Emaciation, Congestive Heart Failure, and Systemic Amyloidosis in Severe Recessive Dystrophic Epidermolysis Bullosa: Possible Internal Complications Due to Skin-Derived Inflammatory Cytokines Derived from the Injured Skin

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
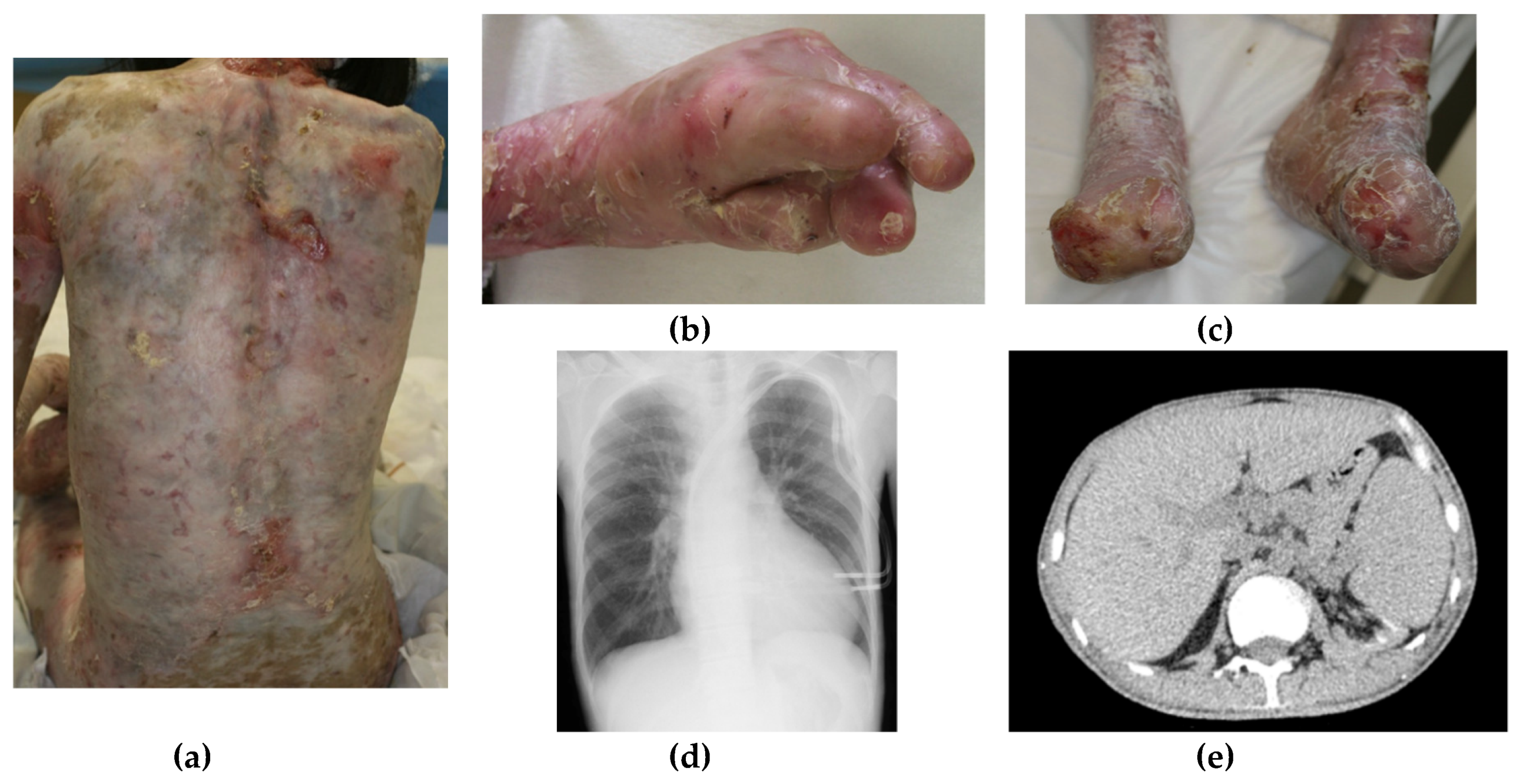
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Li, K.P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef]
- Fine, J.D.; Hall, M.; Weiner, M.; Li, K.P.; Suchindran, C. The risk of cardiomyopathy in inherited epidermolysis bullosa. Br. J. Dermatol. 2008, 159, 677–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Stein, A.; Cash, S.; DeLeoz, J.; Devries, D.T.; Suchindran, C.; National Epidermolysis Bullosa Registry. Inherited epidermolysis bullosa and the risk of death from renal disease: Experience of the National Epidermolysis Bullosa Registry. Am. J. Kidney Dis. 2004, 44, 651–660. [Google Scholar] [CrossRef]
- Csikós, M.; Orosz, Z.; Bottlik, G.; Szöcs, H.; Szalai, Z.; Rozgonyi, Z.; Hársing, J.; Török, E.; Bruckner-Tuderman, L.; Horváth, A.; et al. Dystrophic epidermolysis bullosa complicated by cutaneous squamous cell carcinoma and pulmonary and renal amyloidosis. Clin. Exp. Dermatol. 2003, 28, 163–166. [Google Scholar] [CrossRef]
- Gündüz, K.; Vatansever, S.; Türel, A.; Sen, S. Recessive dystrophic epidermolysis bullosa complicated with nephrotic syndrome due to secondary amyloidosis. Int. J. Dermatol. 2000, 39, 151–153. [Google Scholar] [CrossRef]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Luthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Kobayashi, Y. Selective release of a processed form of interleukin 1 alpha. Cytokine 1994, 6, 597–601. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 signaling pathway and its role in kidney disease: An update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Tanaka, M.; Tsutsui, H.; Kupper, T.S.; Asahi, K.; Okamura, H.; Nakanishi, K.; Suzuki, M.; Kayagaki, N.; Black, R.A.; et al. Skin-specific caspase-1-transgenic mice show cutaneous apoptosis and pre-endotoxin shock condition with a high serum level of IL-18. J. Immunol. 2000, 165, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Nakanishi, T.; Saito, H.; Maruyama, J.; Isoda, K.; Yokochi, A.; Imanaka-Yoshida, K.; Tsuda, K.; Kakeda, M.; Okamoto, R.; et al. Persistent release of IL-1s from skin is associated with systemic cardio-vascular disease, emaciation and systemic amyloidosis: The potential of anti-IL-1 therapy for systemic inflammatory diseases. PLoS ONE 2014, 9, e104479. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Mizutani, H.; Tsutsui, H.; Noben-Trauth, N.; Yamanaka, K.; Tanaka, M.; Izumi, S.; Okamura, H.; Paul, W.E.; Nakanishi, K. IL-18 induction of IgE: Dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 2000, 1, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Tsutsui, H.; Murakami, T.; Yumikura-Futatsugi, S.; Yamanaka, K.; Tanaka, M.; Iwakura, Y.; Suzuki, N.; Takeda, K.; Akira, S.; et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 11340–11345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Okada, K.; Nakanishi, T.; Mizutani, K.; Matsushima, Y.; Kondo, M.; Habe, K.; Mizutani, H.; Seo, N. Skin inflammation leads immunoglobulin G aggregation and deposition in multiple organs. J. Dermatol. Sci. 2017, 88, 146–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, S.; Guez, S.; Orenti, A.; Tadini, G.; Scuvera, G.; Corti, L.; Scala, A.; Biganzoli, E.; Berti, E.; Principi, N. Autoimmunity and cytokine imbalance in inherited epidermolysis bullosa. Int. J. Mol. Sci. 2016, 17, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horai, R.; Asano, M.; Sudo, K.; Kanuka, H.; Suzuki, M.; Nishihara, M.; Takahashi, M.; Iwakura, Y. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 1998, 187, 1463–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsushima, Y.; Mizutani, K.; Goto, H.; Nakanishi, T.; Kondo, M.; Habe, K.; Isoda, K.; Mizutani, H.; Yamanaka, K. Emaciation, Congestive Heart Failure, and Systemic Amyloidosis in Severe Recessive Dystrophic Epidermolysis Bullosa: Possible Internal Complications Due to Skin-Derived Inflammatory Cytokines Derived from the Injured Skin. Dermatopathology 2020, 7, 41-47. https://doi.org/10.3390/dermatopathology7020007
Matsushima Y, Mizutani K, Goto H, Nakanishi T, Kondo M, Habe K, Isoda K, Mizutani H, Yamanaka K. Emaciation, Congestive Heart Failure, and Systemic Amyloidosis in Severe Recessive Dystrophic Epidermolysis Bullosa: Possible Internal Complications Due to Skin-Derived Inflammatory Cytokines Derived from the Injured Skin. Dermatopathology. 2020; 7(2):41-47. https://doi.org/10.3390/dermatopathology7020007
Chicago/Turabian StyleMatsushima, Yoshiaki, Kento Mizutani, Hiroyuki Goto, Takehisa Nakanishi, Makoto Kondo, Koji Habe, Kenichi Isoda, Hitoshi Mizutani, and Keiichi Yamanaka. 2020. "Emaciation, Congestive Heart Failure, and Systemic Amyloidosis in Severe Recessive Dystrophic Epidermolysis Bullosa: Possible Internal Complications Due to Skin-Derived Inflammatory Cytokines Derived from the Injured Skin" Dermatopathology 7, no. 2: 41-47. https://doi.org/10.3390/dermatopathology7020007