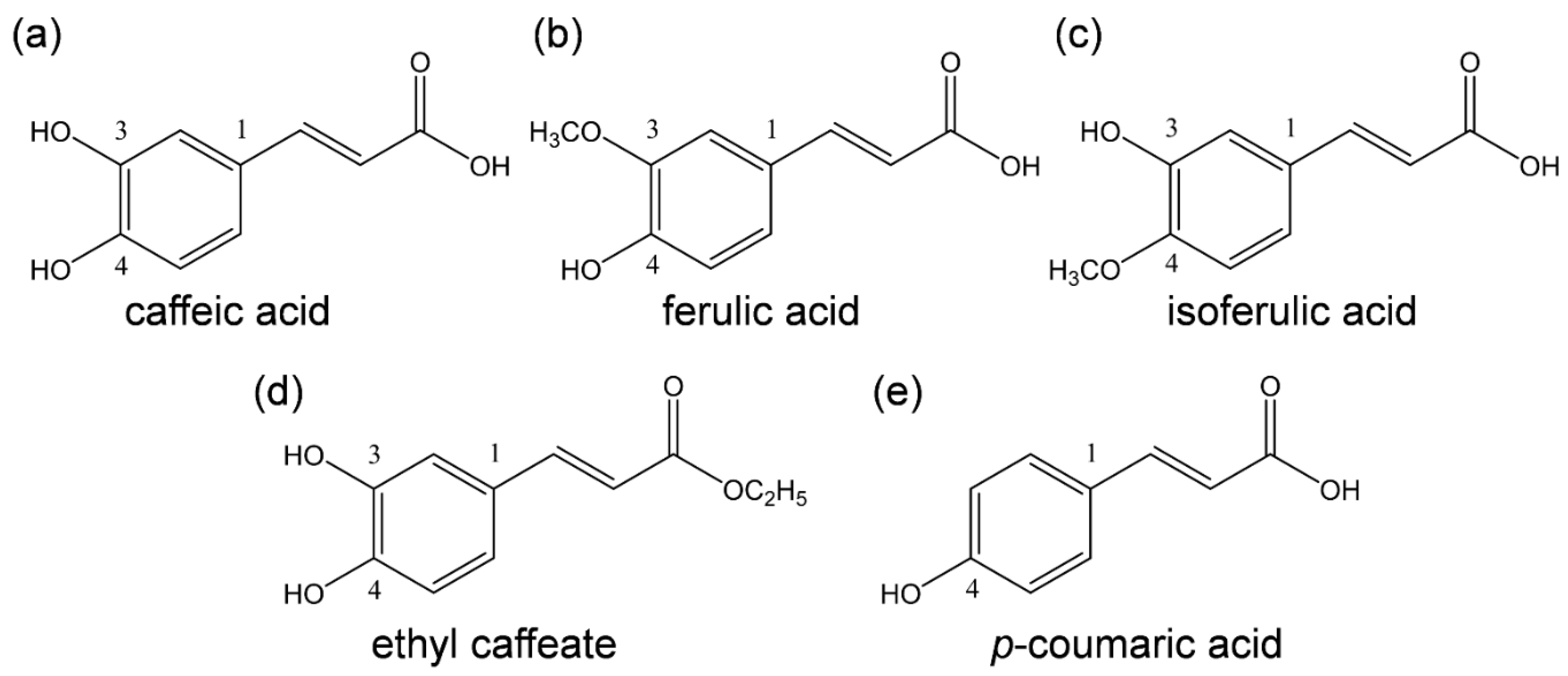
Highest-grade (2E)-3-(3,4-dihydroxyphenyl)prop-2-enoic acid (caffeic acid, CafH2(COOH), >98.0%), (2E)-3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic acid (ferulic acid, >99.0%), (E)-3-(3-hydroxy-4-methoxyphenyl)prop-2-enoic acid (isoferulic acid, >97.0%), ethyl (E)-3-(3,4-dihydroxyphenyl)prop-2-enoate (ethyl caffeate, Et-CafH2, >95.0%), (E)-3-(4-hydroxyphenyl)-2-propenoic acid (p-coumaric acid, >98.0%), and sodium methoxide (CH3ONa, 95.0%) were purchased from Sigma-Aldrich Inc. (Tokyo, Japan) and were used as received. DMF (spectrograde, 99.7%) was used as the solvent for electrochemical and electrolytic ESR/UV–vis spectral measurements, and ferrocene (Fc) used as a potential reference compound, were purchased from Nacalai Tesque Inc. (Kyoto, Japan) and used as received. Dinitrogen (N2) gas (99.0%) and O2 gas (99.0%) were obtained from Medical Sakai Co., Ltd. (Gifu, Japan). Tetrapropylammonium perchlorate (TPAP, >98.0%) used as a supporting electrolyte was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan) and prepared as described previously [27].
2.2. Cyclic Voltammetry and In Situ Electrolytic ESR/UV–Vis Spectrum Measurements
Cyclic voltammetry was performed in a standard three-electrode system: working electrode, a 1.0-mm-diameter glassy carbon (GC); counter electrode, a coiled platinum (Pt, 99.99%); reference electrode, a silver/silver nitrate (Ag/AgNO3) (containing an acetonitrile solution of 0.1 mol dm−3 tetrabutylammonium perchlorate and 0.01 mol dm−3 AgNO3). All measurements were performed at 25 °C using an ECstat-301 electrochemical analyzer (EC-frontier Co., Ltd., Kyoto, Japan) and electrochemical software (Supplementary Materials, Table S1). Before the experiments, the working electrode was polished with alumina paste on a polishing wheel, rinsed with deionized water and acetone, and dried. Calibration of the reference electrode was referenced to the ferrocenim ion/ferrocene couple (Fc+/Fc), and all reported potentials are referenced to the potential of the Fc+/Fc couple. A JES-FA200 X-band spectrometer (JEOL Ltd., Tokyo, Japan) and an OCEAN HDX spectrometer (OptoSirius Co., Ltd., Tokyo, Japan) were used for ESR and UV–vis spectral measurements, respectively. The controlled-potential electrolysis was performed at room temperature in two types of cells: an in situ electrolytic ESR cell with a 0.5-mm-diameter straight Pt wire sealed in a glass capillary as the working electrode (5.0-mm-length), and an optically transparent thin-layer electrochemical (OTTLE) cell (path length: 1.0 mm) with a Pt-mesh working electrode (10.0 mm × 20.0 mm) sandwiched between two pieces of quartz glass (Supplementary Materials, Figure S1). The working electrodes (planar glassy carbon, Pt wire, and Pt mesh) were chosen because they do not cause side reactions such as electrode-absorption with solutions or substrates. Samples were prepared in a glove box completely filled with N2 gas to prevent contamination by moisture. The DMF solutions were saturated with O2 by air-bubbling the gas for ca. 2–3 min, and the gas was passed over the solutions during the measurements to maintain a constant concentration of O2 at 4.8 × 10−3 mol dm−3.
2.3. Theoretical Calculations
All calculations were performed at the DFT level with the Becke three-parameter Lee–Yang–Parr (B3LYP) hybrid functional implemented in the Gaussian 16 Program package [28]. This functional was chosen because it obtains good geometries of the reactants, transition states (TS), and products in PCET reactions between phenolic compounds and free radicals [29]. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were computed from geometry optimization with frontier orbital theory. In the calculations for geometry optimization, vibrational frequency calculation, intrinsic reaction coordinate (IRC) calculation, and population analysis of each compound, we employed the standard split-valence triple ζ basis sets augmented by the polarization 3df,2p and diffusion orbitals 6-311+G(3df,2p). The polarized continuum model (PCM) under the default settings of Gaussian 16 was used to estimate the solvent contribution of DMF to the standard Gibbs free energies, which is widely employed in thermodynamic characteristic studies of solvation. The zero-point energies and thermal correction, together with the entropy, were used to convert the internal energies to standard Gibbs energies at 298.15 K. The natural bond orbital (NBO) technique was used for electron and spin calculations in the population analysis [30].
3. Results and Discussion
3.1. Cyclic Voltammetry of O2/O2•− in the Presence of HCAs
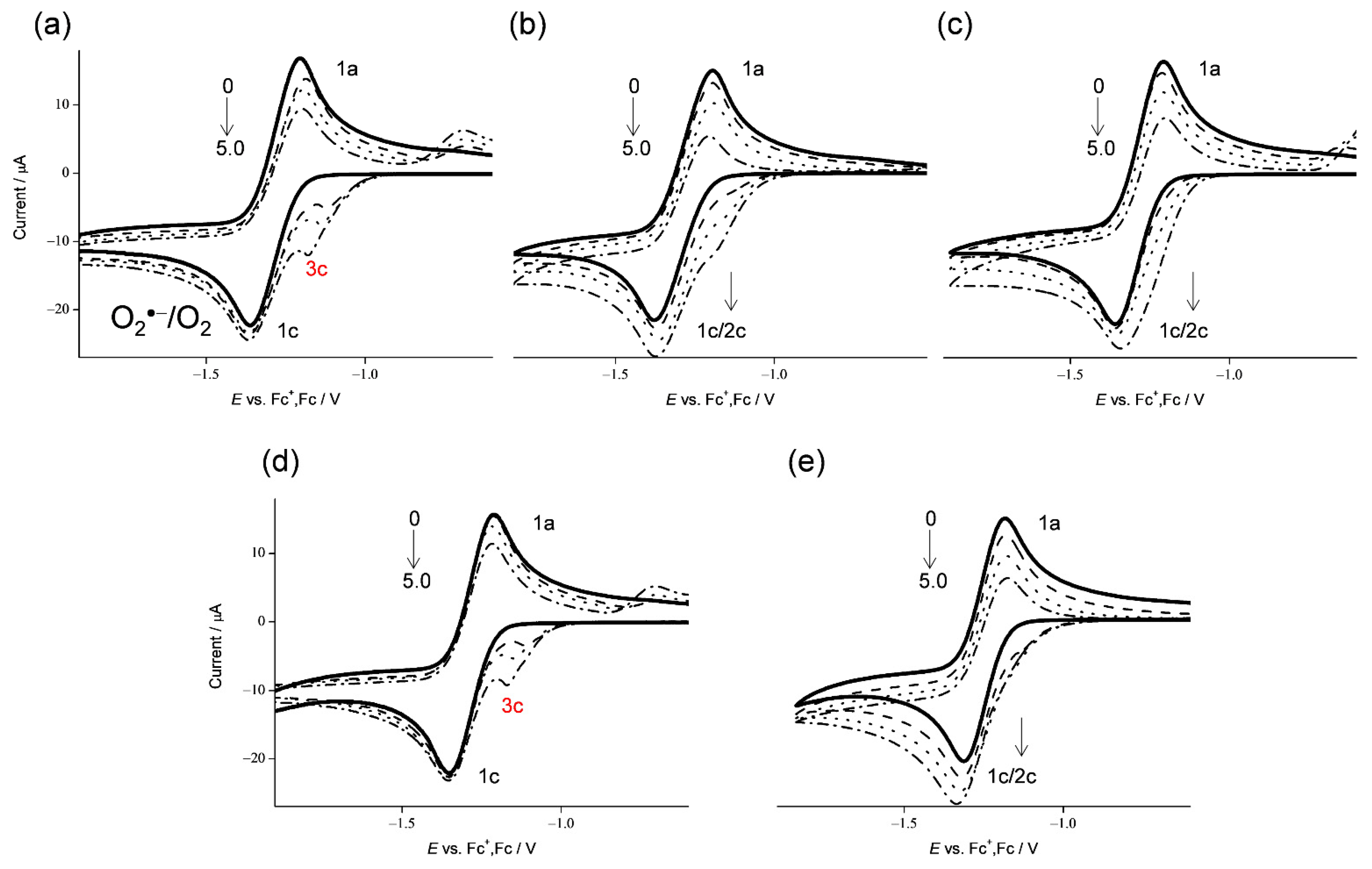
Reactivities of HCAs shown in Scheme 1 toward electrogenerated O2•− were demonstrated in the CVs of 4.8 × 10−3 mol dm−3 of O2 in the presence of HCAs in DMF (Figure 1). In aprotic solvents such as DMF, the relationship between ET and PT is well demonstrated in electrochemical and electrolytic-spectral measurements, avoiding interference of solvent-derived protons. In the CVs, one-electron reduction of O2 (Equation (1)) is demonstrated in a quasi-reversible redox reaction with the initial cathodic scan generating O2•− and following reoxidation to O2 in the returned anodic scan (1c/1a, bold lines in Figure 1). The generated O2•− is lowly reactive toward aprotic DMF. The reversible CVs investigated here became irreversible in the presence of phenolic HCAs (Figure 1a–e) at various concentrations (0 to 5.0 × 10−3 mol dm−3). As the CVs of bubbled N2 showed no peak over the potential range, the loss of reversibility in the CVs of O2/O2•− was attributed to an acid–base reaction; specifically, the O2•− acts as a Brønsted base forming HO2• along the initial PT (Equation (2)).
The initial PT and consequent reduction of HO2• (Equation (3)) gave rise to bielectronic CVs involving its cathodic current (2c) in the presence of ferulic acid, isoferulic acid, and p-coumaric acid (Figure 1b,c,e, respectively). This bielectronic CV did not appear in the presence of CafH2(COOH) and Et-CafH2 (Figure 1a,d, respectively) and was replaced by a prepeak (3c), demonstrating scavenging of HO2• by ET from the deprotonated anions (CafH(COOH)− and Et-CafH−) (Equation (4)) forming substrate radicals (CafH(COOH)• and Et-CafH•) and hydroperoxyl anion (HO2−). Consequently, a monoelectronic CV appeared.
O2 + e− ↔ O2•− (E° = −1.284 V vs. Fc+/Fc)
O2•− + CafH2 → HO2• + CafH(COOH)− (the initial PT)
HO2• + e− → HO2− (E° = −0.4 to −0.2 V vs. Fc+/Fc)
HO2• + CafH(COOH)− → HO2− + CafH(COOH)• (ET)
HO2− + CafH(COOH)• → H2O2 + Caf(COOH)•− (the second PT)
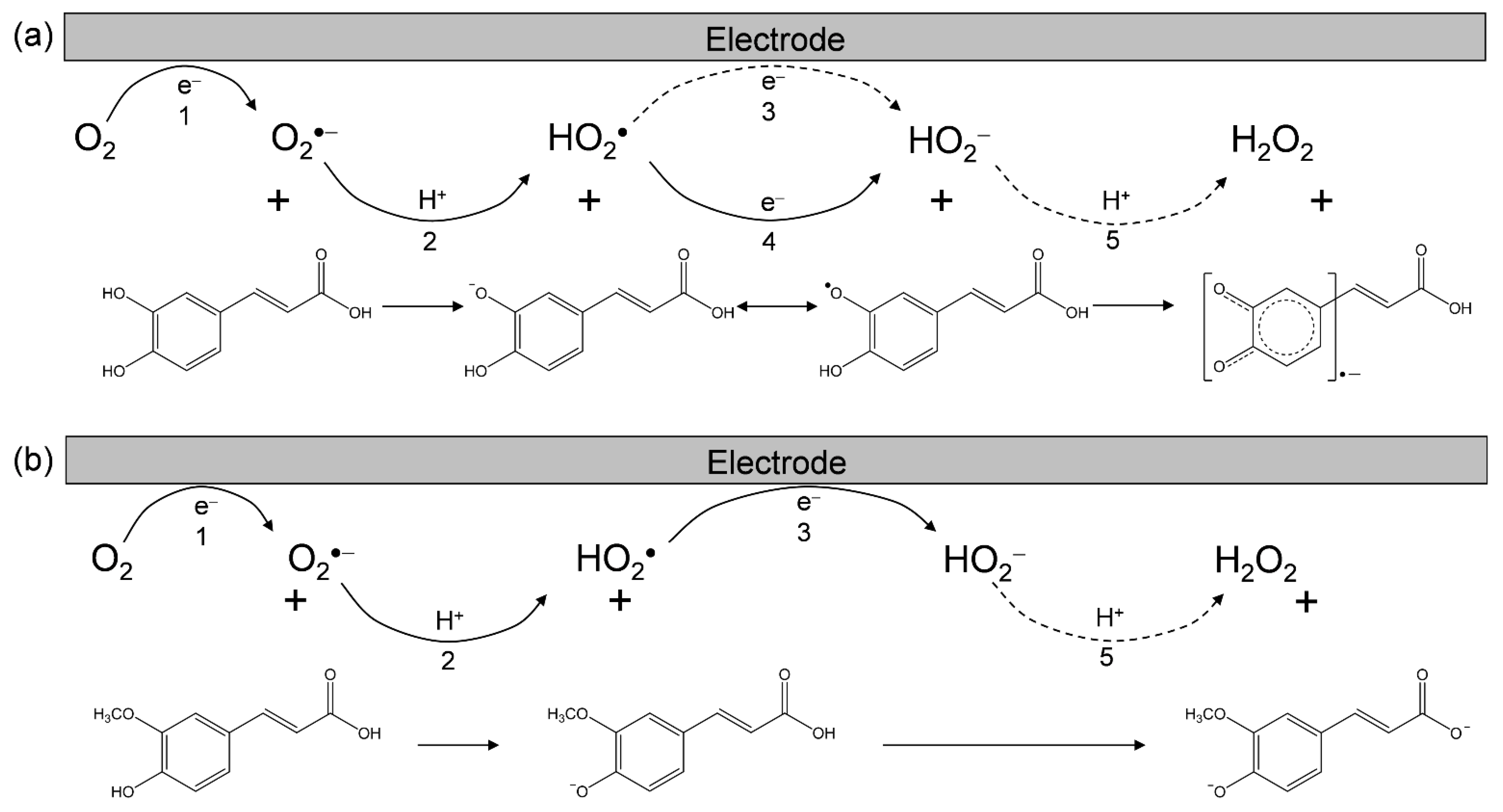
Based on the CVs, we rationalized that O2•− formation associated with PT from HCA leads to the irreversible bielectronic reduction of O2 to H2O2 driven by the exergonic reduction of HO2•/HO2−. Additionally, we divided the CVs of O2/O2•− in the presence of HCAs (Figure 1a–e) into two typical curves: type A, an irreversible two-electron process observed in electro–chemical–electro reactions (Equations (1)–(3)); and type B, an irreversible one-electron process observed in electro–chemical–chemical reactions (Equations (1), (2), (4) and (5)) leading to the scavenging of O2•−. Figure 2 summarizes Equations (1)–(5) showing the electrochemical mechanisms of O2/O2•− in the presence of (a) CafH2(COOH) and (b) ferulic acid.
In this scenario, the CVs (Figure 1) recorded in the presence of ferulic acid, isoferulic acid, and p-coumaric acid were type A, demonstrating the absence of O2•− scavenging and the appearance of a cathodic-current of HO2•. Conversely, the CVs in the presence of CafH2(COOH) and Et-CafH2 were type B, demonstrating the scavenging of O2•−/HO2•. The prepeak in the CVs of type B is mainly ascribed to radical anions (Caf(COOH)•− and Et-Caf•−), which are formed through the PCET from the CatH2 moiety of the substrate. The slightly different cathodic prepeaks in Figure 1a,d can be explained by the reduction of HO2• via PT from the acryloyl group of CafH2(COOH). These CV results suggest a PCET reaction between O2•− and its CatH2 moiety, which is independent of PT from the acryloyl group.
3.2. In Situ Electrolytic ESR/UV–Vis Spectral Analyses of O2/O2•− in the Presence of HCAs
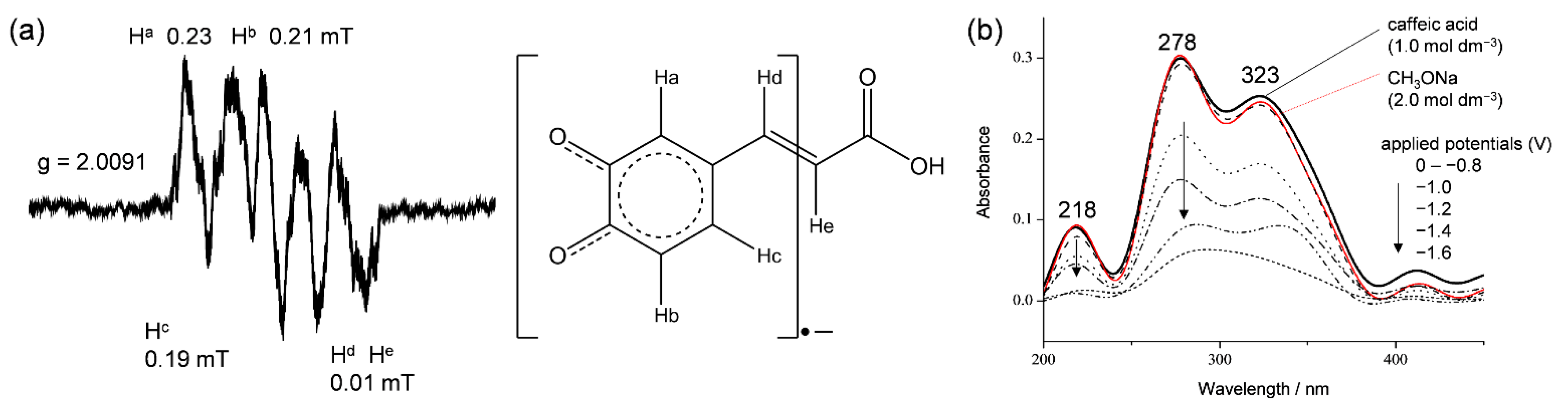
To confirm the O2•− scavenging by HCAs, the solutions of the CV experiment were analyzed by electrolytic ESR using an in situ cell (scanning time = 4 min) and by UV–vis spectral measurements using an OTTLE cell (Supplementary Materials, Figure S1). These spectra were acquired under a potential applied at −1.3 V, which corresponds to the electrogeneration of O2•− (Equation (1)). ESR spectra were obtained only in the presence of CafH2(COOH) and Et-CafH2, suggesting that O2•− was scavenged in these cases (Figure 3a). An almost identical ESR spectrum was obtained in the presence of Et-CafH2 (data not shown). In these ESR spectra, the fine coupling constants for hydrogen (aH/mT) were assigned to hydrogens of CatH2 (Ha, Hb, Hc: 0.23, 0.21, 0.19 mT) in the substrate radical anions (Caf(COOH)•− and Et-Caf•−) formed through the PCET. This result demonstrates that radical spins are barely distributed on the carboxyl group of CafH2(COOH) and the ethyl ester group of Et-CafH2. The in situ ESR system, which is sensitive to hyperfine couplings, showed no clear splitting derived from acryloyl hydrogens (Hd, He: 0.01 mT).
Figure 3b shows the in situ electrolytic UV–vis spectra of the CV solution containing CafH2(COOH) (1.0 × 10−3 mol dm−3). These spectra were measured in the absence and presence of O2 (in the former case, O2 was removed by purging with N2). The spectrum of CafH2(COOH) alone presented characteristic absorption bands centered at 218, 278, and 323 nm. This spectrum was unchanged under an applied potential of 1.0 to −2.0 V vs. Fc+/Fc without O2 (data not shown), demonstrating that CafH2(COOH) was not electrolyzed without deprotonation. Additionally, the spectrum did not alter in the presence of 2.0 × 10−3 mol dm−3 CH3ONa without applying potentials (red line in Figure 3b). As CafH2(COOH) is deprotonated by the Brønsted base CH3ONa, this spectrum was attributed to a mixture of CafH2(COOH), CafH(COOH)−, and CafH2(COO)−. Under O2 (4.8 × 10−3 mol dm−3), the spectrum diminished at applied cathodic potentials above −1.0 V. From these spectral changes, the homogeneous reaction between CafH2(COOH) and the electrogenerated O2•− was inferred to involve the initial PT and the subsequent ET forming Caf(COOH)•−. This radical product was detectable in the in situ electrolytic ESR system, conversely undetectable in the UV–vis spectra, showing a decomposition of the radical product (Caf(COOH)•−). Analogously to the CV results, the spectra revealed that the initial PT (Equation (2)) and the following reactions involving ET between HO2• and CafH(COOH)− (Equation (4)) rapidly underwent base-catalyzed oxidation. The CV and spectral measurements imply that O2•− was successfully scavenged by CafH2(COOH) through the PCET over the time scale of the CV measurements.
3.3. DFT Optimization of the Stable Structure of CafH2(COOH) and Its Deprotonated Anion
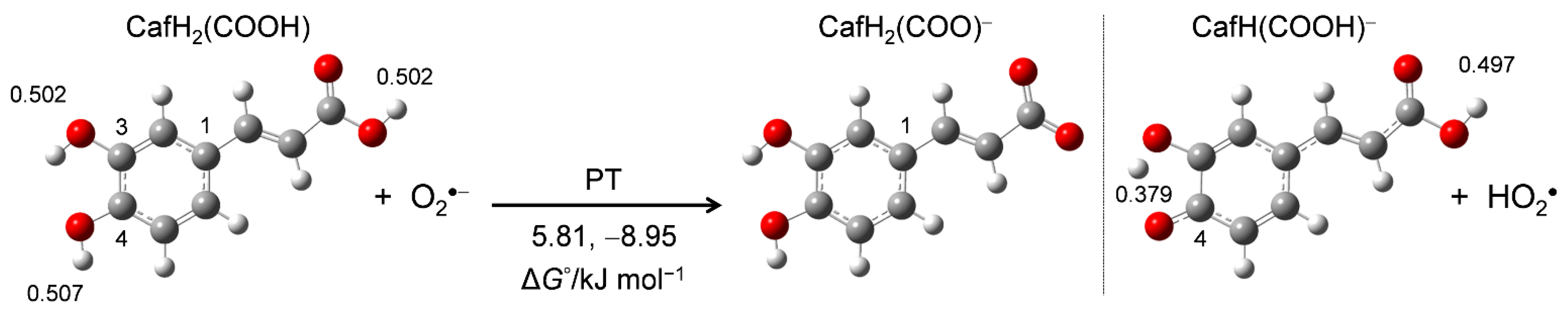
To clarify the mechanism of the PCET between CafH2(COOH) and O2•− in DMF, we performed DFT calculations using the B3LYP hybrid functional employing the PCM method. First, the stable structures of CafH2(COOH) and the conformers of its deprotonated anions (CafH2(COO)−, CafH(COOH)−) along the initial PT route were obtained from energy scanning of the dihedral angle around the acryloyl group. Figure 4 shows the optimized structures along with their calculated standard Gibbs free energy changes (ΔG°/kJ mol−1, 298.15 K) along the PT route. The distributed charges on the OH protons of CafH2(COOH) were obtained in an NBO analysis (Supplementary Materials, Table S2).
Comparing the charge distributions on the three OH protons of CafH2(COOH) (0.502 on 3OH, 0.507 on 4OH, and 0.502 on 1OH of the acryloyl group), one observes that the 4OH proton was more reactive in the acid–base reaction of O2•− than the other OH protons. The ΔG°s of the initial PT also show the higher reaction feasibility of CafH(COOH)− (−8.95 kJ mol−1) than of CafH2(COO)− (5.81 kJ mol−1). According to these calculation results, PT is initiated at the OH of the catechol moiety, although the 3OH and 4OH are indistinguishable along the dihedral rotation. The electrochemical results indicate the occurrence of PT from the three OHs of CafH2(COOH), but only 3OH and 4OH of the CatH2 moiety are involved in the O2•− scavenging reaction.
3.4. Change in HOMO-LUMO Energies during PCET between CafH2(COOH) and O2•−
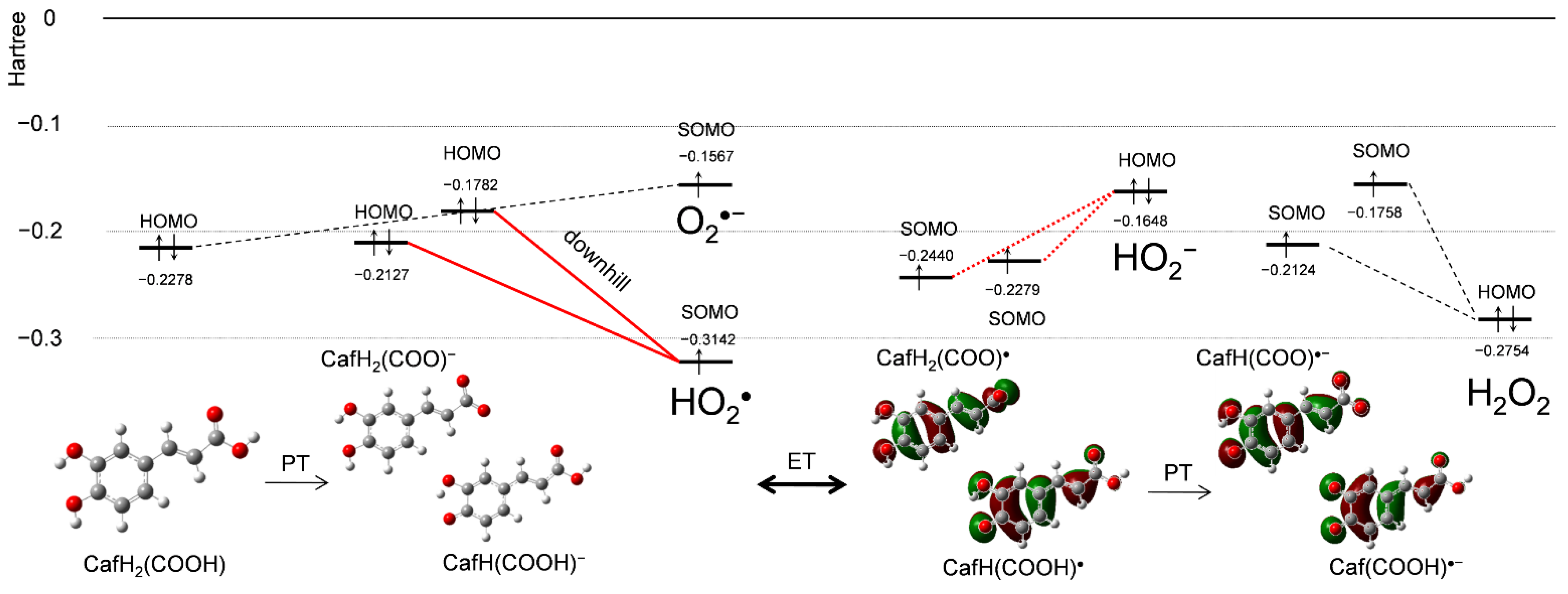
Figure 5 shows changes in HOMO-LUMO (the singly occupied molecular orbital, SOMO) energies (Hartree/a.u.) during the PCET between CafH2(COOH) and O2•− in DMF, supplemented by a frontier molecular-orbital analysis. After the initial PT, the reactant species CafH2(COOH), O2•−, CafH(COOH)−, (CafH2(COO)−), and HO2• are involved in the solution. The SOMO energy of HO2• (−0.3142) is much lower than the HOMO energies of CafH2(COOH) (−0.2278) and its anions (CafH(COOH)−, −0.1782; CafH2(COO)−, −0.2127), indicating that the electron acceptor is HO2• rather than O2•−. Figure 1a demonstrates that HO2• was scavenged, and thus, the electron donor is CafH(COOH)−/CafH2(COO)− where the downhill energy relationship during the ET is shown (bold red lines in Figure 5). These HOMO-LUMO changes occur during the PT between CafH2(COOH) and O2•− forming CafH(COOH)−/CafH2(COO)− and HO2•, implying that the initial reaction is PT. Next, the HOMO-LUMO relationship between the products is reversed after the subsequent ET (red dotted lines in Figure 5). However, since H2O2 formed after the second PT has a lower HOMO (−0.2754) than HO2− (−0.1648), the reverse ET cannot proceed, so the ET direction is dominantly determined by the subsequent PT. Judging from the HOMO-LUMO relationship for O2•− scavenging by CafH2(COOH), the net PCET involves two PTs and one ET.
3.5. Change in Free Energies during PCET between CafH2(COOH) and O2•−
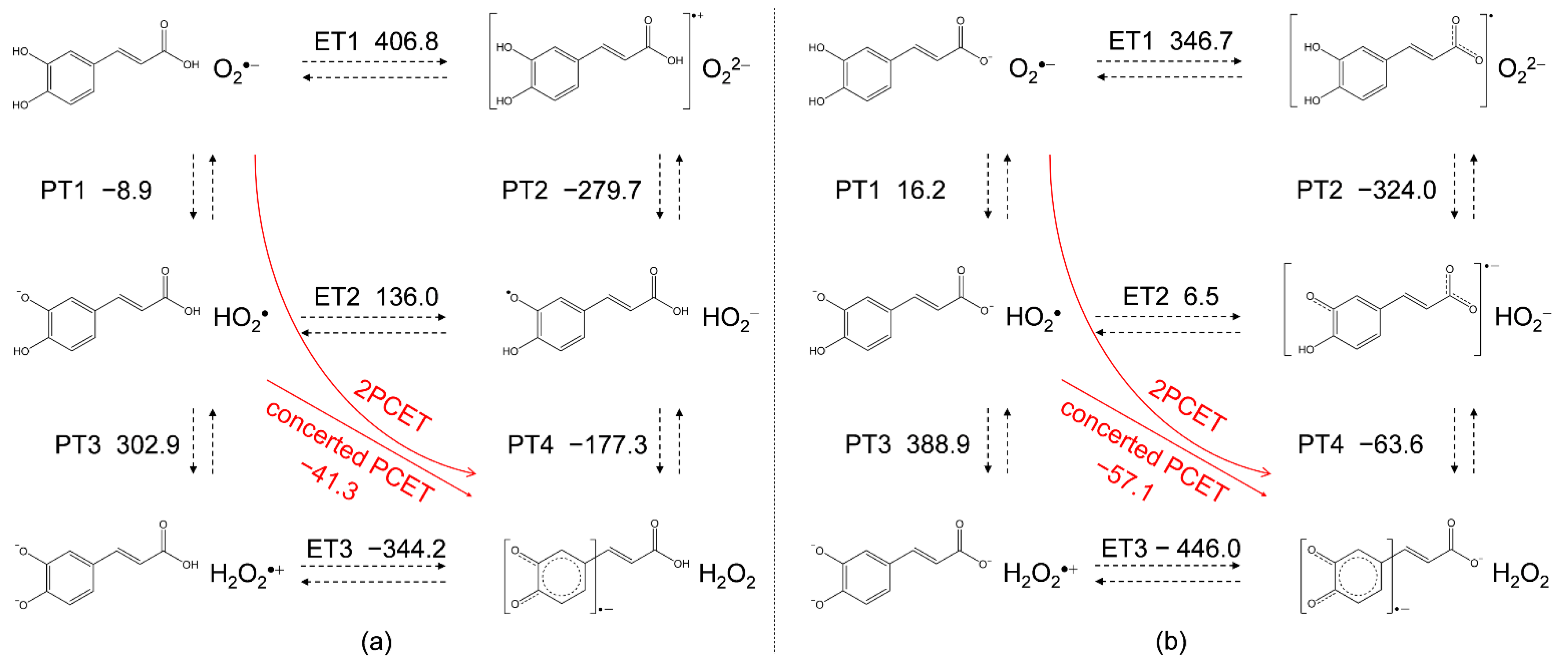
Vibrational frequency calculations were performed to obtain changes in standard Gibbs free energy (ΔG°/kJ mol−1, 298.15 K) along the PCET in DMF. Figure 6 shows the equilibrium schemes and ΔG° of the six diabatic electronic states in the PCET involving two PTs and one ET between CafH2(COOH) and O2•−, and CafH2(COO)− formed after the deprotonation of acryloyl OH and O2•−. The main drivers of these sequential processes are the ΔG°s of the individual reactions, the acid–base interaction and the redox potentials of the components. Since ET1 (a, 406.8; b, 346.7) is strongly endergonic, PT1 (a, −8.9; b, 16.2) dominantly occurs forming CafH(COOH)−/CafH2(COO)− and HO2•. In the following pathway (the lower panels in Figure 6a,b), both PT3 (a, 302.9; b, 388.9) and ET2 (136.0, 6.5) are uphill endergonic, so the sequential PCET is unlikely to proceed. Efficient O2•− scavenging then requires a concerted PCET or HAT reaction involving one ET and one PT, corresponding to movement along the diagonal of the lower panels (red straight arrows). Notably, PT4 in Figure 6b is exergonic (−63.6), meaning that two PTs from the CatH2 moiety must be coupled to one ET for successful O2•− scavenging. That is, PT from the acryloyl OH does not associate with ET. Then, another plausible pathway is a concerted ET and two PTs in one step after preforming the HB complexes between O2•− and the CatH2 moiety of CafH2(COOH)/CafH2 (COO)− without generating high energy intermediates (red curved arrows). We refer to this pathway as the concerted two-proton-coupled electron transfer (2PCET) mechanism [16,29].
For a comparative study, the ΔG°s of the PCET of the other compounds were also calculated (Table 1). Thermodynamically, the total ΔG° of the net PCET obtained by summing the ΔG°s of the two PTs and one ET is the energetic driving force. However, in case the PCET occurs along a pathway involving an infeasible single PT/ET, the total ΔG° cannot embody the energetic driving force. Along the plausible pathways, the concerted PCET (ET2–PT4/PT3–ET3) after the initial PT1, and 2PCET, was exergonic for all compounds. Therefore, the total ΔG° embodies the exergonic driving force similar to the case of CafH2(COOH) (concerted, −41.3; total, −50.2). These ΔG°s cannot explain the higher reactivities of CafH2(COOH) and Et-CafH2 than the other compounds toward electrogenerated O2•−. In the electrochemical results (Figure 1 and Figure 2), ferulic acid, isoferulic acid, and p-coumaric acid did not show O2•−/HO2• scavenging ability, indicating that the CatH2 moiety of CafH2(COOH) and Et-CafH2 confers the superior kinetics of the O2•− scavenging on CV time scales.
3.6. Potential-Energy Surfaces of the PCET between CafH2(COOH) and O2•−
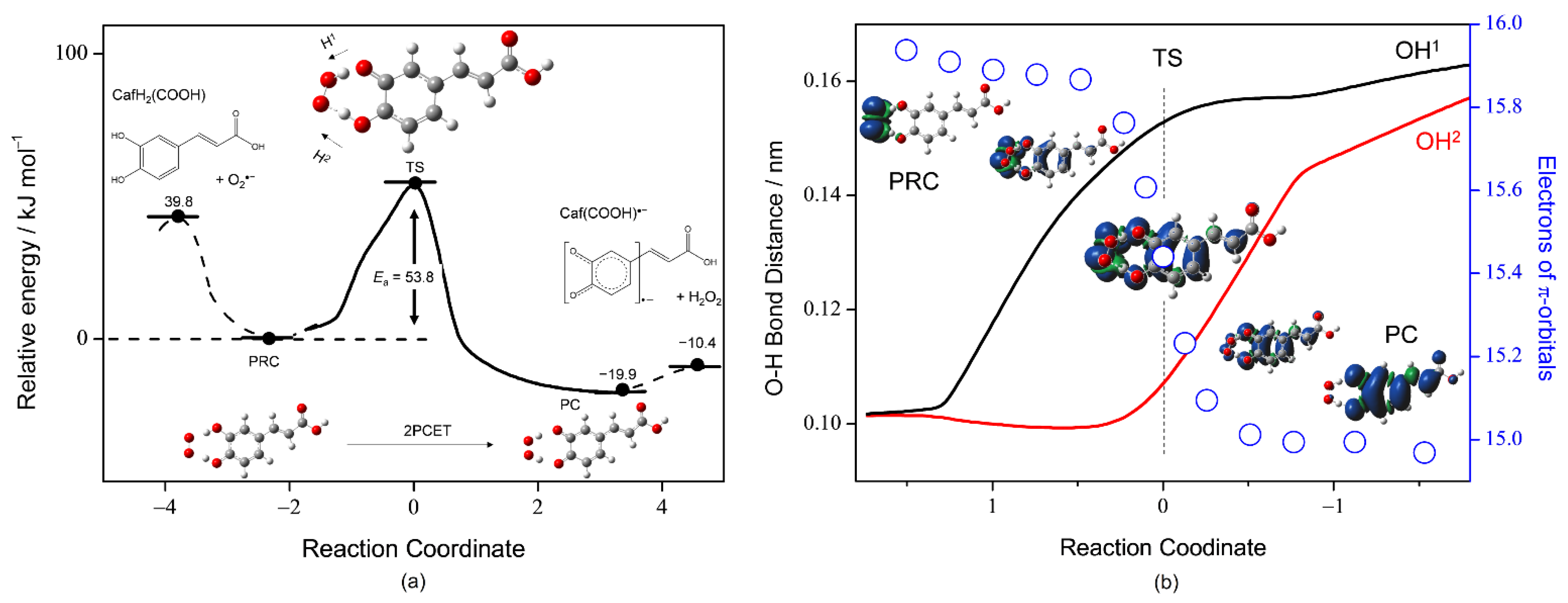
To gain more insight into the PCET mechanism of O2•− scavenging by CafH2(COOH) in DMF, we investigated the potential-energy surfaces with DFT and NBO analyses. The reaction is assumed to involve three elementary steps: (i) formation of the prereactive HB complex (PRC) from the free reactants (FRs), (ii) PCET reaction to the product complex (PC) via a TS, and (iii) dissociation of the PC yielding free products (FPs). First, we performed a geometry optimization of the stable HB complexes (PRC, intermediate complex, and PC) along the PCET reaction (Figure 7a). Then, optimized structures of the plausible PRC (CafH2(COOH)–O2•−) and PC (Caf(COOH)•−–H2O2) formed from the FRs and the FPs via two HBs (step i) were obtained. After optimization, the ΔG° was reduced by 39.8 kJ mol−1 (the ΔG° of the PRC was set to zero in Figure 7a). Then, an energy profile (ΔG°, kJ mol−1) along the IRC for the 2PCET, which forms the PC (step ii). The IRC shows that 2PCET occurs between the CatH2 moiety of CafH2(COOH) and O2•− in a one-kinetic process via a TS with low activation energy (Ea) at 53.8 kJ mol−1 without generating any intermediates such as HO2•, HO2−, CafH(COOH)−, and CafH(COOH)•.
The dependence of OH bond distances (OH1, black line; OH2, red line) on the number of electrons in the π-orbital of the planar molecule of CafH2(COOH) (blue circles) along the IRC are shown in Figure 7b. Spin density distributions localized on the radicals before and after the TS are also demonstrated, where the radical localized on O2•− in the initial PRC is transferred to Caf(COOH)•− in the resulting PC. The changes in spin on the electron-donor side (CafH2(COOH)) correlated well with the changes in the π-electrons of CafH2(COOH). Furthermore, the changes in structures coupled with π-electron transfer occur simultaneously with sequential lengthening of the two OH bond distances (OH1/OH2) of the CatH2 moiety. First, one phenolic proton (H1) is attracted by O2•−. At the TS, this attraction results in nearly complete deprotonation (OH1) with one-half of the π-electrons transferred from CafH2(COOH) to O2•−. The second PT (H2) accelerates the ET forward from the TS, eventually forming the PC.
For a comparative study, the potential-energy surfaces of the PCET with an IRC and TS between O2•− and the other substrates—CafH2(COO)−, Et-CafH2, and CatH2—were also investigated (Supplementary Materials, Figure S2). The 2PCET mechanisms were similarly mediated by the CatH2 moiety and generated no intermediates. Table 2 lists the ΔG° of FR, PC, and FP, and Ea (the geometries of the TS are given in Tables S3–S5 of the Supplementary Materials). The Eas of CafH2(COOH) (53.8 kJ mol−1), CafH2(COOH)− (50.0 kJ mol−1), Et-CafH2 (53.2 kJ mol−1), and CatH2 (52.5 kJ mol−1) were very similar and as low as the hydrogen-bonding energy. In Table 2, the relationship in which the difference in the Ea values is proportional to the difference in their enthalpy of reaction shown as the ΔG°s of the PC suggests that the 2PCET mechanism of the same framework (between CatH2 moiety and O2•−) follows the Bell–Evans–Polanyi principle [31], justifying the conclusion that “CafH2(COOH) scavenges O2•− through the 2PCET” and the methods adopted in this study. The ΔG°s of dissociating the PC to the FPs were −10.4 kJ mol−1 for CafH2(COOH), −10.8 kJ mol−1 for CafH2(COO)−, and −10.0 kJ mol−1 for Et-CafH2, 55–56 kJ mol−1 lower than that for CatH2 (45.3 kJ mol−1). Thus, the superior O2•−-scavenging ability of CafH2(COOH) is mainly attributable to the ΔG°s of dissociating the PC (step iii) that promote the net PCET reaction, rather than to the kinetics of 2PCET via the TS (step ii).
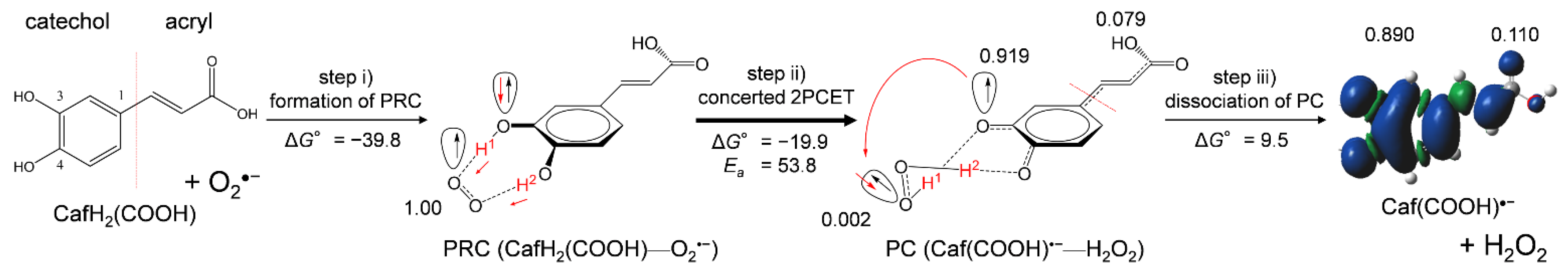
Collectively, DFT results clarified that scavenging of O2•− by CafH2(COOH) in DMF is governed by 2PCET involving two PTs and one ET in the formed PRC via two HBs, which corresponds to moving along the red curved arrows in Figure 6a. Figure 8 shows the net mechanism of O2•− scavenging by CafH2(COOH) in DMF. In the 2PCET mechanism, ET occurs between the π orbitals of oxygen orthogonal to the molecular frameworks of the donor and acceptor, and PT occurs between the σ orbitals of oxygen along the HBs [16]. The numbers of spin distributed on the oxygen species (O2•−, H2O2), the CatH2 moiety, and the acryloyl group, along step iii, the dissociation of the PC (9.5 kJ mol−1), are also given in Figure 8. Notably, the spins are distributed on the acryloyl group of the PC (0.079) and on the dissociated Caf(COOH)•− (0.110) that expands the π-conjugated plane, demonstrating that the radical product (PC and Caf(COOH)•−) is more stabilized by the acryloyl group than the orthoquinone radical generated from CatH2. These results are in good correlation with the ΔG°s of dissociating PC to FP (Table 2), suggesting that the acryloyl group of CafH2(COOH), CafH2(COO)−, and Et-CafH2, thermodynamically promotes step iii, the dissociation of PC. These results imply that the acryloyl group play a promoting role in the net 2PCET mechanism (Figure 8) between O2•− and the catechol moiety via the dissociation (step iii) for efficient O2•− scavenging abilities of CafH2(COOH).
4. Conclusions
In the present study, we investigated the reactivities of HCAs involving CafH2(COOH) toward the electrogenerated O2•− in DMF. In the CV and spectral measurements, it was confirmed that O2•− was successfully scavenged by CafH2(COOH) and Et-CafH2 through PCET mediated by the CatH2 moiety over the time scale of the CV measurements. Then, DFT calculations clarified that the concerted 2PCET mechanism involving two PTs and one ET via the CatH2 moiety (not involving PT from the acryloyl group) embodies the superior kinetics of O2•− scavenging by CafH2(COOH) and Et-CafH2. The net mechanism involves the initial formation of PRC followed by 2PCET and dissociation of the PC into FPs. The acryloyl group contributes to the thermodynamic stability of the product radical, promoting the dissociation of the PC. Efficient O2•− scavenging abilities of CafH2(COOH) and Et-CafH2 are derived from both the CatH2 moiety and the acryloyl group.
Although the presented results are specific to chemical reactions in aprotic DMF solvent, the PCET theory is adaptable to biological processes in biotic structures such as lipid bilayers. Therefore, we hope that the findings will reveal the mechanistic actions of O2•− scavenging by HCAs and the health benefits of CafH2(COOH), thus securing its pharmacological use as a phytoalexin.
Supplementary Materials
The following is available online at https://www.mdpi.com/article/10.3390/electrochem3030024/s1, Table S1: CV parameters, Figure S1: In situ electrolytic ESR/UV-vis system, Table S2: Optimized geometry of caffeic acid, Figure S2: Energy profiles along IRC of 2PCET between CafH2(COO−) and O2•−, Tables S3–S6: Optimized geometry of TS.
Author Contributions
Conceptualization, T.N.; methodology, B.U.; resources, T.N.; writing—original draft preparation, T.N.; writing—review and editing, B.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Grant-in-Aid for Scientific Research, grant number 19K16338 from Japan Society for the Promotion of Science (JSPS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, I. Antioxidant activity of caffeic acid (3,4-dihydroxycinnamic acid). Toxicology 2006, 217, 213–220. [Google Scholar] [CrossRef]
- Hong Chen, J.; Ho, C.-T. Antioxidant Activities of Caffeic Acid and Its Related Hydroxycinnamic Acid Compounds. J. Agric. Food Chem. 1997, 45, 2374–2378. [Google Scholar] [CrossRef]
- Zielińska, D.; Zieliński, H.; Laparra-Llopis, J.M.; Szawara-Nowak, D.; Honke, J.; Giménez-Bastida, J.A. Caffeic acid modulates processes associated with intestinal inflammation. Nutrients 2021, 13, 554. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Itagaki, S.; Kurokawa, T.; Ogura, J.; Kobayashi, M.; Hirano, T.; Sugawara, M.; Iseki, K. In vitro and in vivo antioxidant properties of chlorogenic acid and caffeic acid. Int. J. Pharm. 2011, 403, 136–138. [Google Scholar] [CrossRef]
- Nasr Bouzaiene, N.; Kilani Jaziri, S.; Kovacic, H.; Chekir-Ghedira, L.; Ghedira, K.; Luis, J. The effects of caffeic, coumaric and ferulic acids on proliferation, superoxide production, adhesion and migration of human tumor cells in vitro. Eur. J. Pharmacol. 2015, 766, 99–105. [Google Scholar] [CrossRef]
- Simić, A.; Manojlović, D.; Šegan, D.; Todorović, M. Electrochemical behavior and antioxidant and prooxidant activity of natural phenolics. Molecules 2007, 12, 2327–2340. [Google Scholar] [CrossRef] [Green Version]
- Masek, A.; Chrzescijanska, E.; Latos, M. Determination of antioxidant activity of caffeic acid and p-coumaric acid by using electrochemical and spectrophotometric assays. Int. J. Electrochem. Sci. 2016, 11, 10644–10658. [Google Scholar] [CrossRef]
- Hotta, H.; Ueda, M.; Nagano, S.; Tsujino, Y.; Koyama, J.; Osakai, T. Mechanistic study of the oxidation of caffeic acid by digital simulation of cyclic voltammograms. Anal. Biochem. 2002, 303, 66–72. [Google Scholar] [CrossRef]
- VanBesiena, E.; Marques, M.P.M. Ab initio conformational study of caffeic acid. J. Mol. Struct. THEOCHEM 2003, 625, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, C.; da Silva Miranda, F.; Gonçalves, N.S.; Spinelli, A. Antioxidant activity of phenolic and related compounds: A density functional theory study on the O-H bond dissociation enthalpy. Redox Rep. 2004, 9, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, G.; Russo, N.; Toscano, M. Antioxidant properties comparative study of natural hydroxycinnamic acids and structurally modified derivatives: Computational insights. Comput. Theor. Chem. 2016, 1077, 39–47. [Google Scholar] [CrossRef]
- Amić, A.; Marković, Z.; Klein, E.; Dimitrić Marković, J.M.; Milenković, D. Theoretical study of the thermodynamics of the mechanisms underlying antiradical activity of cinnamic acid derivatives. Food Chem. 2018, 246, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Nsangou, M.; Fifen, J.J.; Dhaouadi, Z.; Lahmar, S. Hydrogen atom transfer in the reaction of hydroxycinnamic acids with {radical dot}OH and {radical dot}HO2 radicals: DFT study. J. Mol. Struct. THEOCHEM 2008, 862, 53–59. [Google Scholar] [CrossRef]
- Singh, P.S.; Evans, D.H. Study of the electrochemical reduction of dioxygen in acetonitrile in the presence of weak acids. J. Phys. Chem. B 2006, 110, 637–644. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Concerted two-proton-coupled electron transfer from catechols to superoxide via hydrogen bonds. Electrochim. Acta 2016, 208, 304–309. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Quinone-hydroquinone π-conjugated redox reaction involving proton-coupled electron transfer plays an important role in scavenging superoxide by polyphenolic antioxidants. Chem. Lett. 2010, 39, 162–164. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Importance of proton-coupled electron transfer from natural phenolic compounds in superoxide scavenging. Chem. Pharm. Bull. 2015, 63, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Uno, B. Structural properties of 4-substituted phenols capable of proton-coupled electron transfer to superoxide. Int. J. Adv. Res. Chem. Sci. 2016, 3, 11–19. [Google Scholar] [CrossRef]
- Biela, M.; Rimarčík, J.; Senajová, E.; Kleinová, A.; Klein, E. Antioxidant action of deprotonated flavonoids: Thermodynamics of sequential proton-loss electron-transfer. Phytochemistry 2020, 180, 112528. [Google Scholar] [CrossRef]
- Nanni, E.J.; Birge, R.R.; Hubbard, L.M.; Morrison, M.M.; Sawyer, D.T. Oxidation and dismutation of superoxide ion solutions to molecular oxygen. singlet vs. triplet state. Inorg. Chem. 1981, 20, 737–741. [Google Scholar] [CrossRef]
- Nanni, E.J.; Stallings, M.D.; Sawyer, D.T. Does superoxide ion oxidize catechol, α-tocopherol, and ascorbic acid by direct electron transfer? J. Am. Chem. Soc. 1980, 102, 4481–4485. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. Electrocatalytic oxygen reduction reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications; Springer: Berlin, Germany, 2008; pp. 89–134. ISBN 9781848009356. [Google Scholar]
- Fridovich, I. Superoxide dismutase. In Encyclopedia of Biological Chemistry, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 352–354. ISBN 9780123786319. [Google Scholar]
- Nakayama, T.; Honda, R. Electrochemical and Mechanistic Study of Superoxide Elimination by Mesalazine through Proton-Coupled Electron Transfer. Pharmaceuticals 2021, 14, 120. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R.; Kuwata, K.; Usui, S.; Uno, B. Electrochemical and mechanistic study of reactivities of α-, β-, γ-, and δ-tocopherol toward electrogenerated superoxide in N,N-dimethylformamide through proton-coupled electron transfer. Antioxidants 2022, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Uno, B. Electronic spectra of the electrogenerated 1,4-benzoquinone π-dianion and the strongly hydrogen-bonded charge-transfer complex with methanol. Bull. Chem. Soc. Jpn. 1999, 72, 1213–1217. [Google Scholar] [CrossRef]
- Frisch, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; ISBN 9781935522027. [Google Scholar]
- Quintero-Saumeth, J.; Rincón, D.A.; Doerr, M.; Daza, M.C. Concerted double proton-transfer electron-transfer between catechol and superoxide radical anion. Phys. Chem. Chem. Phys. 2017, 19, 26179–26190. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, X.; Zhai, Y.; Zhang, L.; Zhang, P.; Sheng, L. A theoretical investigation on Bell-Evans-Polanyi correlations for hydrogen abstraction reactions of large biodiesel molecules by H and OH radicals. Combust. Flame 2020, 214, 394–406. [Google Scholar] [CrossRef]
Scheme 1.
Structures of HCAs considered in this study. (a) (2E)-3-(3,4-Dihydroxyphenyl)prop-2-enoic acid (caffeic acid), (b) (2E)-3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic acid (ferulic acid), (c) (E)-3-(3-hydroxy-4-methoxyphenyl)prop-2-enoic acid (isoferulic acid), (d) ethyl (E)-3-(3,4-dihydroxyphenyl)prop-2-enoate (ethyl caffeate), and (e) (E)-3-(4-hydroxyphenyl)-2-propenoic acid (p-coumaric acid).
Scheme 1.
Structures of HCAs considered in this study. (a) (2E)-3-(3,4-Dihydroxyphenyl)prop-2-enoic acid (caffeic acid), (b) (2E)-3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic acid (ferulic acid), (c) (E)-3-(3-hydroxy-4-methoxyphenyl)prop-2-enoic acid (isoferulic acid), (d) ethyl (E)-3-(3,4-dihydroxyphenyl)prop-2-enoate (ethyl caffeate), and (e) (E)-3-(4-hydroxyphenyl)-2-propenoic acid (p-coumaric acid).

Figure 1.
CVs of 4.8 × 10−3 mol dm−3 O2 in the presence of (a) CafH2(COOH), (b) ferulic acid, (c) isoferulic acid, (d) Et-CafH2, and (e) p-coumaric acid, in DMF containing 0.1 mol dm−3 TPAP. All CVs were recorded with a GC electrode at a scan rate of 0.1 V s−1. Concentrations (×10−3 mol dm−3) are 0, 1.0, 3.0, and 5.0 (arrows indicate the concentration changes).
Figure 1.
CVs of 4.8 × 10−3 mol dm−3 O2 in the presence of (a) CafH2(COOH), (b) ferulic acid, (c) isoferulic acid, (d) Et-CafH2, and (e) p-coumaric acid, in DMF containing 0.1 mol dm−3 TPAP. All CVs were recorded with a GC electrode at a scan rate of 0.1 V s−1. Concentrations (×10−3 mol dm−3) are 0, 1.0, 3.0, and 5.0 (arrows indicate the concentration changes).

Figure 2.
Electrochemical mechanisms of O2/O2•− in the presence of (a) CafH2(COOH), and (b) ferulic acid in DMF. 1 one-electron reduction of O2/O2•−, 2 the initial PT from substrate to O2•−, 3 exergonic reduction of HO2•/HO2−, 4 ET from substrate anion to HO2•, 5 the second PT to HO2−.
Figure 2.
Electrochemical mechanisms of O2/O2•− in the presence of (a) CafH2(COOH), and (b) ferulic acid in DMF. 1 one-electron reduction of O2/O2•−, 2 the initial PT from substrate to O2•−, 3 exergonic reduction of HO2•/HO2−, 4 ET from substrate anion to HO2•, 5 the second PT to HO2−.

Figure 3.
(a) ESR and (b) UV–vis spectra of a O2/O2•− solution exposed to CafH2(COOH) in DMF under an applied potential of −1.3 V vs. Fc+/Fc.
Figure 3.
(a) ESR and (b) UV–vis spectra of a O2/O2•− solution exposed to CafH2(COOH) in DMF under an applied potential of −1.3 V vs. Fc+/Fc.

Figure 4.
Optimized structures of CafH2(COOH) and its deprotonated anion conformers (CafH2(COO)− and CafH(COOH)−) along the PT route to O2•− in DMF, calculated by DFT-B3LYP/PCM/6-311+G(3df,2p). The ΔG°s (kJ mol−1, 298.15 K) of PT and the NBO-determined charges distributed on the three OH protons of CafH2(COOH) are also indicated.
Figure 4.
Optimized structures of CafH2(COOH) and its deprotonated anion conformers (CafH2(COO)− and CafH(COOH)−) along the PT route to O2•− in DMF, calculated by DFT-B3LYP/PCM/6-311+G(3df,2p). The ΔG°s (kJ mol−1, 298.15 K) of PT and the NBO-determined charges distributed on the three OH protons of CafH2(COOH) are also indicated.

Figure 5.
Changes in the HOMO−LUMO energies (Hartree/a.u.) along PCET mechanism between CafH2(COOH) and O2•− and their corresponding chemical species in DMF, obtained by DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
Figure 5.
Changes in the HOMO−LUMO energies (Hartree/a.u.) along PCET mechanism between CafH2(COOH) and O2•− and their corresponding chemical species in DMF, obtained by DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.

Figure 6.
Six diabatic electronic states of PCET between (a) CafH2(COOH) and O2•−, and (b) CafH2(COO)− and O2•− in DMF, involving two PTs and one ET. The ΔG°s (kJ mol−1, 298.15 K) of PT1–PT4 and ET1–ET3 were calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
Figure 6.
Six diabatic electronic states of PCET between (a) CafH2(COOH) and O2•−, and (b) CafH2(COO)− and O2•− in DMF, involving two PTs and one ET. The ΔG°s (kJ mol−1, 298.15 K) of PT1–PT4 and ET1–ET3 were calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.

Figure 7.
(a) Energy profile (kJ mol−1) along the intrinsic reaction coordinate of the 2PCET between CafH2(COOH) and O2•− in DMF, along with the structures of free reactants, prereactive HB complex (PRC), transition state (TS), product complex (PC), and free products (Caf(COOH)•−, H2O2). (b) Changes in OH bond distances (nm, left vertical axes; OH1, black line; OH2, red line) and the number of electrons (open circles) on the π-orbitals of CafH2(COOH) (right vertical axes). All calculations were conducted by the DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level. Spin distributions and the electrons of π-orbitals were obtained by NBO analysis.
Figure 7.
(a) Energy profile (kJ mol−1) along the intrinsic reaction coordinate of the 2PCET between CafH2(COOH) and O2•− in DMF, along with the structures of free reactants, prereactive HB complex (PRC), transition state (TS), product complex (PC), and free products (Caf(COOH)•−, H2O2). (b) Changes in OH bond distances (nm, left vertical axes; OH1, black line; OH2, red line) and the number of electrons (open circles) on the π-orbitals of CafH2(COOH) (right vertical axes). All calculations were conducted by the DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level. Spin distributions and the electrons of π-orbitals were obtained by NBO analysis.

Figure 8.
Net PCET mechanism between CafH2(COOH) and O2•− in DMF with the ΔG° and Ea values (kJ mol−1, 298.15 K). Step (i) initial formation of PRC; step (ii) concerted 2PCET; step (iii) dissociation of the PC. ΔG° and Ea were calculated using the DFT-(U)B3LYP/PCM/6-311+G(3df,2p) method. The numbers are spins distributed on the oxygen species (O2•−, H2O2), the CatH2 moiety, and the acryloyl group obtained by NBO analysis.
Figure 8.
Net PCET mechanism between CafH2(COOH) and O2•− in DMF with the ΔG° and Ea values (kJ mol−1, 298.15 K). Step (i) initial formation of PRC; step (ii) concerted 2PCET; step (iii) dissociation of the PC. ΔG° and Ea were calculated using the DFT-(U)B3LYP/PCM/6-311+G(3df,2p) method. The numbers are spins distributed on the oxygen species (O2•−, H2O2), the CatH2 moiety, and the acryloyl group obtained by NBO analysis.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
ΔG°s (kJ mol−1, 298.15 K) of PCET between O2•− and HCAs (CafH2(COOH), ferulic acid, isoferulic acid, Et-CafH2, and p-coumaric acid) in DMF, calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
Table 1.
ΔG°s (kJ mol−1, 298.15 K) of PCET between O2•− and HCAs (CafH2(COOH), ferulic acid, isoferulic acid, Et-CafH2, and p-coumaric acid) in DMF, calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
| Compounds | PT1 | PT2 | PT3 | PT4 | ET1 | ET2 | ET3 | Concerted 1 | Total 2 |
|---|---|---|---|---|---|---|---|---|---|
| CafH2(COOH) | −8.9 | −279.7 | 302.9 | −177.3 | 406.8 | 136.0 | −344.2 | −41.3 | −50.2 |
| Ferulic acid | 6.2 | −476.9 | 313.1 | −79.6 | 532.2 | 49.0 | −343.6 | −30.5 | −24.3 |
| Isoferulic acid | 4.5 | −471.2 | 298.2 | −77.1 | 517.0 | 41.2 | −334.1 | −35.9 | −31.4 |
| Et-CafH2 | −5.4 | −365.8 | 371.3 | −85.9 | 402.9 | 42.5 | −414.6 | −43.3 | −48.7 |
| p-Coumaric acid | 7.7 | −353.7 | 301.9 | −79.4 | 420.1 | 58.6 | −322.8 | −20.8 | −13.1 |
1 Concerted values are the summed ΔG°s of ET2 and PT4 (ET3 and PT3). 2 Total values are the summed ΔG°s of two PTs and one ET.
Table 2.
ΔG° and Ea values (kJ mol−1, 298.15 K) of the 2PCET between O2•− and the substrates (CafH2(COOH), CafH2(COO)−, Et-CafH2, and CatH2) in DMF, calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
Table 2.
ΔG° and Ea values (kJ mol−1, 298.15 K) of the 2PCET between O2•− and the substrates (CafH2(COOH), CafH2(COO)−, Et-CafH2, and CatH2) in DMF, calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
| Reactants 1 | FR | TS (Ea) | PC | FP |
|---|---|---|---|---|
| CafH2(COOH) (+O2•−) | 39.8 | 53.8 | −19.9 | −10.4 |
| CafH2(COO)− (+O2•−) | 30.1 | 50.0 | −27.6 | −10.8 |
| Et-CafH2 (+O2•−) | 38.7 | 53.2 | −20.3 | −10.0 |
| CatH2 (+O2•−) | 71.6 | 52.5 | −20.9 | 45.3 |
1 ΔG°s (kJ mol−1) of PRC were set as a zero point.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nakayama, T.; Uno, B. Reactivities of Hydroxycinnamic Acid Derivatives Involving Caffeic Acid toward Electrogenerated Superoxide in N,N-Dimethylformamide. Electrochem 2022, 3, 347-360. https://doi.org/10.3390/electrochem3030024
AMA Style
Nakayama T, Uno B. Reactivities of Hydroxycinnamic Acid Derivatives Involving Caffeic Acid toward Electrogenerated Superoxide in N,N-Dimethylformamide. Electrochem. 2022; 3(3):347-360. https://doi.org/10.3390/electrochem3030024
Chicago/Turabian StyleNakayama, Tatsushi, and Bunji Uno. 2022. "Reactivities of Hydroxycinnamic Acid Derivatives Involving Caffeic Acid toward Electrogenerated Superoxide in N,N-Dimethylformamide" Electrochem 3, no. 3: 347-360. https://doi.org/10.3390/electrochem3030024