1. Introduction
South African cassava mosaic virus (SACMV) belongs to the family
Geminiviridae and genus
Begomovirus, whose members infect a wide range of plant hosts [
1,
2]. SACMV is composed of a bipartite genome (single-stranded circular DNA-A and DNA-B components) [
1]. DNA-A ORFs of bipartite begomoviruses encode three proteins in the antisense direction—namely, the Rep-associated (AC1), replication enhancer Ren (AC2), and transcriptional activator TrAP (AC3) proteins [
3]. Additionally, an overlapping AC4 ORF encodes for a putative host RNA-silencing suppressor protein. Conversely, in the sense direction, the structural coat protein (CP) is encoded by AV1 [
3], and AV2 is a reported RNA-silencing suppressor and pathogenicity determinant [
4]. DNA-B BC1 ORF encodes the cell-to-cell movement protein (MP) from the complementary strand, whereas the nuclear shuttle protein (NSP) is encoded from the virion sense strand by the BV1 ORF [
3]. In plants, viruses have to travel through the cell wall via the plasmodesmata [
5], and the MP is required for cell-to-cell and long-distance movement through its interaction with the NSP [
6]. Geminiviruses also utilise host membranes such as the endoplasmic reticulum (ER) and cytoskeleton to traffic from the nucleus to the plasmodesmata [
7]. There are two models suggested for the active role of geminivirus MP and NSP during cell-to-cell transport. The ‘couple-skating’ model suggests that the MP binds to the ssDNA/dsDNA–NSP complex on the cytoplasmic side of nuclear membranes or microsomal vesicles and subsequently traffics the nucleoprotein complex into the proximal cell along the endoplasmic reticulum, and via microtubules (forms the desmotubule) and filamentous actin through the plasmodesmata [
8,
9]. The ‘relay-race’ model suggests that, after NSP-mediated nuclear export, viral ssDNA or dsDNA associates with the MP, which further transports the viral DNA-MP through the plasmodesmata into an adjacent cell, with the assistance of other host accessory proteins [
8,
9].
The deduced 307 amino acid sequence of the SACMV MP [
1] and functional studies [
9,
10] have informed a putative model of the MP protein structural domains—namely, the N-terminus and central anchor domains that putatively are both required for localisation to the cell periphery, and a C-terminal oligomerisation domain. The nuclear shuttle protein (NSP)-binding site is predicted to locate in the central region of the MP [
10]. The secondary and tertiary structures of geminiviral MPs have not yet been elucidated. Since the MP of SACMV is a novel protein in that little homology exists with other characterised proteins in the database (NCBI), the elucidation of the protein structure is crucial to understanding the function of the protein in the cytoplasmic movement of the geminivirus DNA complex, and transit from cell to cell. Since the MP does not have any predicted enzymatic function, but rather the binding capability to other cellular proteins, this study was not focused on kinetics but rather on the elucidation of the basic structural characteristics related to protein–protein interactions. The molecular weight, as well as secondary and tertiary structures (protein folding) of the full-length SACMV MP, was characterised for the first time in this study.
2. Materials and Methods
2.1. Vector Construction
The MP gene sequence (923 nucleotides/307 amino acids [
Figure S1]) was sent to GenScript Inc, Piscataway, NJ, USA, where the sequence was codon harmonised for expression into
E. coli and cloned into expression vector system pET-28a (+) (
Figure S2) using
NdeI and
NotI restriction sites. The codon harmonisation was carried out via a single nucleotide change (cysteine to guanine) in the codon translating for Alanine (
Figure S4).
2.2. Recombinant Protein Expression
E. coli BL21 (DE3) cells were transformed with the codon-harmonised MP sequence in a pET-28a (+) vector. Single colonies were cultured overnight into SOB media comprising 5% (w/v) yeast extract (Sigma-Aldrich, St. Louis, MO, USA), 20% (w/v) tryptone (Sigma-Aldrich, St. Louis, MO, USA), 0.5% (w/v) NaCl (Sigma-Alrich, USA), and 1 M KCl (Merck, Darmstadt, Germany), supplemented with 1 M MgCl2 (Sigma-Aldrich, St. Louis, MO, USA), 1 M MgSO4 (Sigma-Aldrich-USA, St. Louis, MO, USA), and 50 μg mL−1 kanamycin (ThermoFisher Scientific, New York, NY, USA) at 37 °C, shaking at 200 rpm. The overnight cultures were diluted 1/10 with fresh SOB media supplemented with 1 M MgCl2, 1 M MgSO4, and 50 μg mL−1 kanamycin at 37 °C, shaking at 200 rpm until the OD600 was approximately 0.45. The cultures were then induced with IPTG to a final concentration of 0.25 mM and cultured for an additional 4 h (37 °C, 200 rpm). Cells were harvested via centrifugation (20,000× g, 10 min, 4 °C). Approximately 1 g of wet cell pellet was obtained per one hundred millilitres, and the pellet was resuspended in lysis buffer (50 mM Tris, 5% glycerol (v/v), and 50 mM NaCl; pH 7.5), followed by 6 cycles of sonication (30 s on and 30 s off). Following centrifugation at 20,000× g for 25 min at 4 °C, the supernatant (soluble fraction) and pellet (insoluble fraction) were separated.
2.3. Protein Denaturation, Purification and Refolding, and Purity Assessment
The insoluble cell fraction was subject to denaturation with 8 M urea in 50 mM Tris- HCl (pH 8.5) and 0.05% NaN3 (pH 7.5). The cell solution was subjected to overnight solubilisation via dialysis in 4 M urea (1 L of 4 M urea, 20 mM sodium phosphate, 0.5 M NaCl, and 10 mM imidazole; pH 7.5). The solubilised cell fraction was further subjected to purification using a gravity flow column comprising a nickel-charged NTA Profinity™ IMAC resin (Bio-Rad, Hercules, CA, USA). The IMAC column was equilibrated with dialysate (4 M urea solution). To remove any unbound protein, the resin was washed with 4 M urea, 20 mM sodium phosphate, 0.5 M NaCl, and 25 mM imidazole (pH 7.5), and eluted with 4 M urea containing 250 mM imidazole (pH 7.5). Fractions containing eluted proteins were pooled and subject to refolding via stepwise dialysis (three changes: first change, 16 h; 50 mM Tris-HCl, pH 10; 0.5 M l-arginine, 2% (v/v); Triton X-100 (Merck, Darmstadt, Germany); and 10 mM DTT; remaining changes, 16 h each; 50 mM Tris-HCl, pH 10; and 0.5 M l-arginine). The protein concentration following protein refolding was determined spectrophotometrically using a theoretically estimated molar extinction coefficient of 31,650 M−1 cm−1.
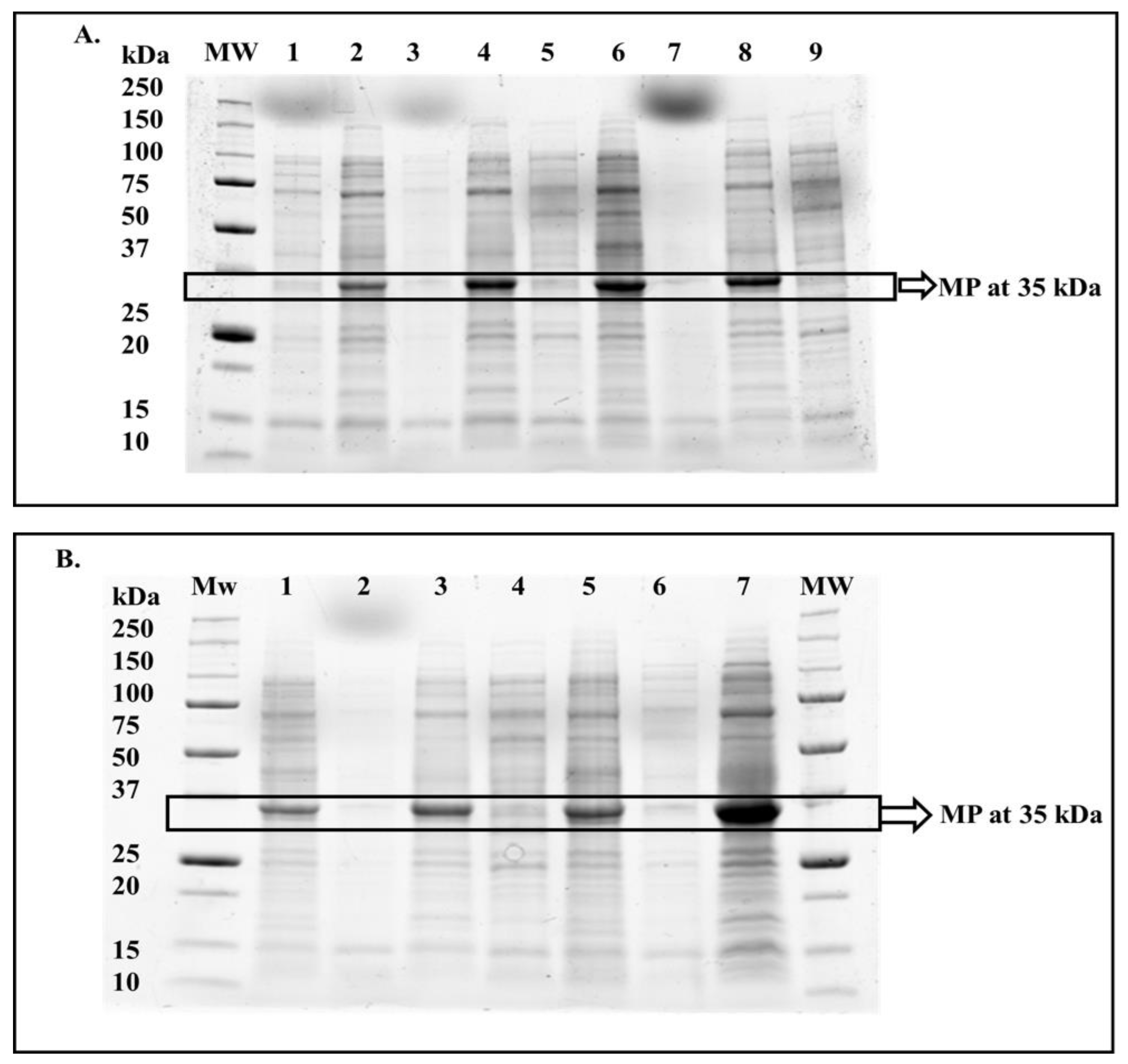
All expressed protein fractions and all eluted protein fractions were prepared with 2X SDS buffer (added in a 1:1 ratio) and run on a 12% SDS–PAGE mini-PROTEAN gels (TGX Stain-Free™ Fastcast™ acrylamide kit Bio-Rad Laboratories, Inc. (Hercules, CA, USA)). Based on the SDS–PAGE results, column fractions containing the purified protein of high purity (no contamination) (eluted fractions 1–4) were pooled together and refolded via stepwise dialysis, as described above.
2.4. Secondary Structure Content Determination
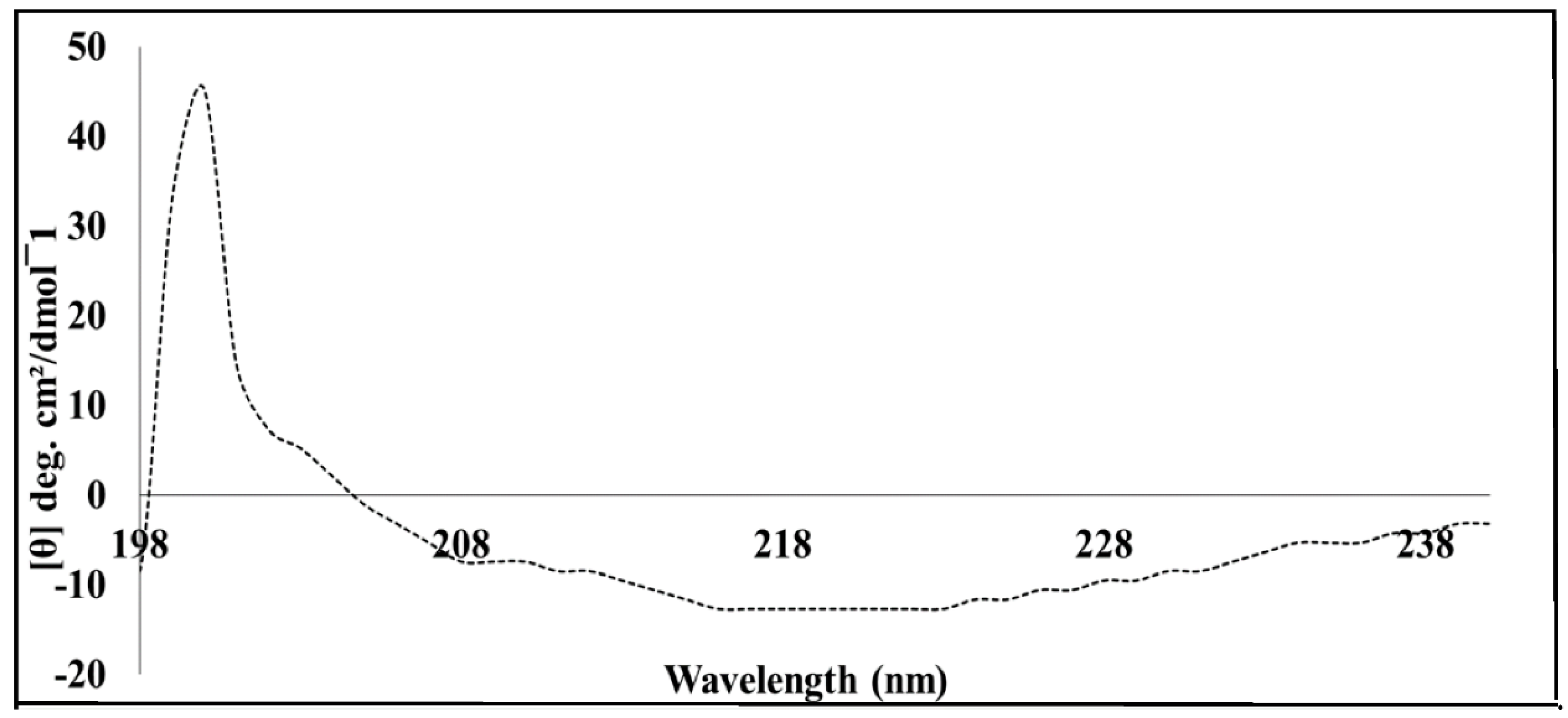
Far-UV circular dichroism (Far-UV CD) measurements of MP were performed at 20 °C in a Jasco J1500 spectrometer. Briefly, the protein sample was diluted to a final concentration of 5 µM using ultra-pure deionised water, which resulted in a final Tris buffer containing 0.5 mM Tris-HCl (pH 7.5). Three independent spectra were collected for both the protein sample (5 µM MP in 0.5 mM Tris-HCl, pH 7.5) and the buffer (as blank). All spectra were corrected for using the Tris-HCl buffer as a baseline. The raw data were converted to mean residue ellipticity [
Θ] (mdeg cm
2 dmol
−1) using the following formula:
where
c is the concentration in g/L,
n is the number of residues, and
l is the path length in cm.
The estimation of secondary structure of BC1 was estimated using the CONTINL algorithm in the Dichroweb online server [
11].
2.5. Extrinsic Fluorescence ANS Binding Assay
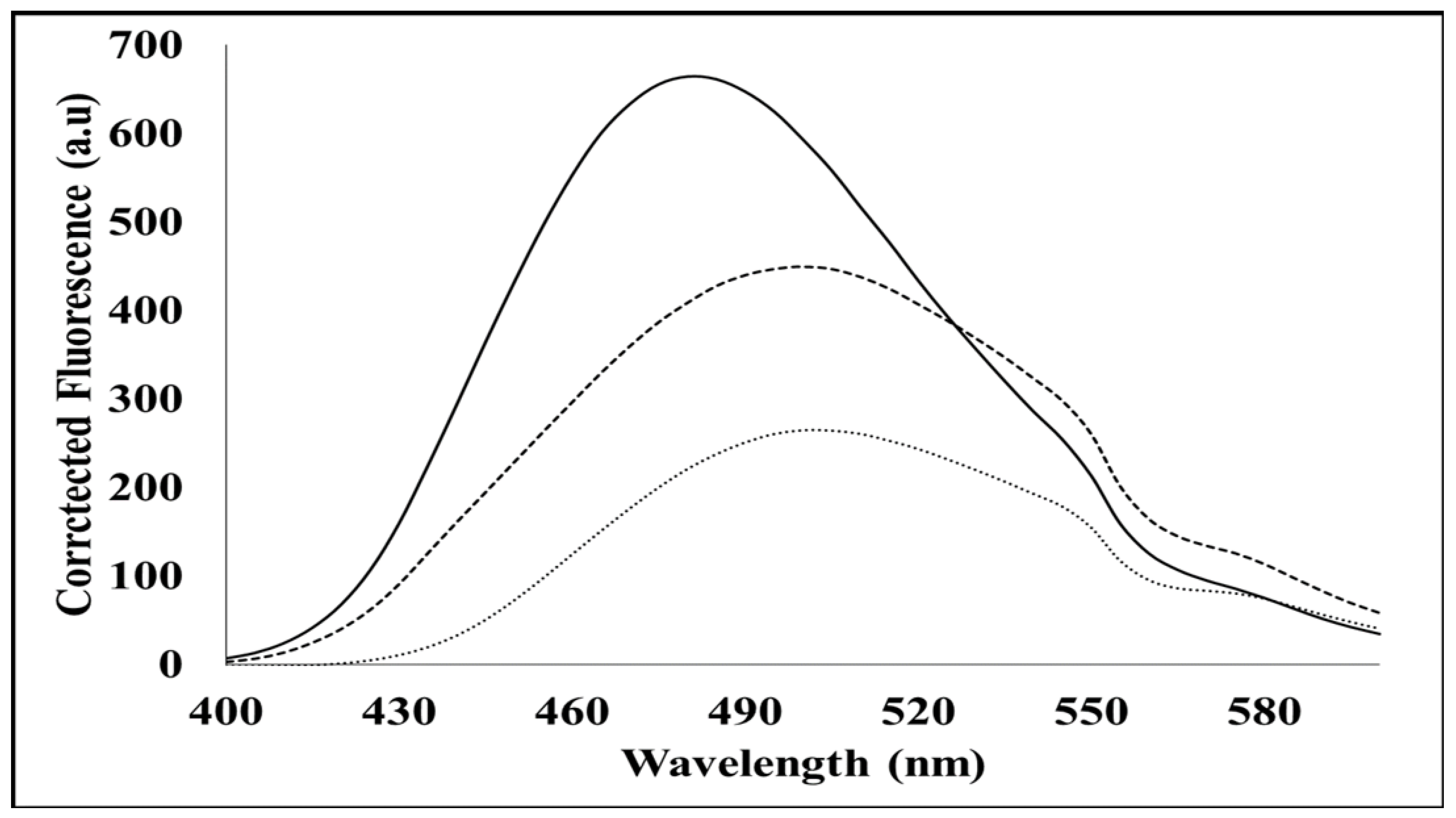
The extrinsic ANS fluorescence spectra for the SACMV MP were measured with a Jasco FP-6300 spectrofluorometer. A 20 mM stock of ANS was resuspended in 100 mM sodium phosphate (pH 6.5), 1 mM EDTA, and 0.02% NaN3, and stored in the dark. The concentration of ANS was determined using UV–Vis absorbance at 350 nm. All measurements were performed in a 50 mM Tris-HCl (pH 10) and 0.5 M l-arginine buffer. Each protein sample (triplicate) was equilibrated in the buffer, and 10 µM protein was used for each measurement. In order to determine what occurs in denatured proteins, measurements were also performed in 8 M urea (Sigma-Aldrich, St. Louis, MO, USA) in 50 mM Tris-HCl (pH 8.5) and 0.05% (w/v) NaN3 (pH 7–7.5). Each sample was excited at 380 nm, and three emission spectra between 400 and 600 nm were collected and averaged.
2.6. Intrinsic Fluorescence Mant-ATP Binding Assay
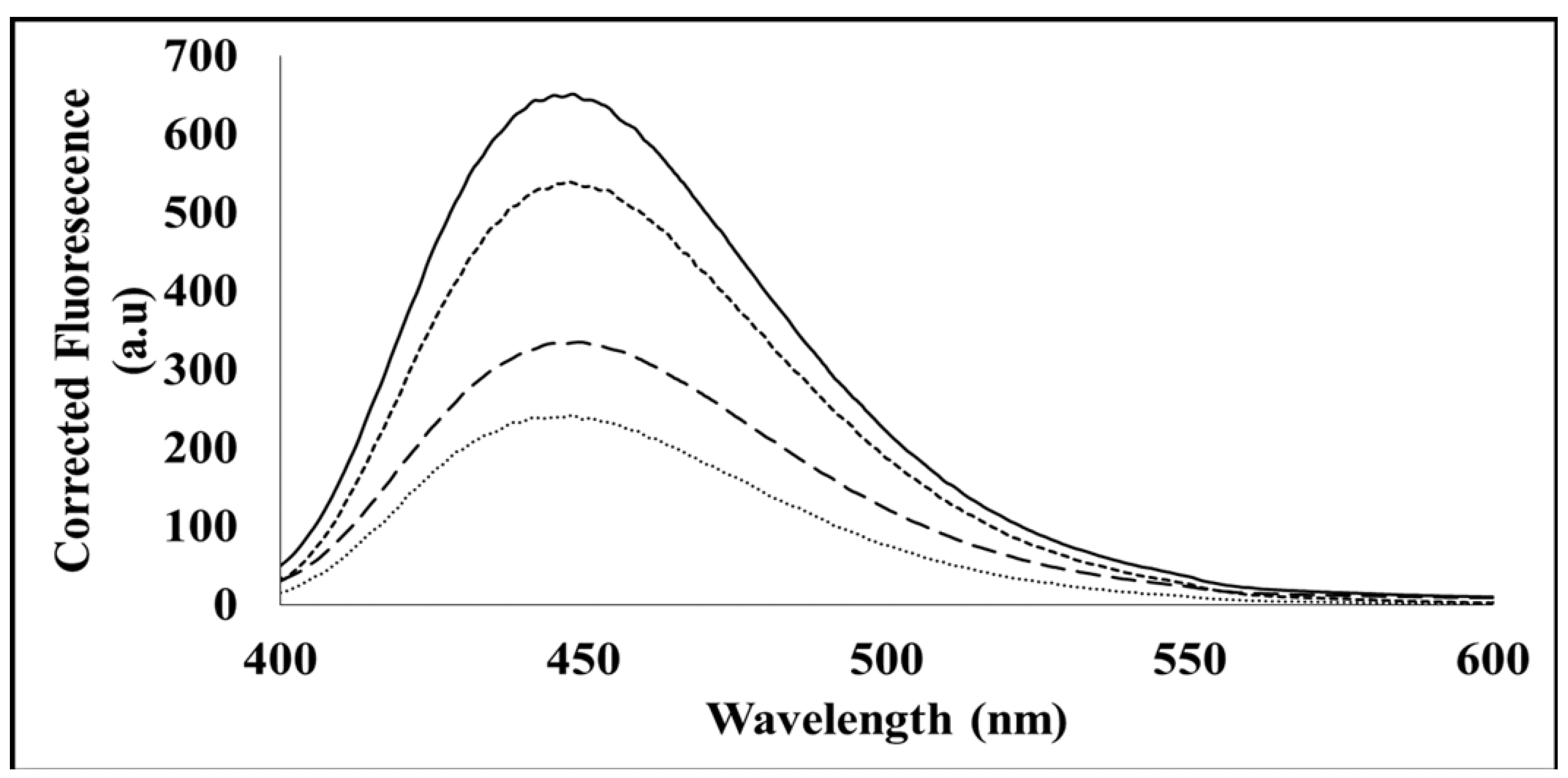
The ability of the hydrophobic sites of the MP to bind mant-ATP was determined using fluorescence spectroscopy. The fluorescence spectra were measured using a Jasco FP-6300 spectrofluorometer. A 100 µM stock of an original 5 mM stock of mant-ATP (Sigma-Aldrich, St. Louis, MO, USA) was resuspended in refolding buffer (50 mM Tris-HCl, pH 10; and 0.5 M l-arginine) and in 8 M urea (8 M Urea in 50 mM Tris-HCl, pH 8.5; and 0.05% NaN3, pH 7–7.5). Per 100 µM of mant-ATP, 35 µM of protein was used (ratio 20:7). Each sample was excited at 360 nm, and three emission spectra between 400 and 600 nm were captured and averaged.
2.7. Phosphatase Activity Assay
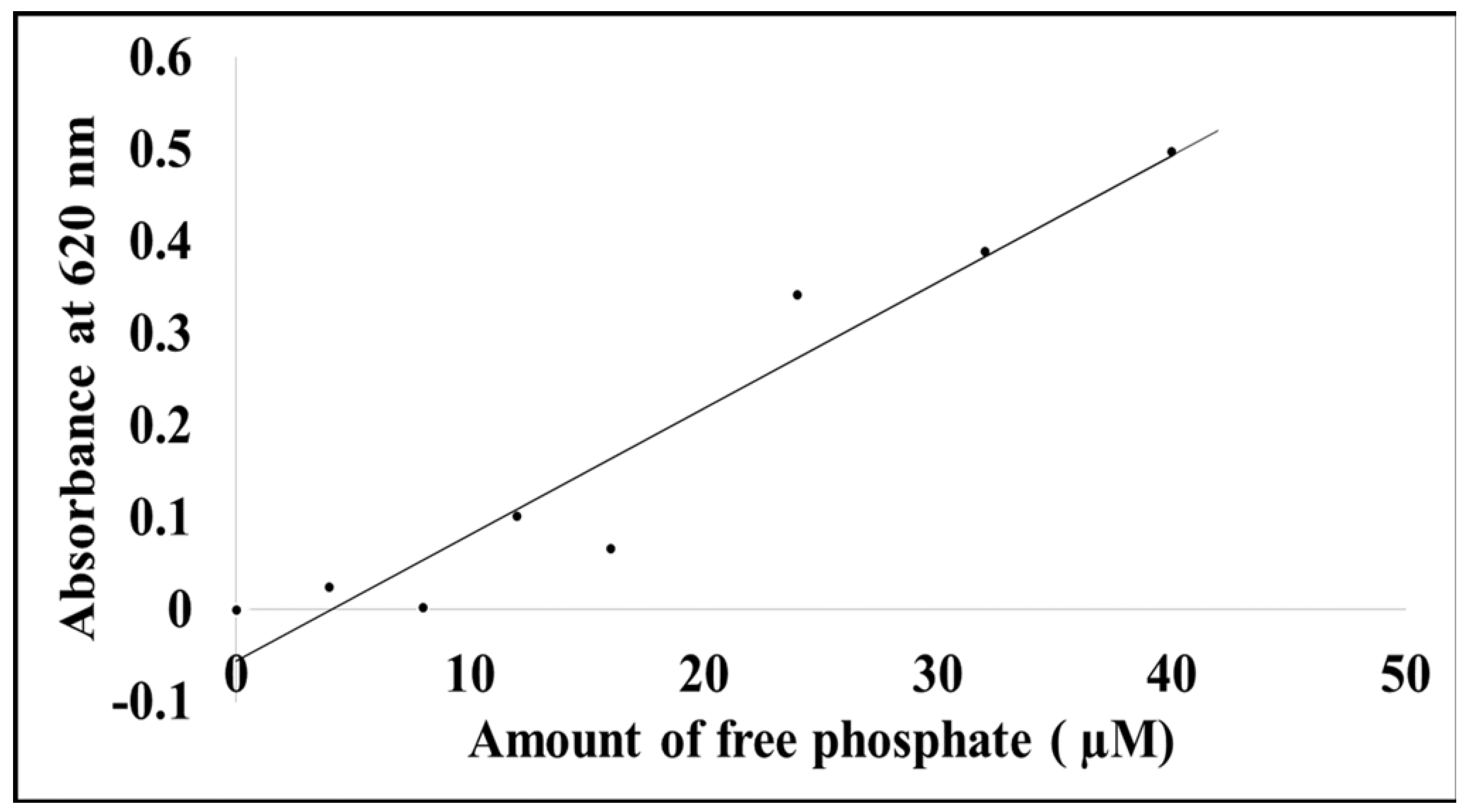
To determine if the MP has phosphatase activity, an assay kit (Sigma-Aldrich, St. Louis, MO, USA) was used to determine the amount of free phosphate released. Experiments were prepared and measured as per the manufacturer’s instructions. All measurements were recorded at 620 nm on a Multiskan spectrophotometric plate reader. All background absorbance was corrected by subtracting background values from the standard phosphate absorbance.
2.8. Intrinsic Tryptophan Fluorescence
Intrinsic tryptophan fluorescence was carried out in order to determine if the protein was refolded and if there were any conformational changes in the presence and absence of ATP. Measurements were performed in the presence and absence of ATP and for the native and denatured forms of the protein. Six samples were analysed using a Jasco FP-6300 spectrofluorometer. Tryptophan residues were excited at 280 nm, and fluorescence spectra were recorded between 300 and 500 nm. Each spectrum is the average of three accumulation replicates in 50 mM Tris-HCl (pH 7.5), containing 0.5 mM l-arginine or 8 M urea. Background fluorescence (free unbound tryptophan residues) was corrected by subtracting background values from the actual protein spectra (protein bound to tryptophan residues).
4. Discussion
While low temperatures usually favour soluble protein expression [
16,
17,
18], this was not the case for the insoluble SACMV MP. According to Burgess-Brown et al. (2008), codon optimisation increases heterologous protein expression in bacteria such as BL21 (DE3), and high expression levels of the insoluble expressed protein were obtained (
Figure 1A,B). Viral proteins are notoriously difficult to purify and solubilise due to the abundance of disordered regions, leading to poorly defined secondary structure elements [
19]. The Rep protein N-terminus of tomato yellow leaf curl virus (TYLCV) and the Rep protein C-terminus of tomato leaf curl Gujarat virus (ToLCGuV) have been partially characterised [
20,
21]. In both these studies, the soluble protein was attained only by means of expression and characterisation of a truncated (single terminus) region, as opposed to the full-length protein. This study is the first to characterise a full-length geminiviral protein. It is proposed that the lack of soluble SACMV full-length MP could be due to the bacterial
E. coli host chosen. Bacteria such as
E. coli, while preferred due to their fast growth rate, are also known for possible misfolding and precipitation of heterologous proteins [
12,
22]. An alternate expression host, yeast is a well-studied eukaryotic system used for the expression of proteins but is more difficult to work with because cell growth is slower, and the level of expression is not always high. Additionally, post-translational modifications may differ in vitro and in planta. However, working with yeast is advantageous, as the system offers a high level of intracellular expression and secretes a few of its own proteins, making expression products easier to purify for downstream characterisation [
12]. While it is notable that no other attempts to express soluble geminiviral proteins in yeast have been reported in the literature, it is suggested that the BC1 gene be codon-optimised for expression in a yeast host such as
Pichia pastoris.
Intrinsically disordered proteins or proteins with intrinsically disordered regions at the N- or C-termini often present difficulties with protein expression, purification, and crystallisation. Therefore, it is advised that disordered regions of a protein of interest are predicted before any structural studies are pursued [
13]. Based on the data retrieved from computational predictions in this study, it appears that SACMV MP is intrinsically disordered, predominantly at the C-terminus. The normalised root-mean-square deviation NRMSD, which is indicative of the goodness of fit of the calculated data to experimental data (Dichroweb) (
Figure S3) (<0.1 acceptable as a good fit), was calculated to be 1.041 for SACMV MP. This high NRMSD value further supports the prediction that MP contains long regions which are intrinsically disordered. Viral proteins often do not contain a well-defined secondary structure such as α-helix or β-strands but instead appear as random coils [
23], further suggesting that MP is an intrinsically disordered protein (IDP) or is a protein with high disordered regions (IDPRs). Viral proteins such as the Nopp140 protein which is a nucleolar protein and shuttles between the nucleolus and cytoplasm in mammalian cells, and the UL56 protein from herpes simplex virus type 2 which is a membrane protein involved in vesicular trafficking, are illustrations of IDPs as most of their protein structure has been deemed disordered [
19,
24]. Proteins that are IDPs, or contain IDPRs, have the ability to take part in protein interactions with other proteins. These protein–protein interactions are especially facilitated in regions high in disorder [
25]. Abutilon mosaic virus (AbMV) movement protein has been found to interact with HSP70 in
Arabidopsis thaliana [
26]. The interaction and binding of chaperone proteins to geminivirus movement proteins, in particular, suggest that these chaperones are needed for interaction with microtubules or cytoskeleton proteins.
Since structural studies on geminiviral MPs are absent from the literature, it is not known if ATP or other biomolecules bind to these proteins to form a complex in order to facilitate movement in the cell. In some geminiviruses, such as the Squash leaf curl virus (SqLCV), the MP traps the NSP–ssDNA complexes in the cytoplasm to move them to adjacent cells [
26], suggesting the MP is capable of binding to other proteins. Since MPs are involved in the cell-to-cell movement of ss/ds DNA complexes, as proposed by the ‘couple-skating’ and ‘relay-race’ models, it can be predicted that MPs would bind ATP. The intracellular or intercellular movement of complexes along the plasma membranes would require energy. Indeed, the phosphate assay demonstrated that SACMV denatured MP was able to hydrolyse ATP (
Figure 7), suggesting it must bind. However, whether this occurs with the native form of the MP in cells in planta is not known.
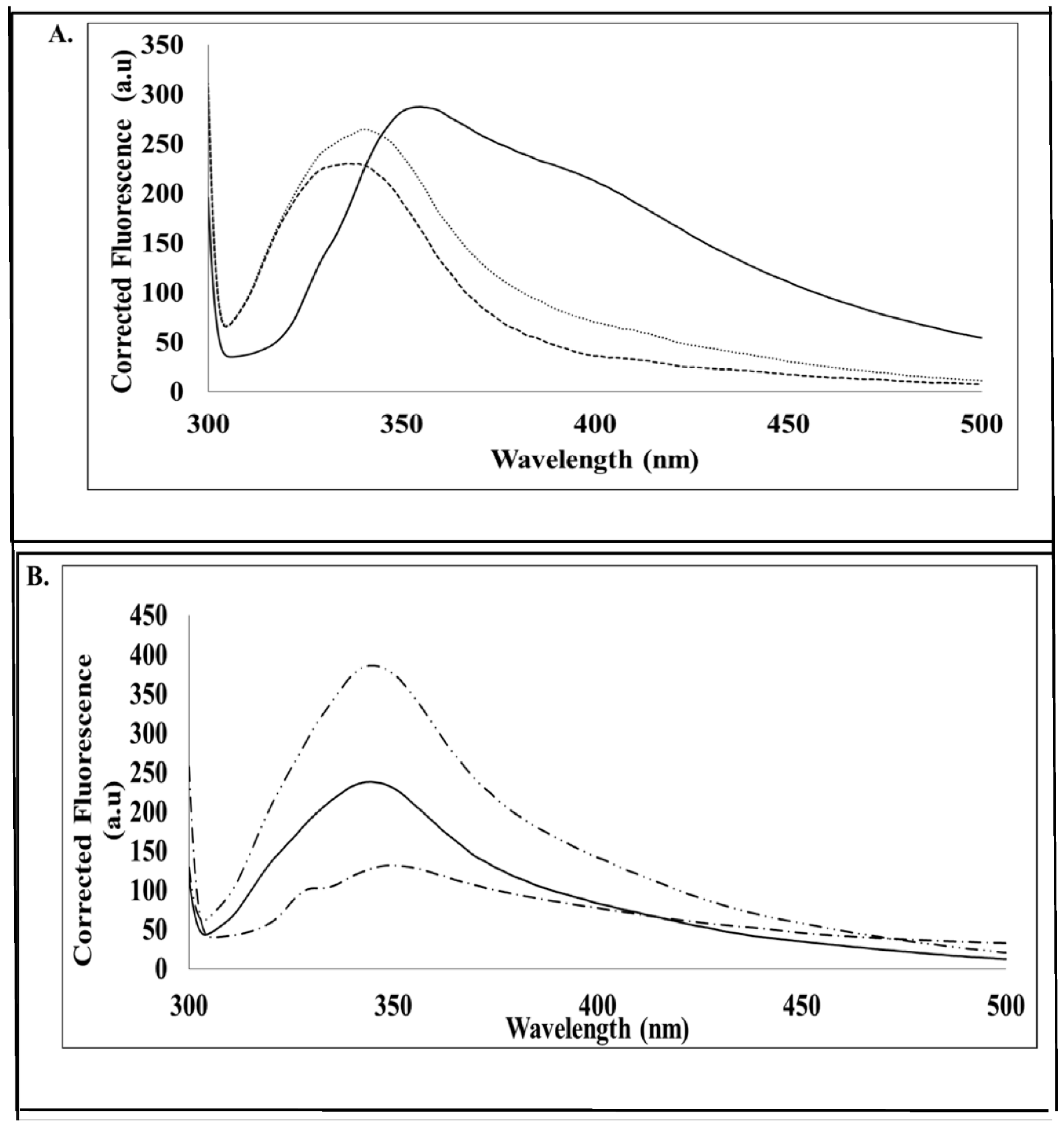
In order to confirm ATP binding, the presence of hydrophobic binding regions was investigated using an intrinsic mant-ATP assay. It can be proposed that in the denatured form in vitro, the hydrophobic regions of the MP are buried by hydrophilic sites, and therefore, mant-ATP molecules are unable to bind. Failure of the mant-ATP to bind to the denatured MP was observed by the decrease in quantum yield and lack of blue shift (λ
max) (
Figure 5). This suggests that in vivo, the MP may occur in a different conformation in its native form, allowing ATP binding to the predicted ATP binding site. ATP-binding domains on geminiviral MPs have not been predicted to date. Proteins that bind ATP are usually involved in cellular movement and membrane transport and are responsible for transporting substrates across extra and intracellular membranes [
15]. Based on the ‘couple-skating model’ and the proposed functions of the domains of MPs, as well as in vitro ATPase activity demonstrated in this study, it is suggested that SACMV MP is able to bind ATP in order to localise to the cell periphery, but further in vivo studies need to be performed. A number of plant virus movement proteins have been shown to possess ATPase activity such as the MP of potato virus X and tobacco mosaic virus (TMV) [
27]. More recent studies carried out on the geminivirus beta-satellite-encoded βC1 protein have also displayed ATPase activity [
28]. Heat shock proteins such as heat shock protein 70 (HSP70) have also been shown to possess ATPase activity, and since HSP70 chaperones form interactive complexes with geminivirus MPs [
8,
9], they may perform the ATPase function during movement. It is proposed that the ATPase activity of SACMV MP and putative interactions of SACMV DNA–MP–NSP complex with HSP70 would facilitate cytoplasmic movement from the nucleus to the cell periphery. In planta, direct binding of ligands such as ATP to the native form of SACMV MP in the cell may not lead to conformational change. After the binding of a different ligand or DNA–protein complex to the MP in the cell, such as an HSP, the MP may change conformation, allowing the transfer of the DNA/DNA–protein complex to adjacent cells. This is supported by the active role of MP in cellular movement [
8,
9]. Size exclusion–high-performance liquid chromatography (SE–HPLC) was carried out on full-length MPs (data not shown); however, the correct retention time expected for protein elution was not attained. The protein appears to bind to the stationary phase of the column and elutes extremely late off the column, giving the incorrect retention time and resulting in incorrect molecular weight. It can be proposed that, due to the SACMV MP being highly intrinsically disordered and containing large hydrophobic regions, the protein remains bound to the non-polar stationary phase of the column, rather than moving through with the polar mobile phase, resulting in the protein eluting at the incorrect retention time [
12]. The non-elution of the protein off the non-polar stationary phase of the SE–HPLC column further suggests that the protein may be aggregating and may form a high-order oligomer post refolding.
5. Conclusions
In conclusion, this research on the SACMV MP revealed, for the first time, the proposed secondary structure of a denatured refolded full-length monomeric geminivirus cell-to-cell movement protein in vitro. Due to the lack of attainment of soluble protein, denaturation-refolding was required. Results establish that the BC1-encoded MP from DNA-B is highly insoluble in solution and has a secondary structure which predominantly consists of β-strands. Its predicted large unfolded domain at the C-terminus presents some interesting speculations as to the function of the MP C-terminus. A study on Abutilon mosaic virus has revealed a homo-oligomerisation of the C-terminus of the MP and suggests that the virus possesses a host-plant species-independent capacity to bind to plasma membranes, likely occurring at the C-terminus. AbMV MP-mediated intracellular sorting is dependent on an ‘anchor domain’ (amino acids 117 to 180), which is necessary to traffic the MP complex to the cell periphery. We propose that SACMV MP has the ability to bind ATP in its native form due to ATPase activity, as demonstrated by the in vitro results herein. Viral MPs change in conformation in the presence of ATP and adopt a different conformation when denatured, compared with their native state. This suggests that, in the plant cell in vivo, native forms of the SACMV MP, or alternatively MPs that form a high-order oligomer, may undergo conformational changes when bound to biomolecules such as ATP. Indirect binding of other ligands with ATPase activity, such as HSP70, has been shown in other geminivirus studies, and recently Arabidopsis homologs HSP90 (AT5G56030) and mitochondrial HSP60 (AT3G23990) were demonstrated to interact with SACMV MP via Y2H and co-IP in cassava infected with SACMV. Since the MP has the proposed ability to bind ATP, further kinetic research may be carried out in future studies. In addition, further structural and molecular characterisation studies of geminivirus cell-to-cell movement proteins will unravel more in planta features of their functions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






