Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes
by
 , , and
, , and
Benny Danilo Belviso
1 ,
,
Rosanna Caliandro
2,
Shabnam Majidi Salehi
3,
Gianluca Di Profio
3,* and
and
Rocco Caliandro
1,*
1
Institute of Crystallography, CNR, via Amendola 122/o, Bari 70126, Italy
2
Faculty of Science and Technology, Free University of Bozen, Piazza Università 5, Bolzano 39100, Italy
3
Institute on Membrane Technology, CNR, via P. Bucci 17/C, Rende 87036, Italy
*
Authors to whom correspondence should be addressed.
Crystals 2019, 9(5), 253; https://doi.org/10.3390/cryst9050253
Submission received: 22 March 2019
/
Revised: 14 May 2019
/
Accepted: 15 May 2019
/
Published: 17 May 2019
(This article belongs to the Special Issue Protein Crystallography)
Abstract
:Protein crystallization is a powerful purification tool. It is the first step for crystallographic structural investigations, and can be preparatory for biotechnological applications. However, crystallizing proteins is challenging and methods to control the crystallization process are needed. Ionic-liquid hydrogel composite membranes (IL-HCMs) have been used here as material capable of supporting protein crystallization and hosting grown crystals. We found that IL-HCMs affect the selection mechanism of glucose isomerase (GI) polymorphs and make GI crystals grow completely immersed into the hydrogel layer. X-ray diffraction studies show that IL ions do not bind to the protein, likely because IL molecules are constrained in the polymeric framework. Our GI crystal structures have been compared with many existing GI crystal structures using multivariate analysis tools, allowing a comprehensive overview of factors determining structural similarities, i.e., temperature variations and external stresses exerted during or after crystal growth, such as dehydration or presence of hydrogel of a different nature. GI crystals grown on IL-HCM fit perfectly in this framework, showing typical features induced by external forces. Overall, protein crystallization by IL-HCMs show potential for biotechnological applications, as it could constitute a natural means for containing crystallized enzymes in working conditions.
1. Introduction
Ionic liquids (ILs) are liquids consisting of positive (cation) and negative (anion) charges bound together by electrostatic interactions. The electrical mobility of ILs is generally lower than that of their corresponding ions free in aqueous solution, but it is still too high to allow the molecular packing observed in crystalline solid salts. Therefore, ILs can be considered as a sea of charges that do not organize into a regular structure, while still maintaining strong associations with each other. ILs have been extensively used as additives in protein crystallization [1,2,3,4,5,6] and many screenings using them are already on the market, i.e., the Ionic Liquid Screen, HR2-214 of Hampton Research.
The effect of ILs on protein intermolecular contacts has been understood via simulation studies. Free ionic monomers in the solvent offer the possibility to tune specific interactions, in particular anionic hydrogen bonding and cationic surfactant effects. As a consequence, polar and apolar domains can form in pure ILs, which critically influence protein crystallization. For example, longer cationic alkyl chains can enforce surfactant effects, thus positively affecting stability and activity of proteins, and providing the opportunity to crystallize membrane proteins. Simulations have shown that the concentration of cations at the protein surface exceeds that of anions, since cations do not contribute to the anionic hydrogen-bonded network with water. Excess cations interact with the protein surface at negatively charged areas, through strong Coulomb interactions, and at non-polar surface areas, through dispersion interactions mediated by their alkyl chains [7]. Instead anions strongly interact with positively charged areas through hydrogen bonds and Coulomb interactions. Both cations and anions have favorable solvation sites at the polar protein surface, and interact stronger with protein than water, with a higher residence time for anions and higher mobility for cations. As a result of the complex framework of IL-protein surface interactions, protein solubility is affected enough to induce protein precipitation and crystallization [7]. The impact of ILs on the crystallization process is even more pronounced than for precipitation, as they contribute to slowing down of the vapor transfer rate and to control crystal growth velocity [8]. For example, IL-based additives were found to improve crystal size, X-ray diffraction resolution, tolerance to impurities during the crystallization, and, more importantly, to enable polymorph selection, thus making the crystallization process more selective [7].
ILs are very attractive for technological applications, because the huge combination of salts forming ILs, the possibility to functionalize anion and cation, as well as the variety of gel preparation methods make such materials particularly adaptable and tailorable to the technical requirements of final applications [9]. Among these materials, IL hydrogels have very interesting properties for biotechnological applications, due their ability to incorporate active species such as enzymes for biocatalysis [10].
On the other hand, protein crystallization has been recently found to be enhanced by using composite materials formed by hydrophobic membranes and hydrogel layers [11]. Extensive literature reports about the advantages of using hydrophobic membranes to support protein crystallization. Membranes have the role of controlling vapor diffusion and inducing heterogeneous crystallization, where specific properties of the membrane surface are crucial in guiding the crystallization process [12]. The role of hydrogels in influencing crystal growth and affecting lattice properties of crystals in their final state is well known. Hydrogel composite membranes (HMCs) conjugate the advantages of membranes and hydrogels and have proven to be effective in enhancing nucleation and promoting crystal growth. More importantly, the use of hydrogel layers has paramount advantages in view of biotechnological exploitation of protein crystals, because, the hydrogel layer preserves embedded protein crystals from osmotic stresses (pH, T, P changes, effect of radicals), thus increasing their lifetime. Moreover, it has been found that enzymatic activity is enhanced by using proteins in the crystal form, so that lower quantities of enzymes can be used to obtain the same macroscopic effects (for example reducing the bacterial load in a food) of the same enzyme dissolved in solution or immobilized on films in the molecular form [13]. In summary, both crystallization and protein performance greatly benefit from the use of HMCs.
In this framework, in the present work we have explored the possibility of using HCMs made of hydrophobic membranes and IL-based hydrogels (IL-HCMs), both to support protein crystallization and to preserve crystal properties. Ionic-liquid hydrogels have been developed and properly layered on hydrophobic material by obtaining a composite material suitable for crystallization experiments at lab scale [14]. IL-HCMs have been tested by using a model protein for polymorphic studies such as glucose isomerase (GI) and, once structurally solved, our crystals have been compared with GI crystals grown on standard sitting-drop setup in the same crystallization conditions, and with other GI crystal structures present in the Protein Data Bank (PDB, www.rscb.org) [15].
2. Materials and Methods
2.1. IL-HCM Preparation
IL-HCMs have been produced as done in ref. [14] for membrane-contactors applications. Commercial polypropylene (PP) flat sheet membranes (Accurel PP 2E HF, nominal pore size 200 nm, overall porosity 70%) were purchased from Membrana GmbH (Wuppertal, Germany). [2-(Methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (SPE, cod. 537284), N,N′-Methylenebisacrylamide (MBA, cod. 146062), 2-hydroxy-2-methyl propiophenone (cod. 405655), and 1-Butyl-3-methylimidazolium hexafluorophosphate (IL, cod. 18122), used as monomer, cross-linker, photoinitiator, and ionic liquid additive, respectively, were bought from Sigma-Aldrich. Polypropylene membrane sheets were conditioned by soaking in methanol for 24 h at room temperature. Thereafter, they were dried between tissue paper immediately before use. The hydrogel solution (pre-polymerization solutions) was prepared by dissolving the monomer (10 %wt) and the cross-linker (1 %wt) in water, by magnetically stirring at 50 °C until complete dissolution. To this solution, the photoinitiator 0.3 %wt and the ionic liquid at different amount (0-1-5-10-15 %wt of monomer), were added. Solutions were then cast onto the PP substrate by rolling on the loaded support with a bar coater at 100 µm thickness. Photo-initiated graft polymerization was carried out immediately under an UV/Vis irradiation lamp (GR.E, 500 W) in a vented exposition chamber (Helios Italquartz, Italy), for 15 min. Thereafter, IL-HCMs were washed and stored in distilled water at room temperature for membrane crystallization tests. Composite membranes samples are designated as IL-HCM 0, IL-HCM 1, IL-HCM 2, IL-HCM 3, and IL-HCM 4 for ionic liquid content 0, 1, 5, 10 and 15 %wt of the monomer, respectively.
2.2. Protein Crystallization
Glucose Isomerase from S. rubiginosus (HR7-102, Hampton Research, Aliso Viejo, CA, USA), supplied as a crystal suspension in 6 mM TRIS hydrochloride buffer pH 7, 0.91 M Ammonium sulfate and 1 mM magnesium sulfate, was dialyzed against 10 mM HEPES buffer pH 7, and 1 mM Magnesium chloride. Before crystallization, protein was concentrated to 26 mg/mL and a solution of ammonium sulfate of 1.5 M has been prepared as reservoir solution. Crystallization experiments were performed by equilibrating 10 μL drop (1:1 volume ratio of protein solution and reservoir) against 0.5 mL of reservoir solution. Crystallization experiments were carried out by vapor diffusion technique using Cryschem sitting-drop 24-well plates: in the case of reference material, standard setup for sitting drop has been used, whilst in the case of tests for IL-HCMs, the drop was placed on support disk made of IL-HCM that, in turn, has been laid on the sitting-drop bridge. Crystallization tests were carried out at 20° C with three replications for each condition to test the reproductively of the results. Protein crystals were observed by optical microscope (DM 2500M, Leica Microsystems, Wetzlar, Germany) equipped with a video camera.
2.3. X-ray Diffraction Data Collection and Analysis
Diffraction properties of protein crystals were checked by using the X-ray beam generated at Diamond Light Source of Oxford, beamline I04. Data collections were carried out in cryogenic conditions (100 K), and at beam energy of 12658 eV. The XDS program [16] was used to perform data reduction, while POINTLESS and AIMLESS programs [17] were used to find space group symmetry and to scale and merge diffraction data. Crystal structures were solved by molecular replacement (MR), by using the REMO program [18] included in the package SIR2014 [19], and the crystal structures with PDB codes 4jkl and 4zbc as MR models for the respective P21212 and I222 structures. Matching between experimental and calculated electron densities was improved by performing an automatic building procedure on the MR structure in rebuilt-in-place mode, by using Autobuild wizard [20] included in PHENIX [21]. Final refinements to add water molecules were performed by using the program BUSTER v.2 [22], where water molecules having thermal factor > 80 Å2, 2F0-Fc < 1 σ, and distance from first-neighboring atom > 4 Å are removed. As a final step, water molecules were manually inspected by using the program COOT [23], and empty electron density map bumps have been filled with glycerol and sulfate ions, as they are both present in the crystallization solution. Structural models modified manually have been refined by using REFMAC5 [24]. IL-1 and REF-2 crystal structures were deposited in PDB (entry 6quk and 6quf, respectively), as the best representative of GI crystallized in the same conditions with and without IL-HCMs.
2.4. Structural Comparisons
New GI crystal structures obtained by crystallographic analysis here reported, denoted IL-1,2,3 and REF-1,2, were compared with other known GI crystal structures, selected in the PDB by querying with the sequence of our structure (GI from S. rubiginosus) and selecting the structures having I222 or P21212 space groups with a very significant E cut off (0.001). A total of 81 GI crystal structures have been so selected (67 having I222 crystal symmetry and 14 having P21212 crystal symmetry), including GIs from S. diastaticus (2) and from S. olivochromogenes (9). These have been compared with IL-1,2,3 and REF-1,2 crystal structures, so that the total number of crystal structures considered for mutual comparison is 86. To produce a consistent set of structural models, alternative conformations of residues have been removed, together with full residues Arg10, Asp175 and Ile176, which are missing in some crystal structures. A first comparison concerned crystal cell parameters, which has been carried out by using principal component analysis (PCA). A second comparison concerned Cartesian coordinates of the Cα atoms of GI monomers, which have been used to calculate the root mean square deviation (RMSD) between each pair of structures. The (86 × 86) data matrix containing the RMSD values for each pair of structures has been used as a distance matrix for hierarchical clustering based on the group-average method. The program RootProf [25] has been used for PCA and clustering analysis. RMSD calculations have been carried out by the program Superpose [26]. Structural modifications have been performed by using VMD [27] scripts.
3. Results
3.1. Glucose Isomerase Crystallization by Using IL-HCMs
IL-HCMs of different composition (IL-HCM 1-4) were prepared by varying the amount of ionic liquid and used as supports for the crystallization drop in sitting-drop geometry. Crystallization supported by bare membrane (IL-HCM 0) and by standard plastic sitting-drop cradle (REF) was carried out for comparison (see Section 2.2). Among proteins usually used for testing new crystallization methods or screenings, we have chosen GI, whose crystallization in standard conditions, in the presence of ILs as additives [7] and of hydrogels [28], is well known.
GI crystals grew overnight with very fast kinetics both with and without IL-HCM supports and influence of IL-HCM properties on GI crystallization was assessed by calculating the probability of the nucleation event and the density of crystals in the drop. Three replicates for each support were carried out, thus enabling calculation of the nucleation probability (i.e., the fraction of wells where crystallization occurs after one day) and of the density of crystal (i.e., the average number of crystals obtained for each condition in the three replicates). Results, shown in Figure 1, indicate that IL-HCMs have superior crystallization properties than bare membranes, and that the effect on crystallization is strongly IL-dependent. Particularly, IL-HCM 2 provides both the highest nucleation probability and density of crystals, suggesting it is the best nucleant for the GI protein among the tested IL-HCMs.
Another interesting feature of crystals grown in the presence of IL-HCMs is that they appear completely immersed in the IL hydrogel (Figure 2c). Based on our past experience in protein crystallization on hydrogels [11,29], we can definitely infer that this feature is peculiar of IL-HCMs. In fact, by using non-IL hydrogels, crystals usually grow above or partially immersed in the hydrogel layer, while they are systematically and completely immersed in it in the case of IL-HCMs. This latter feature has made extremely difficult to fish crystals for X-ray diffraction studies, but could be very interesting for in situ or in-plate diffraction setups that are being developing in new-generation beamlines, as they allow at the same time immobilization of protein crystals and passage of small molecules through the hydrogel.
3.2. GI Crystallographic Analysis
Despite difficulties in monitoring crystals completely immersed in the hydrogel by using the microscope in reflection model (transmission mode is inhibited by membrane opacity), we managed to fish crystals after digging around them with the micro cutter. By using this strategy, we collected good X-ray diffraction patterns for three GI crystals (IL-1, IL-2, IL-3) taken from the same well, containing an IL-HCM 2 support. For comparison, two GI crystals obtained by using the standard sitting-drop support and the same crystallization cocktail as that used for IL-HCMs were considered (REF-1, REF-2). The crystallographic analysis of these five crystals, reported in Table 1, shows the following systematic effects induced by IL-HCMs:
(i) IL-HCM-grown crystals have lower data resolution and increased mosaicity compared to GI crystals obtained on standard sitting-drop supports. Although in many cases the presence of gel positively affects resolution and mosaicity [30,31], it is not rare to observe that specific hydrogels deteriorate diffraction data quality [13,30], which was interpreted as an effect of the inclusion of the hydrogel in the crystal lattice during crystallization [11,32].
(ii) There is a higher variability of crystallographic parameters among IL-1, IL-2, and IL-3 crystals than that between REF-1 and REF-2 crystals.
(iii) IL-HCM affects the polymorphism of the GI protein, since IL-1, IL-2, IL-3 have P21212 symmetry, while REF-1 and REF-2 have I222 symmetry. It is well known that GI crystallizes in many space groups, among which I222 and P21212 are those observed in the same conditions as used in our experiment (GI from S. rubiginosus; ammonium sulfate, HEPES, and pH 7 as crystallization cocktail). Such polymorphs show very different crystal shape (polyhedral and rectangular prism for I222 and P21212, respectively) and, more importantly, a different solubility according to the concentration of ammonium sulfate: I222 being more stable at concentrations lower than 1.5-1.9 M, P21212 becoming the favored space group at higher concentrations [33]. This behavior is in agreement with results of our experiment in the case of standard sitting-drop support, since we obtained I222 symmetry by using 1.5 M of precipitant. However, in the presence of IL-HCMs and at the same precipitant concentration (1.5 M), GI crystals show P21212 symmetry instead of the expected I222 one, indicating that IL-HCMs strongly affect the relative stability of these two polymorphs. Under high-ionic force, protein is surrounded by ions that affect protein–protein and protein–solvent interactions, by favoring short-range over long-range interactions. On these basis, Gillespie et al. [34] suggested that GI crystal packing is kept by short-range forces, since a high-ionic salt is required for crystallization and a polymorphic change is driven by its initial concentration. Since ILs change the ammonium sulfate concentration at which polymorphism switches, it is very likely that they affect balancing between long- and short-range interactions. This feature agrees with the ability of the above-mentioned ILs to affect kinetically and thermodynamically the crystallization process.
From a crystallographic point of view, lowering of the I222 to the P21212 symmetry (the number of equivalent positions in the crystal cell goes from 8 to 4) is caused by a slight (~2°) tilt of one of the molecular 2-fold axes from the crystallographic axis [35], which induces the appearance of screw axes. Such an effect is often connected with the use of different types of hydrogels during crystallization or with the execution of dehydration protocols [28,36,37]. For some P21212 structures present in the PDB, a strong pseudo-I222 symmetry was found, where deviation from the true I222 was only hinted [38]. A similar effect of inducing a space group transition was observed for lipase A crystals soaked with a solution containing imidazolium IL as additive [39]. The symmetry transition, occurred at higher IL concentrations, was there attributed to stronger IL-protein interactions.
Despite the important involvement of ILs in the crystallization process, no IL ions were observed in IL-1, IL-2 and IL-3 crystal structures, suggesting that IL in the hydrogel form does not interact permanently with protein residues. This is similar to what was found in [8], were IL were used as additives and had the effect of producing subtle changes in solution conditions, but contrary to what was found in other crystallographic investigations about the effect of ILs on protein crystals [40,41], where several IL molecules were reconstructed in the crystal structure, forming hydrophobic and cation-π interactions with surface residues. This strong interaction between protein and IL monomers was recognized as the structural evidence for the mechanism of enzyme denaturation by ILs [39]. Thus, we could hypothesize that having constrained ILs into a hydrogel framework, as that used in the case of IL-HCMs, allows influencing the crystallization process without affecting protein functionality.
3.3. Comparison of Glucose Isomerase Structural Models
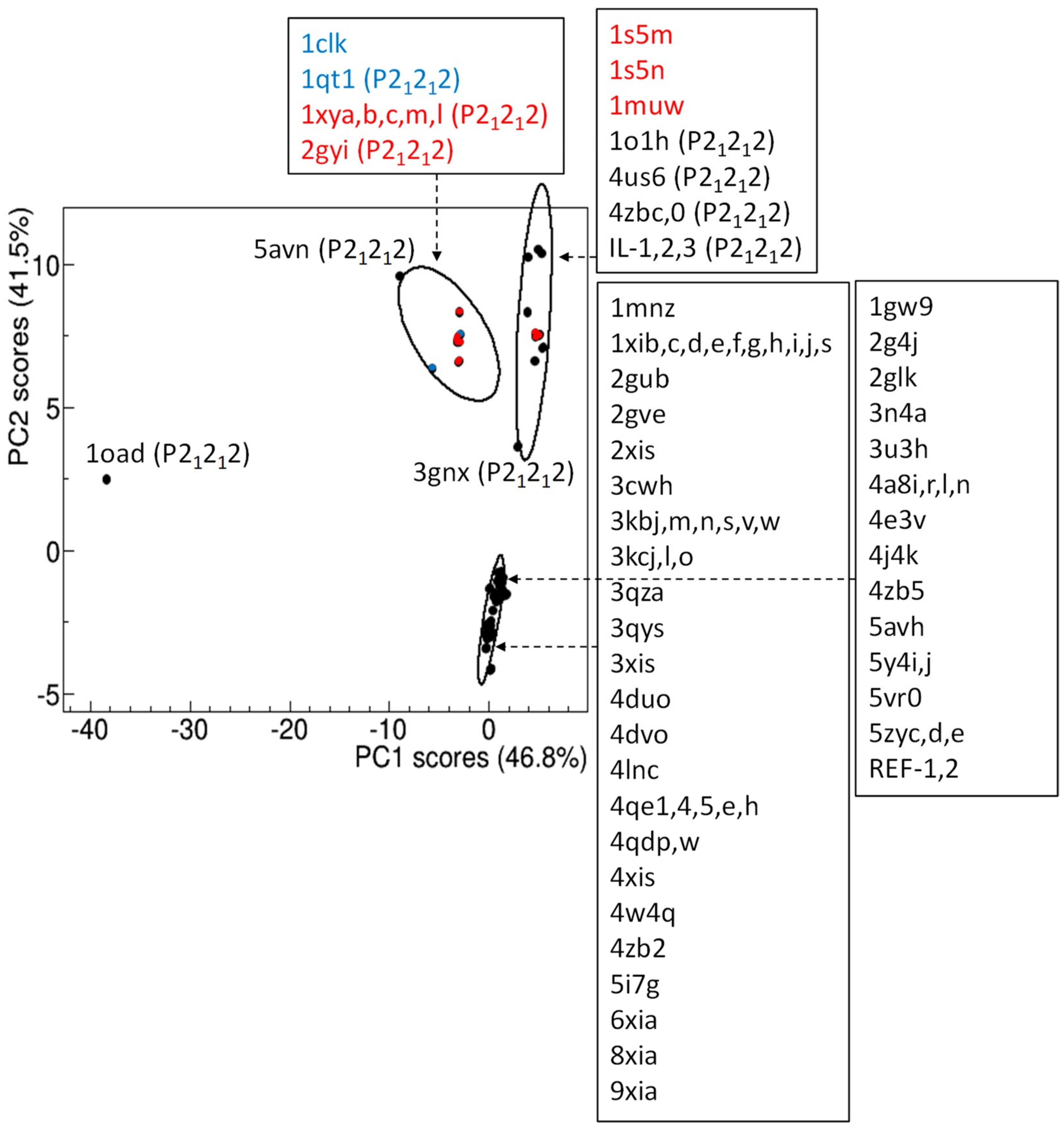
Known glucose isomerase structural models can be conveniently compared by using the length of their crystal cell axes. Each model is identified by crystal cell parameters a, b, c, which are determined with very high precision from X-ray diffraction experiments, as a result of the indexing procedure applied to diffraction patterns. The 86x3 matrix formed by the a, b, c values of each of the 86 GI crystal structures considered for comparison has been processed by principal component analysis (PCA), to reduce dimensionality of the problem and assess the separation among structural models in the 2D space defined by the first two principal components. Data points, representing structural models, can be grouped according to their position in the PC2 vs PC1 score plot (Figure 3). It can be observed that PC2 is able to distinguish data according to crystal symmetry, as structural models with I222 symmetry are associated with negative PC2 scores, while positive PC2 scores are mostly populated by P21212 polymorphs. Some crystal structures, namely 1clk, 1s5m, 1s5n and 1muw, deviate from this trend, as they have I222 symmetry and positive PC2 scores. Notably, they are the only known I222 glucose isomerase from an organism different from S. rubiginosus: 1clk is from S. diastaticus and 1s5m, 1s5n, 1muw from S. olivochromogenes. Moreover, S. diastaticus GI has been also solved in P21212 [42], and the corresponding PDB structure (1qt1) lies in the same cluster of 1clk. Such analysis suggests that 1s5m, 1s5n, and 1muw could also be solved in P21212. A hierarchical clustering suggests the presence of three clusters and one outlier (1oad), which notably represents the first crystal structure found in the P21212 form [43]. IL-1, IL-2 and IL-3 belong to the same cluster, which is slightly separated from a second cluster, mostly populated by P21212 GI polymorphs from organisms different from S. rubiginosus. A well-separated cluster is formed by I222 polymorphs, which seems to be arranged into two subgroups (Figure 3). As common features, proteins of the upper subgroup diffracted in cryogenic conditions; those of the bottom subgroup diffracted at room temperature.
Among the P21212 polymorphs, those clustered together with our IL-grown crystal structures have strong perturbations of the crystal lattice as characteristic feature: 4us6 has been crystallized in a short peptide supramolecular hydrogel [36], 4zbc and 4zb0 crystals have been both subjected to dehydration, and collected respectively in cryogenic and room-temperature conditions [37], and 1o1h has been derivatized in Kr [44]. None of the crystal structures belonging to the other P21212 cluster were subjected to such perturbations. A remarkable example is given by four GI crystal structures crystallized in the same conditions [37]: native crystals collected at room temperature (4zb5) and in cryogenic conditions (4zb2) belong to different subgroups of the I222 cluster, while dehydrated crystals collected at room temperature (4zb0) and in cryogenic conditions (4zbc) belong to the same P21212 cluster.
The PCA applied to the subset of the 75 GI crystal structures from S. rubiginosus (Figure S1, in the Supplementary Materials) confirms the above results.
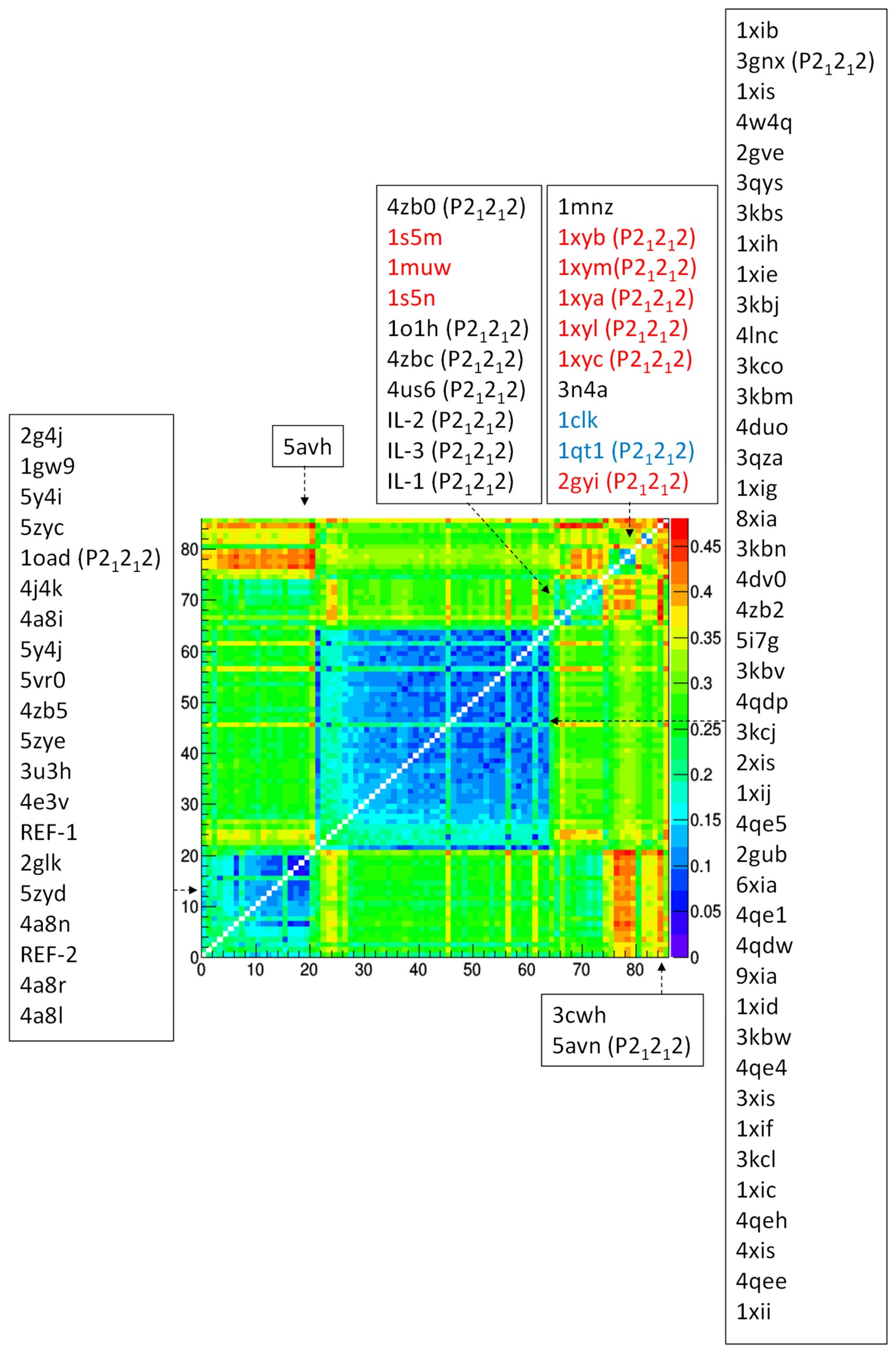
A more detailed comparison, based on the root-mean-square deviation (RMSD) of Cα atoms between pairs of crystal structures at monomer level, highlights a similar grouping. The distance matrix ordered after hierarchical clustering reveals the occurrence of two clusters populated by P21212 polymorphs and two clusters populated by I222 polymorphs (Figure 4). They roughly contain the same list of crystal structures as shown in Figure 3. As an example, 4zb2 and 4zb5 still belong to different I222 clusters. Dehydrated forms 4zb0 and 4zbc also belong to different clusters, however 4zb0 can be found in the same cluster of 4zb2, although they have different crystal packing. This confirms that changes induced by dehydration occur at the level of biological assembly, rather than at individual monomer level [35].
Similar results are obtained by restricting the analysis on the 75 GI crystal structures from S. rubiginosus (Figure S2).
To explore the effect of X-ray diffraction data resolution on structural comparison, root-mean-square deviation (RMSD) analysis has been repeated on a subset of 74 GI crystal structures, obtained by removing outliers in the data resolution distribution (Figure S3). Results, reported in Figure S4, are very similar to those reported in Figure 4, indicating that data resolution (and related coordinate errors) has a negligible effect on RMSD comparison.
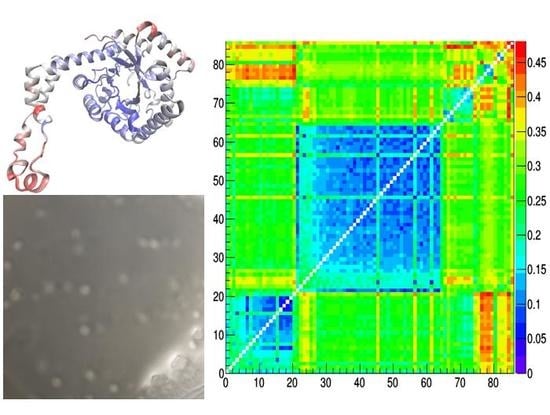
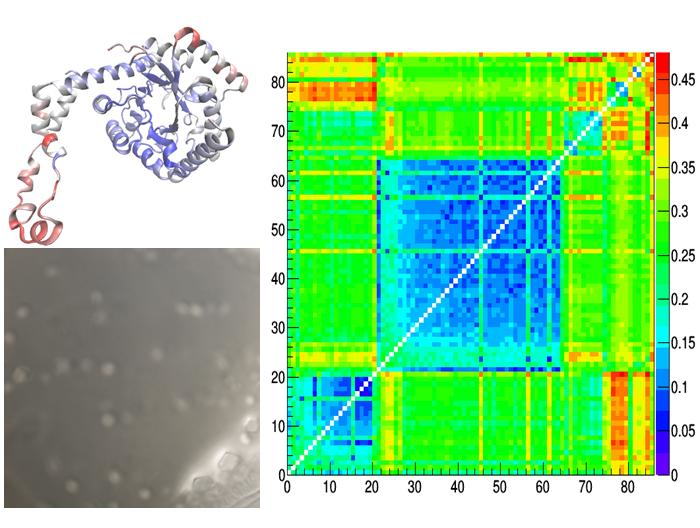
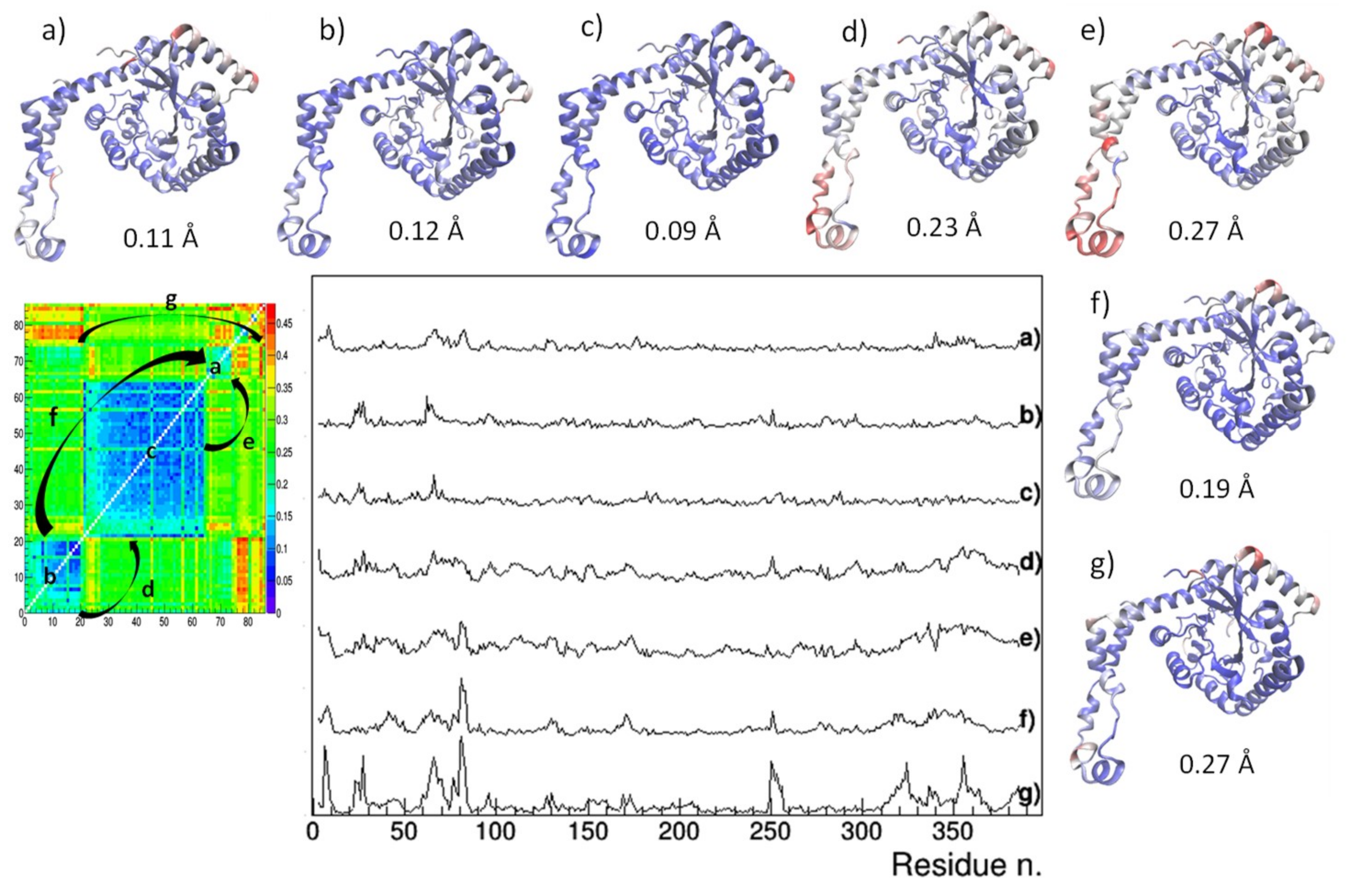
At structural level (Figure 5), crystal structures from the two I222 clusters differs mainly in the C-term loop (residues Arg334 to Met370), in residues Asp65 to Glu67 lying on an exposed α-helix and in the loop residues Asn250 to Ile252 (case d of Figure 5). Such conformational changes can be attributed to the temperature effect, since, as discussed for Figure 3, the two clusters separates crystal structures determined in cryogenic conditions from those determined at room temperature. On the other hand, conformational changes characterizing different polymorphic forms are located at the beginning of the exposed α-helix (residues Asp79 to Thy82), a region which is likely affected by stresses produced by crystal packing (cases e, f, g of Figure 5). The amplitude of conformational changes is lower when comparing crystal structures of the IL-populated cluster with those of the larger I222 cluster, containing crystal structures obtained at room temperature (case e of Figure 5). In fact, the average RMSD value is 0.19 Å when calculated between our IL-1,2,3 and REF-1,2 crystal structures, whereas it is 0.15 Å within IL-1,2,3 structures and 0.10 Å within REF-1,2 structures. Larger deviations (~0.27 Å) occur between structures of the IL cluster and those of the smaller I222 cluster (that containing crystal structures obtained in cryogenic conditions), where the conformational changes due to crystal packing and induced by temperature are contemporary present (case f of Figure 5).
It is worth comparing crystals grown on IL-HCMs with those grown on high-strength agarose hydrogel. These latter diffracted X-rays at atomic resolution and showed a resistance to osmotic stress due to soaking with buffer-free solutions [28]. However two polymorphs were obtained in the same crystallization condition, having PEG6000 as precipitant: 5avh (I222) and 5avn (P21212). They are both outliers in RMSD analysis (Figure 4), i.e., their structural features differ from those of other crystals in the respective clusters. Moreover, their RMSD is 0.27 Å, with huge deviations in regions affected by crystal packing (case g of Figure 5). Thus, it can be argued that IL-HCMs more effectively direct the crystallization path than high-strength agarose hydrogels, as they produce a unique crystal form in the same crystallization condition, and less disturb the crystal structure, as very small deviations have been found with respect to crystals of the same crystal form grown without hydrogel.
4. Discussion
We have demonstrated that IL can be used in protein crystallization not only as additives, but also as constituents of hybrid materials, the IL-HCMs, composed of hydrophobic membranes supporting hydrogels layers, which are able to drive, favor, and sustain protein crystallization [11,13,29]. IL-HCMs have proved compatible with protein crystallization, and a new feature with respect to commonly used hydrogels has emerged, as due to stronger interactions between protein and IL monomers: protein crystals grow completely immersed in the IL hydrogel layer. This discloses the possibility of using IL-HCMs as supports for enzymes in the solid state in biotechnological applications, were protein crystals need to be preserved from external, mechanical, and chemical stresses.
GI has been used to test IL-HCM-driven crystallization, and crystal structures from crystals grown in the same conditions with and without IL-HCMs have been determined. We have proven that IL-HCMs affect the selection mechanism of GI polymorphs, and that this effect is not the result of a strong and stable interactions between IL and protein molecule, since no IL-ions have been found bound to the protein. These results demonstrate that IL-HCMs are active during the crystallization process and preserve crystals after their growth, but do not directly interact with residues.
The new structural models achieved have been compared to the multitude of GI models already present in the PDB, by using multivariate analysis based on crystal cell parameters and backbone conformation. These analyses highlighted that structural models obtained by crystals grown in IL-HCMs can be distinguished from those obtained by standard crystallization setup. More generally, crystal cell and backbone conformation can distinguish crystal structures obtained by crystals grown in the presence of hydrogels or subjected to hydration after growth, indicating that mechanical or chemical forces acting during or after crystallization can affect the final average protein conformation represented in the crystal, inducing small deviations with respect to the equilibrium structural model. These analyses also highlight that perturbed crystals have a higher variability in crystal cell and backbone conformations and mostly occur in the GI P21212 polymorph. This agrees with the higher variability of crystallographic parameters observed for crystals grown on IL-HCM with respect to reference crystals (Table 1). Whether the observed variations in the P21212 GI crystals are a factor of the crystallization medium, are induced by mechanical effects acting on the crystals after their growth, or represent an inherent variability of GI crystallized in this space group is an open question, which could be investigated by in situ X-ray diffraction experiments.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/9/5/253/s1, Figure S1: Comparison of the 75 experimental structural models of glucose isomerase from S. rubiginosus by principal component analysis applied on crystal cell parameters. Figure S2: Comparison of the 75 experimental structural models of glucose isomerase from S. rubiginosus, carried out by using the root-mean-square deviation of their Cα atoms. Figure S3: Distribution of data resolution of the 86 glucose isomerase crystal structures considered for comparative analysis. Figure S4: Comparison of the 74 experimental structural models of glucose isomerase obtained after selection on data resolution, carried out by using the root-mean-square deviation of their Cα atoms.
Author Contributions
G.D.P. and Roc.C. conceived and supervised the project; G.D. conceived the ionic-liquid hydrogel composite membranes work; S.M.S. optimized and prepared ionic-liquid hydrogel composite membranes; B.D.B. performed crystallization experiments; Ros.C. and Roc.C. performed X-ray diffraction experiments; B.D.B and Ros.C. determined and refined crystal structures; Roc.C. wrote the manuscript; all authors commented on the manuscript.
Funding
This research received no external funding.
Acknowledgments
Authors wish to thank the Diamond Light Source for access to beamline I04 (Proposal No. MX15832-1), that contributed to the results presented here.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pusey, M.L.; Paley, M.S.; Turner, M.B.; Robers, R.D. Protein Crystallization Using Room Temperature Ionic Liquids. Cryst. Growth Des. 2007, 7, 787–793. [Google Scholar] [CrossRef]
- Li, X.; Xu, X.; Dan, Y.; Feng, J.; Ge, L.; Zhang, M. The crystallization of lysozyme in the system of ionic liquid [BMIm][BF4]-water. Cryst. Res. Technol. 2008, 43, 1062–1068. [Google Scholar] [CrossRef]
- Hekmat, D.; Hebel, D.; Joswig, S.; Schmidt, M.; Weuster-Botz, D. Advanced protein crystallization using water-soluble ionic liquids as crystallization additives. Biotechnol Lett. 2007, 29, 1703–1711. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Wang, J. Ionic liquids in the assay of proteins. Anal. Methods 2010, 2, 1222–1226. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, W.; Li, Y.; Zhang, J.; Gu, Q. Korean, A new strategy for protein crystallization: Effect of ionic liquids on lysozyme crystallization and morphology. J. Chem. Eng. 2014, 31, 919–923. [Google Scholar]
- Kowacz, M.; Marchel, M.; Juknaitė, L.; Esperança, J.M.S.S.; Romão, M.J.; Carvalho, A.L.; Rebelo, L.P.N. Ionic-Liquid-Functionalized Mineral Particles for Protein Crystallization. Cryst. Growth Des. 2015, 15, 2994–3003. [Google Scholar] [CrossRef]
- Schroder, C. Proteins in ionic liquids: current status of experiments and simulations. Top. Curr. Chem (Z) 2017, 375–425. [Google Scholar]
- Judge, R.A.; Takahashi, S.; Longenecker, K.L.; Fry, E.H.; Abad-Zapatero, C.; Chiu, M.L. The Effect of Ionic Liquids on Protein Crystallization and X-ray Diffraction Resolution. Cryst. Growth Des. 2009, 9, 3463–3469. [Google Scholar] [CrossRef]
- Marr, P.C.; Marr, A.C. Ionic liquid gel materials: Applications in green and sustainable chemistry. Green Chem. 2016, 18, 105–128. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Li, J.; Li, Z.; He, P.; Liu, H.; Li, J. Highly active horseradish peroxidase immobilized in 1-butyl-3-methylimidazolium tetrafluoroborate room-temperature ionic liquid based sol–gel host materials. Chem. Comm. 2005, 13, 1778–1780. [Google Scholar] [CrossRef] [PubMed]
- Di Profio, G.; Polino, M.; Nicoletta, F.P.; Belviso, B.D.; Caliandro, R.; Fontananova, E.; De Filpo, G.; Curcio, E.; Drioli, E. Tailored Hydrogel Membranes for Efficient Protein Crystallization. Adv. Func. Mat. 2014, 24, 1582–1590. [Google Scholar] [CrossRef]
- Drioli, E.; Di Profio, G.; Curcio, E. Membrane-assisted crystallization technology. In Advances in Chemical and Process Engineering; Imperial College Press: London, UK, 2015; Volume 2. [Google Scholar]
- Mirabelli, V.; Salehi, S.M.; Angiolillo, L.; Belviso, B.D.; Conte, A.; Del Nobile, M.A.; Di Profio, G.; Caliandro, R. Enzyme Crystals and Hydrogel Composite Membranes as New Active Food Packaging Material. Glob. Chall. 2018, 2, 1700089. [Google Scholar] [CrossRef] [Green Version]
- Salehi, S.M.; Santagada, R.; De Pietra, S.; Fontananova, E.; Curcio, E.; Di Profio, G. Ionic Liquid Hydrogel Composite Membranes (IL-HCMs). ChemEngineering 2019, 3, 47. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gillialand, G.; Bhat, T.N.; Weissing, H.; Shindyalov, P.E.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Cryst. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mazzone, A.; Siliqi, D. Molecular replacement: The probabilistic approach of the program REMO09 and its applications. Acta Cryst. A 2009, 65, 512–527. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. D 2008, 64, 61–69. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Bricogne, G.; Blanc, E.; Brandl, M.; Flensburg, C.; Keller, P.; Paciorek, W.; Roversi, P.; Sharff, A.; Smart, O.S.; Vonrhein, C.; et al. BUSTER version 2.X; Global Phasing Ltd.: Cambridge, UK, 2017. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Cryst. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Cryst. D 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Caliandro, R.; Belviso, B.D. RootProf: Software for multivariate analysis of unidimensional profiles. J. Appl. Cryst. 2014, 47, 1087–1096. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. D 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulte, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sugiyama, S.; Maruyama, M.; Sazaki, G.; Hirose, M.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; More, Y.; Matsumura, H. Growth of Protein Crystals in Hydrogels Prevents Osmotic Shock. J. Am. Chem. Soc. 2012, 134, 5786–5789. [Google Scholar] [CrossRef]
- Salehi, S.M.; Manjua, A.C.; Belviso, B.D.; Portugal, C.A.M.; Coelhoso, I.M.; Mirabelli, V.; Fontananova, E.; Caliandro, R.; Crespo, J.G.; Curcio, E.; et al. Hydrogel Composite Membranes Incorporating Iron Oxide Nanoparticles as Topographical Designers for Controlled Heteronucleation of Proteins. Cryst. Growth Des. 2018, 18, 3317–3327. [Google Scholar] [CrossRef]
- Lorber, B.; Sauter, C.; Ng, J.D.; Zhu, D.W.; Giegé, R.; Vidal, O.; Robert, M.C.; Capelle, B. Characterization of protein and virus crystals by quasi-planar wave X-ray topography: A comparison between crystals grown in solution and in agarose gel. J. Cryst. Growth 1999, 204, 357–368. [Google Scholar] [CrossRef]
- Lorber, B.; Sauter, C.; Robert, M.C.; Capelle, B.; Giegé, R. Crystallization within agarose gel in microgravity improves the quality of thaumatin crystals. Acta Cryst. D 1999, 55, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, J.M.; Gavira, J.A.; Otálora, F.; Guash, A.; Coll, M. Reinforced protein crystals. Mater. Res. Bull. 1998, 33, 1593–1598. [Google Scholar] [Green Version]
- Vuolanto, A.; Uotila, S.; Leisola, M.; Visuri, K. Solubility and crystallization of xylose isomerase from Streptomyces rubiginosus. J. Cryst. Growth. 2003, 257, 403–411. [Google Scholar] [CrossRef]
- Gillespie, C.M.; Asthagiti, D.; Lenhoff, A.M. Polymorphic Protein Crystal Growth: Influence of Hydration and Ions in Glucose Isomerase. Cryst. Growth Des. 2014, 14, 46–57. [Google Scholar] [CrossRef]
- Farber, G.K.; Petsko, G.A.; Ringe, D. The 3.0 Å crystal structure of xylose isomerase from Streptomyces olivochromogenes. Protein Eng. 1987, 1, 459–466. [Google Scholar] [CrossRef]
- Conejero-Muriel, M.; Gavira, J.A.; Pineda-Molina, E.; Belsom, A.; Bradley, M.; Moral, M.; Garcia-Lopez Durán, J.D.D.; Luque Gonzȧlez, A.; Diaz-Mochȯn, J.J.; Contreras-Montoya, R.; et al. Influence of the chirality of short peptide supramolecular hydrogels in protein crystallogenesis. Chem. Comm. 2015, 51, 3862–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobley, C.M; Sandy, J.; Sanchez-Weatherby, J.; Mazzorana, M.; Krojer, T.; Nowak, R.P.; Sorensen, T.L. A generic protocol for protein crystal dehydration using the HC1b humidity controller. Acta Cryst. D 2016, 72, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Lavie, A.; Allen, K.N.; Petsko, G.A.; Ringe, D. X-ray Crystallographic Structures of D-Xylose Isomerase-Substrate Complexes Position the Substrate and Provide Evidence for Metal Movement during Catalysis. Biochem. 1994, 33, 5469–5480. [Google Scholar] [CrossRef]
- Nordwald, E.M.; Plaks, J.G.; Snell, J.R.; Scousa, M.C.; Kaar, J.L. Crystallographic Investigation of Imidazolium Ionic Liquid Effects on Enzyme Structure. ChembioChem 2015, 16, 2456–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ji, Y.; Wang, J. Improvement on the crystallization of lysozyme in the presence of hydrophilic ionic liquid. J. Analyst. 2010, 135, 2241–2248. [Google Scholar] [CrossRef]
- Kowacs, M.; Mukhopadhyay, A.; Carvalho, A.L.; Esperança, J.M.S.S.; Romão, M.J.; Rebelo, L.P.N. Hofmeister effects of ionic liquids in protein crystallization: Direct and water-mediated interactions. CrystEngComm 2012, 14, 4912–4921. [Google Scholar] [CrossRef]
- Zhu, X.; Teng, M.; Niu, L.; Xu, C.; Wang, Y. Structure of xylose isomerase from Streptomyces diastaticus No. 7 strain M1033 at 1.85 Å resolution. Acta Cryst D 2000, 56, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Ramagopal, U.A.; Dauter, M.; Dauter, Z. SAD manganese in two crystal forms of glucose isomerase. Acta Cryst. D 2003, 59, 868–875. [Google Scholar] [CrossRef]
- Nowak, E.; Pajikar, S.; Tucker, P.A. Structure of Glucose Isomerase Derivatized with Kr. 2002. [Google Scholar] [CrossRef]
Figure 1.
Number of successful crystallization experiments, proportional to nucleation probability (bars, left vertical axis) and average crystal density (line, fight vertical axis) related to the glucose isomerase crystallization performed on 4 different ionic-liquid hydrogel composite membrane (IL-HCM) supports (IL-HCM 1-4). REF is related to the crystallization performed by using the standard sitting drop setup and IL-HCM 0 is related to the test with bare hydrophobic membrane. Nucleation probability is the fraction of wells producing crystals and average crystal density is the mean of crystals for each well, both with respect the three replicates performed for each support. Error bars in the average crystal density indicate estimated measurement errors, while bar heights indicate the minimum (light blue) and maximum (dark blue) number of successful crystallization experiment, as determined by considering uncertainties in the visualization of crystals.
Figure 1.
Number of successful crystallization experiments, proportional to nucleation probability (bars, left vertical axis) and average crystal density (line, fight vertical axis) related to the glucose isomerase crystallization performed on 4 different ionic-liquid hydrogel composite membrane (IL-HCM) supports (IL-HCM 1-4). REF is related to the crystallization performed by using the standard sitting drop setup and IL-HCM 0 is related to the test with bare hydrophobic membrane. Nucleation probability is the fraction of wells producing crystals and average crystal density is the mean of crystals for each well, both with respect the three replicates performed for each support. Error bars in the average crystal density indicate estimated measurement errors, while bar heights indicate the minimum (light blue) and maximum (dark blue) number of successful crystallization experiment, as determined by considering uncertainties in the visualization of crystals.

Figure 2.
Crystals of glucose isomerase grown overnight on plastic sitting-drop supports (a) and on ionic-liquid based hydrogel membrane composites (b,c) with 5 %wt ionic liquid content of the monomer (IL-HCM 2). Image a is taken with the microscope in transmission mode, images b and c are taken in reflection mode at two different magnifications of the same well.
Figure 2.
Crystals of glucose isomerase grown overnight on plastic sitting-drop supports (a) and on ionic-liquid based hydrogel membrane composites (b,c) with 5 %wt ionic liquid content of the monomer (IL-HCM 2). Image a is taken with the microscope in transmission mode, images b and c are taken in reflection mode at two different magnifications of the same well.

Figure 3.
Comparison of experimental structural models of glucose isomerase by principal component analysis applied on crystal cell parameters. Score plot of the first two principal components, where representative points are clustered according to a hierarchic clustering. 85% confidence level ellipses are shown and PDB codes associated to representative points are reported. Space group are indicated if they are P21212; in the other cases the space group is I222. The percentage of the total data variance explained by each principal component is shown on the axes. Data points and PDB codes corresponding to GI from organisms different from S. rubiginosus are colored in red (S. olivochromogenes) and in blue (S. diastaticus).
Figure 3.
Comparison of experimental structural models of glucose isomerase by principal component analysis applied on crystal cell parameters. Score plot of the first two principal components, where representative points are clustered according to a hierarchic clustering. 85% confidence level ellipses are shown and PDB codes associated to representative points are reported. Space group are indicated if they are P21212; in the other cases the space group is I222. The percentage of the total data variance explained by each principal component is shown on the axes. Data points and PDB codes corresponding to GI from organisms different from S. rubiginosus are colored in red (S. olivochromogenes) and in blue (S. diastaticus).

Figure 4.
Comparison of experimental structural models of glucose isomerase by using the root-mean-square deviation of their Cα atoms. Distance matrix after application of hierarchical clustering, with Protein Data Bank (PDB) codes associated to each row/column reported, ordered according to their occurrence in the matrix and divided according to the clustering results. Space group is indicated if it is P21212, when missing it is I222. PDB codes corresponding to GI from organisms different from S. rubiginosus are colored in red (S. olivochromogenes) and in blue (S. diastaticus).
Figure 4.
Comparison of experimental structural models of glucose isomerase by using the root-mean-square deviation of their Cα atoms. Distance matrix after application of hierarchical clustering, with Protein Data Bank (PDB) codes associated to each row/column reported, ordered according to their occurrence in the matrix and divided according to the clustering results. Space group is indicated if it is P21212, when missing it is I222. PDB codes corresponding to GI from organisms different from S. rubiginosus are colored in red (S. olivochromogenes) and in blue (S. diastaticus).

Figure 5.
Reside-by-residue root-mean-square deviation (RMSD) of Cα atoms between pairs of representative glucose isomerase crystal structures. They are: IL-1 – 4zbc (a), REF-1 – 4zb5 (b), 1xib-5i7g (c), REF-1 – 1xib (d), IL-1 – 1xib (e), IL-1 – REF-1 (f), 5avh – 5avn (g) and are indicated within the distance matrix after application of hierarchic clustering. Structural models colored according to rescaled residue-by-residue RMSD values are shown, together with the average RMSD values.
Figure 5.
Reside-by-residue root-mean-square deviation (RMSD) of Cα atoms between pairs of representative glucose isomerase crystal structures. They are: IL-1 – 4zbc (a), REF-1 – 4zb5 (b), 1xib-5i7g (c), REF-1 – 1xib (d), IL-1 – 1xib (e), IL-1 – REF-1 (f), 5avh – 5avn (g) and are indicated within the distance matrix after application of hierarchic clustering. Structural models colored according to rescaled residue-by-residue RMSD values are shown, together with the average RMSD values.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic parameters of glucose isomerase (GI) crystals grown on IL-HCMs (IL-1,2,3) and, for comparison, without them (REF 1,2), by using the same crystallization condition. Values in parenthesis refer to the outer resolution shell).
Table 1.
Crystallographic parameters of glucose isomerase (GI) crystals grown on IL-HCMs (IL-1,2,3) and, for comparison, without them (REF 1,2), by using the same crystallization condition. Values in parenthesis refer to the outer resolution shell).
| IL-1 | IL-2 | IL-3 | REF-1 | REF-2 | |
|---|---|---|---|---|---|
| Resolution (Å) | 1.58 (1.62–1.58) | 1.77 (1.87–1.77) | 1.98 (2.01–1.98) | 1.19 (1.21–1.19) | 1.19 (1.22–1.19) |
| Space group | P21212 | P21212 | P21212 | I222 | I222 |
| Crystal Cell (Å) a | 85.90 | 81.62 | 81.75 | 92.52 | 92.52 |
| b | 92.60 | 94.92 | 93.35 | 99.12 | 98.17 |
| c | 99.11 | 97.47 | 97.85 | 102.48 | 101.85 |
| Rmerge (%) | 9.9 (66.4) | 9.1 (54.8) | 22.0 (122) | 11.7 (48.0) | 4.2 (16.6) |
| I/σ | 10.2 (3.4) | 9.2 (2.1) | 3.1 (0.9) | 5.3 (1.0) | 13.8 (2.6) |
| Completeness (%) | 95.6 (97.4) | 97.9 (99.6) | 99.7 (96.9) | 75.8 (2.9) | 76.7 (4.9) |
| Multiplicity | 4.2 (4.1) | 4.0 (4.2) | 3.9 (4.1) | 3.4 (1.0) | 3.4 (1.0) |
| Mosaicity (°) | 0.16 | 0.32 | 0.59 | 0.14 | 0.05 |
| B Wilson (Å2) | 9.60 | 19.17 | 22.12 | 9.65 | 8.59 |
| Rwork | 0.167 | 0.170 | 0.220 | 0.171 | 0.107 |
| Rfree | 0.194 | 0.205 | 0.271 | 0.188 | 0.129 |
| Number of water molecules | 929 | 902 | 491 | 584 | 538 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Belviso, B.D.; Caliandro, R.; Salehi, S.M.; Di Profio, G.; Caliandro, R. Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes. Crystals 2019, 9, 253. https://doi.org/10.3390/cryst9050253
AMA Style
Belviso BD, Caliandro R, Salehi SM, Di Profio G, Caliandro R. Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes. Crystals. 2019; 9(5):253. https://doi.org/10.3390/cryst9050253
Chicago/Turabian StyleBelviso, Benny Danilo, Rosanna Caliandro, Shabnam Majidi Salehi, Gianluca Di Profio, and Rocco Caliandro. 2019. "Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes" Crystals 9, no. 5: 253. https://doi.org/10.3390/cryst9050253
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.