Phosphate Groups in the Lipid A Moiety Determine the Effects of LPS on Hepatic Stellate Cells: A Role for LPS-Dephosphorylating Activity in Liver Fibrosis

 ,
,
Abstract
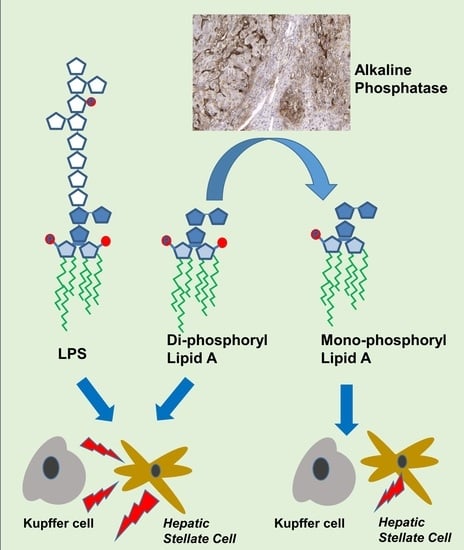
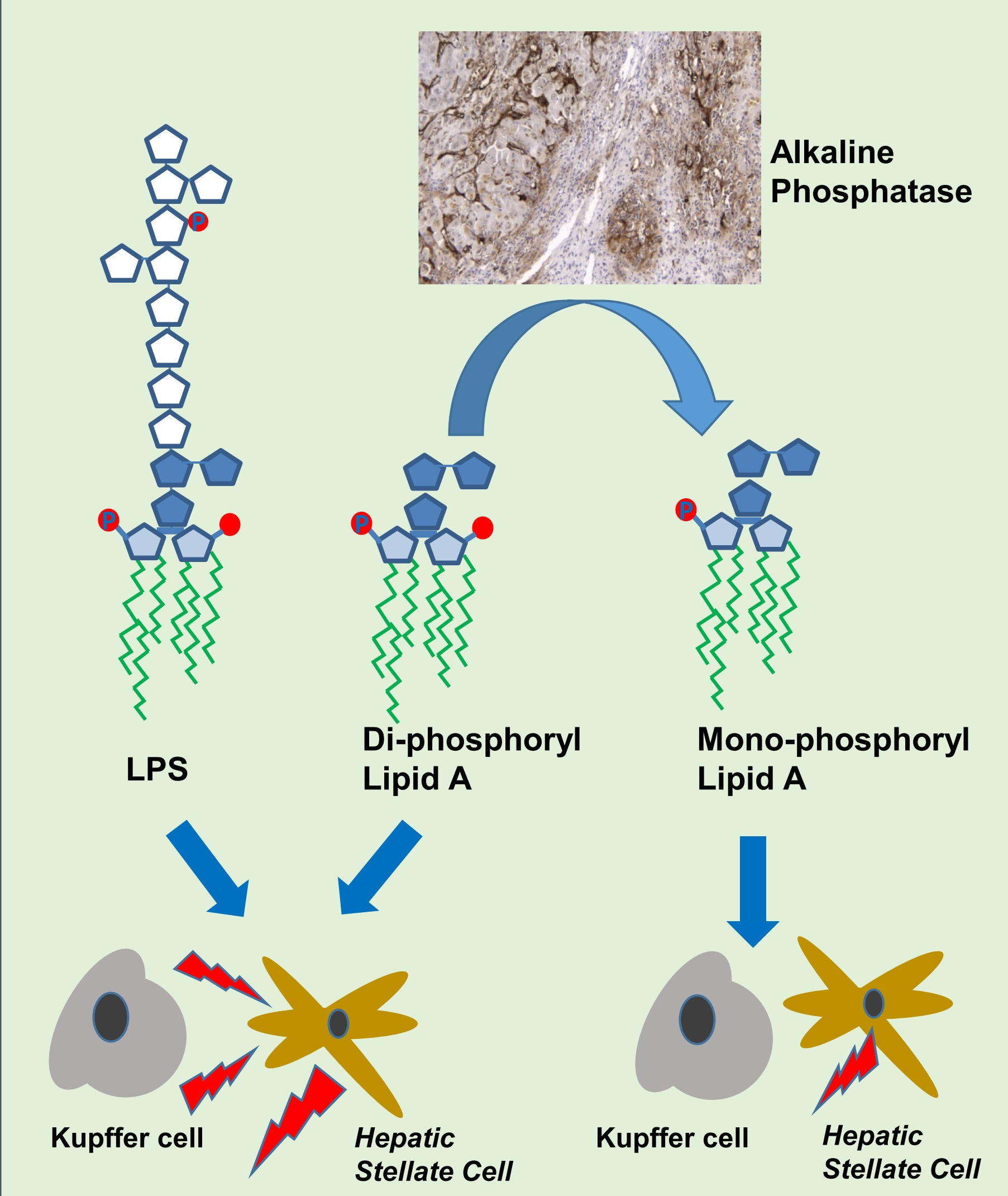
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. In Vitro Studies: Effects of Mono- and Di-Phosphoryl Lipid A on Macrophages and Hepatic Stellate Cells
2.3. NO Assay
2.4. Animal Experiments
2.5. Human Tissue Samples
2.6. Immunohistochemistry, Enzyme Histochemistry and Quantitative Analysis of Sections
2.6.1. Alkaline Phosphatase Staining
2.6.2. Histochemical Detection of LPS-Dephosphorylation
2.6.3. Quantitative Analysis of Alkaline Phosphatase Activity in Tissue and Serum
2.7. Statistical Analysis
3. Results
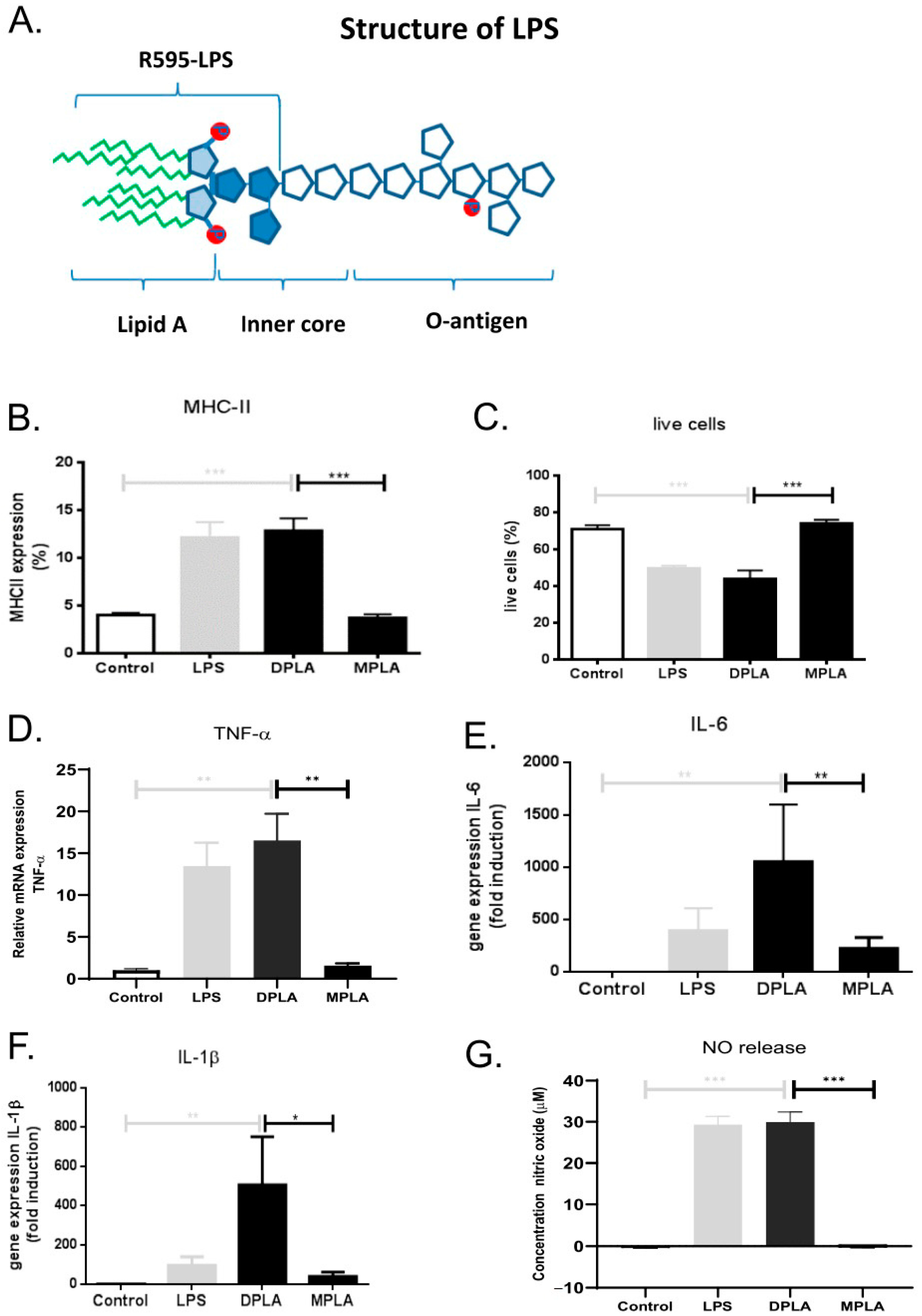
3.1. Effect of Lipid A Molecules on RAW 264.7 Macrophages
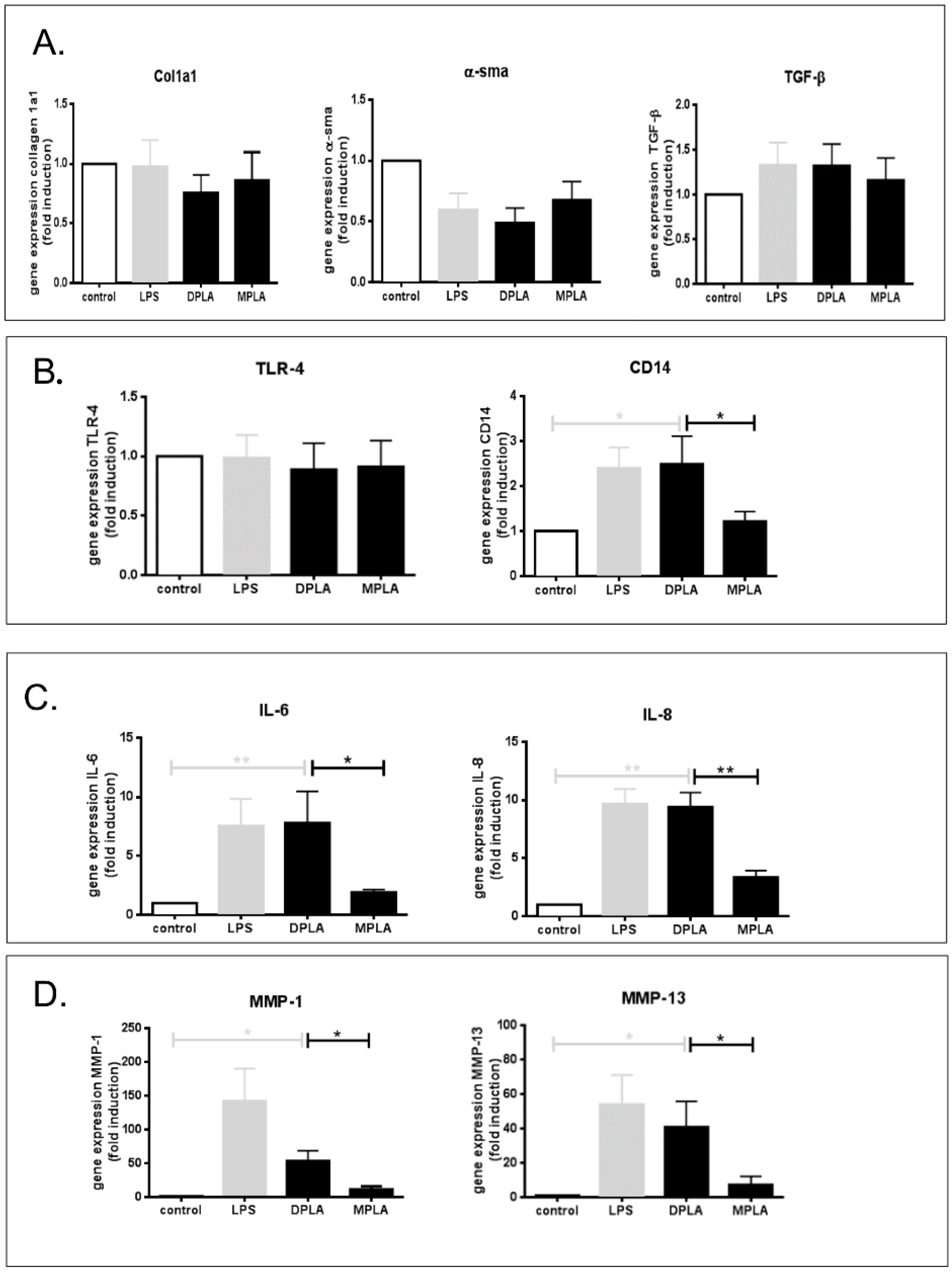
3.2. Effect of Lipid A Molecules on Primary Rat HSCs
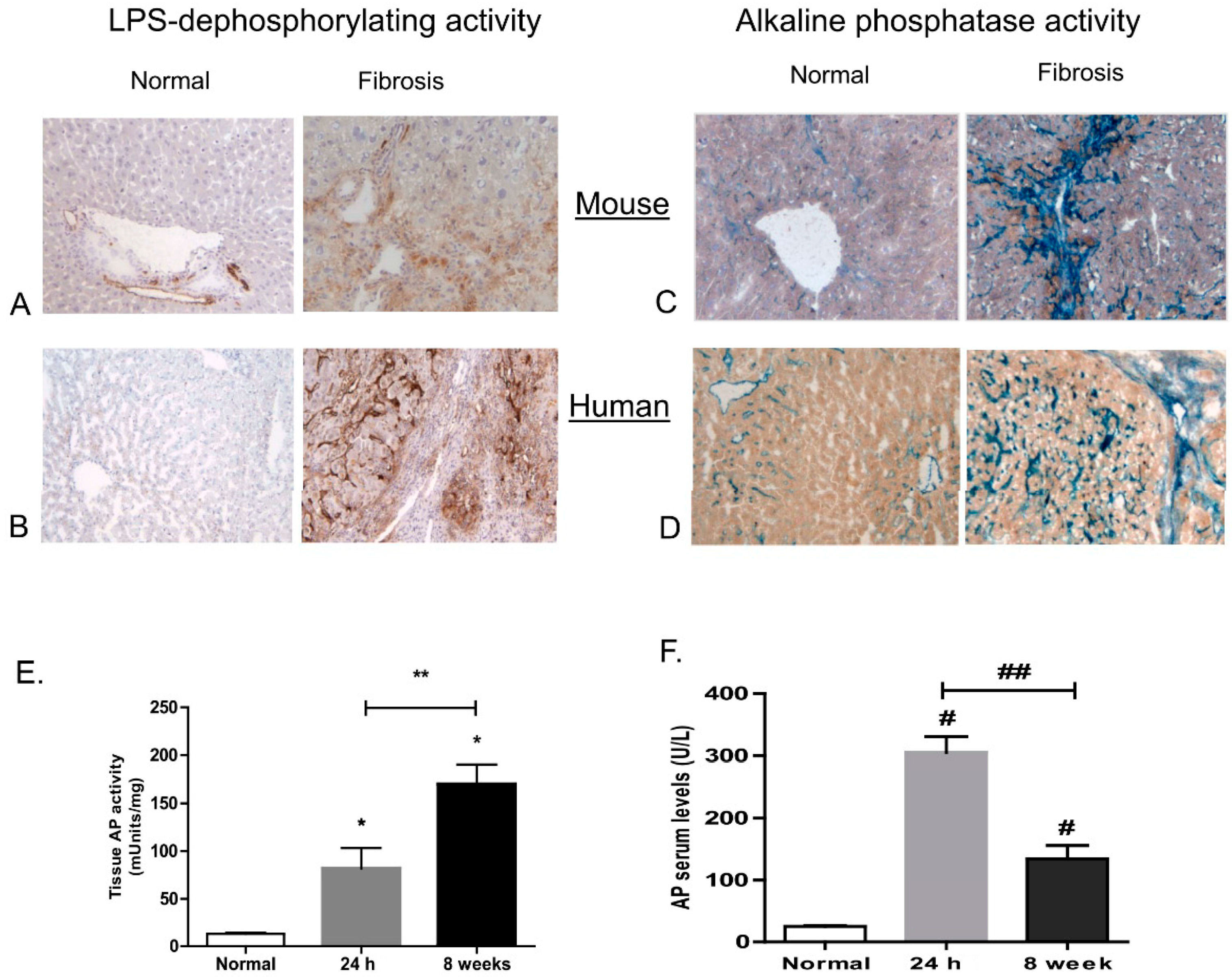
3.3. Dephosphorylation of LPS and Lipid A in Mouse and Human Livers
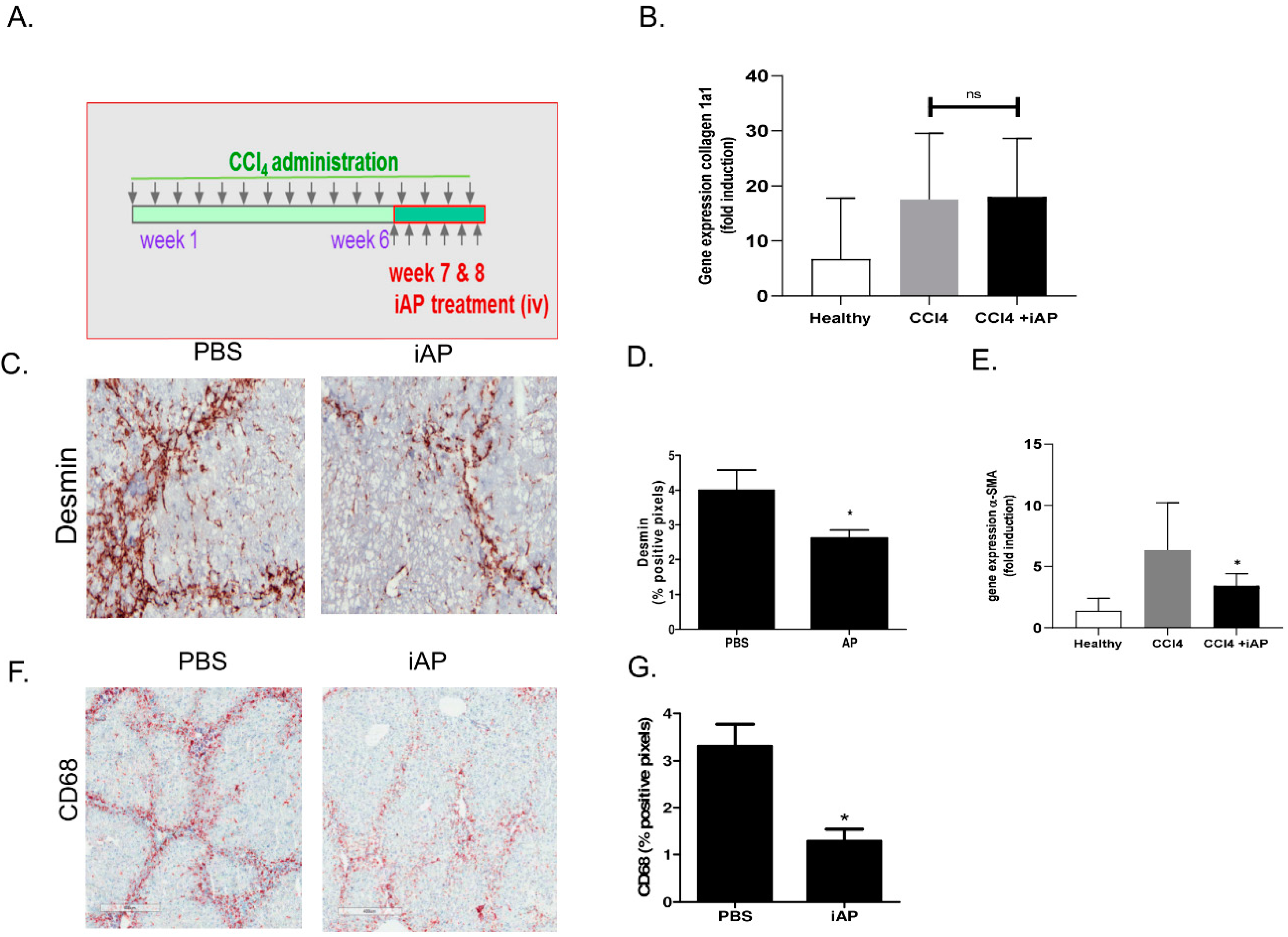
3.4. Effect of Exogenous iAP on Liver Fibrogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poelstra, K.; Bakker, W.W.; Klok, P.A.; Hardonk, M.J.; Meijer, D.K. A physiologic function for alkaline phosphatase: Endotoxin detoxification. Lab. Investig. 1997, 76, 319–327. [Google Scholar] [PubMed]
- Poelstra, K.; Bakker, W.W.; Klok, P.A.; Kamps, J.A.; Hardonk, M.J.; Meijer, D.K. Dephosphorylation of endotoxin by alkaline phosphatase in vivo. Am. J. Pathol. 1997, 151, 1163–1169. [Google Scholar] [PubMed]
- Beutler, B.; Rietschel, E.T. Innate immune sensing and its roots: The story of endotoxin. Nat. Rev. Immunol. 2003, 3, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Albitar-Nehme, S.; Kim, E.; Marr, N.; Novikov, A.; Caroff, M.; Fernandez, R.C. Minor modifications to the phosphate groups and the C3’ acyl chain length of lipid A in two bordetella pertussis strains, BP338 and 18-323, independently affect toll-like receptor 4 protein activation. J. Biol. Chem. 2013, 288, 11751–11760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Q.; Six, D.A.; Liu, Q.; Gu, L.; Wang, S.; Alamuri, P.; Raetz, C.R.H.; Curtiss, R., III. Phosphate groups of lipid A are essential for salmonella enterica serovar typhimurium virulence and affect innate and adaptive immunity. Infect. Immun. 2012, 80, 3215–3224. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Six, D.A.; Roland, K.L.; Gu, L.; Reynolds, C.M.; Wang, X.; Raetz, C.R.H.; Curtiss, R. Salmonella synthesizing 1-dephosphorylated lipopolysaccharide exhibits low endotoxic activity while retaining its immunogenicity. J. Immunol. 2011, 187, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Cullen, T.W.; Schofield, W.B.; Barry, N.A.; Putnam, E.E.; Rundell, E.A.; Trent, M.S.; Degnan, P.H.; Booth, C.J.; Yu, H.; Goodman, A.L. Gut microbiota. antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 2015, 347, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Verweij, W.R.; Bentala, H.; van der Vlag, A.H.; Miek van Loenen-Weemaes, A.; Kooi, K.; Meier, D.K.F.; Poelstra, K. Protection against an escherichia coli-induced sepsis by alkaline phosphatase in mice. Shock 2004, 22, 174–179. [Google Scholar] [CrossRef]
- Bentala, H.; Verweij, W.R.; van der Vlag, A.H.; van Loenen-Weemaes, A.M.; Meijer, D.K.F.; Poelstra, K. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock 2002, 18, 561–566. [Google Scholar] [CrossRef]
- Hwang, S.W.; Kim, J.H.; Lee, C.; Im, J.P.; Kim, J.S. Intestinal alkaline phosphatase ameliorates experimental colitis via toll-like receptor 4-dependent pathway. Eur. J. Pharmacol. 2018, 820, 156–166. [Google Scholar] [CrossRef]
- Lee, C.; Chun, J.; Hwang, S.W.; Kang, S.J.; Im, J.P.; Kim, J.S. The effect of intestinal alkaline phosphatase on intestinal epithelial cells, macrophages and chronic colitis in mice. Life Sci. 2014, 100, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Brands, R.; Wang, Z.; Verdant, C.; Bruhn, A.; Cai, Y.; Raaben, W.; Wulferink, M.; Vincent, J.-L. Beneficial effects of alkaline phosphatase in septic shock. Crit. Care Med. 2006, 34, 2182–2187. [Google Scholar] [CrossRef] [PubMed]
- Pickkers, P.; Heemskerk, S.; Schouten, J.; Laterre, P.-F.; Vincent, J.-L.; Beishuizen, A.; Jorens, P.G.; Spapen, H.; Bulitta, M.; Peters, W.H.M.; et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: A prospective randomized double-blind placebo-controlled trial. Crit. Care 2012, 16, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, M.; Drastich, P.; Konecny, M.; Gionchetti, P.; Urban, O.; Cantoni, F.; Bortlik, M.; Duricova, D.; Bulitta, M. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm. Bowel Dis. 2010, 16, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.; Mehta, R.L.; Murray, P.T.; Hummel, J.; Joannidis, M.; Kellum, J.A.; Arend, J.; Pickkers, P. Study protocol for a multicentre randomised controlled trial: Safety, tolerability, efficacy and quality of life of a human recombinant alkaline phosphatase in patients with sepsis-associated acute kidney injury (STOP-AKI). BMJ Open 2016, 6, e012371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickkers, P.; Mehta, R.L.; Murray, P.T.; Joannidis, M.; Molitoris, B.A.; Kellum, J.A.; Bachler, M.; Hoste, E.A.J.; Hoiting, O.; Krell, K.; et al. Effect of human recombinant alkaline phosphatase on 7-day creatinine clearance in patients with sepsis-associated acute kidney injury: A randomized clinical trial. JAMA 2018, 320, 1998–2009. [Google Scholar] [CrossRef] [Green Version]
- Hummeke-Oppers, F.; Hemelaar, P.; Pickkers, P. Innovative drugs to target renal inflammation in sepsis: Alkaline phosphatase. Front. Pharmacol. 2019, 10, 919. [Google Scholar] [CrossRef]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S1), 38–42. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.A.; Paik, Y.H.; Schnabl, B. Role of gut microbiota in liver disease. J. Clin. Gastroenterol. 2015, 49, S25–S27. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, C.R. Pro-and anti-fibrogenic functions of gram-negative bacterial lipopolysaccharide in the liver. Front. Med. (Lausanne) 2020, 7, 130. [Google Scholar] [CrossRef] [Green Version]
- Nagasaki, A.; Sakamoto, S.; Chea, C.; Ishida, E.; Farusho, H.; Fujii, M.; Takata, T.; Miyauchi, M. Odontogenic infection by porphyromonas gingivalis exacerbates fibrosis in NASH via hepatic stellate cell activation. Sci. Rep. 2020, 10, 4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC-mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelmann, C.; Sheikh, M.; Sharma, S.; Kondo, T.; Loeffler-Wirth, H.; Zheng, Y.B.; Novelli, S.; Hall, A.; Kerbert, A.J.C.; Macnaughtan, J.; et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J. Hepatol. 2020, 73, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 2020, 72, 470–485. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Pradere, J.-P.; Troeger, J.S.; Dapito, D.H.; Mencin, A.A.; Schwabe, R.F. Toll-like receptor 4 and hepatic fibrogenesis. Semin Liver Dis. 2010, 30, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Kaji, K.; Kitade, M.; Kubo, T.; Furukawa, M.; Saikawa, S.; Shimozato, N.; Sato, S.; Seki, K.; Kawaratani, H.; et al. Exogenous Administration of Low-Dose Lipopolysaccharide Potentiates Liver Fibrosis in a Choline-Deficient l-Amino-Acid-Defined Diet-Induced Murine Steatohepatitis Model. Int. J. Mol. Sci. 2019, 20, 2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarneh, S.R.; Kim, B.-M.; Kaliannan, K.; Morrison, S.A.; Tantillo, T.J.; Tao, Q.; Mohamed, M.M.R.; Ramirez, J.M.; Karas, A.; Liu, W.; et al. Intestinal alkaline phosphatase attenuates alcohol-induced hepatosteatosis in mice. Dig. Dis. Sci. 2017, 62, 2021–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelmann, C.; Adebayo, D.; Oria, M.; De Chiara, F.; Novelli, S.; Habtesion, A.; Davies, N.; Andreola, F.; Jalan, R. Recombinant alkaline phosphatase prevents acute on chronic liver failure. Sci. Rep. 2020, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.J.; Betrapally, N.S.; Ghosh, S.A.; Sartor, R.B.; Hylemon, P.B.; Gillevet, P.M.; Sanyal, A.J.; Heuman, D.M.; Carl, D.; Zhou, H.; et al. Gut microbiota drive the development of neuroinflammatory response in cirrhosis in mice. Hepatology 2016, 64, 1232–1248. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.-R.; Jing, Y.-Y.; Liu, W.-T.; Han, Z.-P.; Li, R.; Yang, Y.; Zhu, J.-N.; Li, X.-Y.; Li, P.-P.; Wei, L.-X. Lipopolysaccharide induces the differentiation of hepatic progenitor cells into myofibroblasts via activation of the hedgehog signaling pathway. Cell Cycle 2017, 16, 1357–1365. [Google Scholar] [CrossRef]
- Zou, Y.; Cai, Y.; Lu, D.; Zhou, Y.; Yao, Q.; Zhang, S. MicroRNA-146a-5p attenuates liver fibrosis by suppressing profibrogenic effects of TGFbeta1 and lipopolysaccharide. Cell. Signal. 2017, 39, 1–8. [Google Scholar] [CrossRef]
- Robbe, P.; Draijer, C.; Borg, T.R.; Luinge, M.; Timens, W.; Wouters, I.M.; Melgert, B.N.; Hylkema, M.N. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L358–L367. [Google Scholar] [CrossRef] [Green Version]
- Tuin, A.; Poelstra, K.; de Jager-Krikken, A.; Bok, L.; Raaben, W.; Velders, M.P.; Dijkstra, G. Role of alkaline phosphatase in colitis in man and rats. Gut 2009, 58, 379–387. [Google Scholar] [CrossRef]
- Suzuki, N.; Irie, M.; Iwata, K.; Nakane, H.; Yoshikane, M.; Koyama, Y.; Uehara, Y.; Takeyama, Y.; Kitamura, Y.; Sohda, T.; et al. Altered expression of alkaline phosphatase (ALP) in the liver of primary biliary cirrhosis (PBC) patients. Hepatol. Res. 2006, 35, 37–44. [Google Scholar] [CrossRef]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; et al. Kupffer cells: Inflammation pathways and cell-cell interactions in alcohol-associated liver disease. Am. J. Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef]
- Sharma, A.; Verma, A.K.; Kofron, M.; Kudira, R.; Miethke, A.; Wu, T.; Wang, J.; Gandhi, C.R. Lipopolysaccharide reverses hepatic stellate cell activation via modulation of cMyb, SMADs and C/EBP transcription factors. Hepatology 2020, 72, 1800–1818. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Salzman, N.; Acharya, C.; Takei, H.; Kakiyama, G.; Fagan, A.; White, M.B.; Gavis, E.A.; Holtz, M.L.; Hayward, M.; et al. Microbial functional change is linked with clinical outcomes after capsular fecal transplant in cirrhosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Gabele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Chilton, P.M.; Hadel, D.M.; To, T.T.; Mitchell, T.C.; Darveau, R.P. Adjuvant activity of naturally occurring monophosphoryl lipopolysaccharide preparations from mucosa-associated bacteria. Infect. Immun. 2013, 81, 3317–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuin, A.; Huizinga-Van der Vlag, A.; van Loenen-Weemaes, A.M.M.-A.; Meijer, D.K.F.; Poelstra, K. On the role and fate of LPS-dephosphorylating activity in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G377–G385. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Heithoff, D.M.; Aziz, P.V.; Haslund-Gourley, B.; Westman, J.S.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Nizet, V.; Mahan, M.J.; et al. Accelerated aging and clearance of host anti-inflammatory enzymes by discrete pathogens fuels sepsis. Cell Host Microbe 2018, 24, 500–513.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, D.; John, S. Alkaline phosphatase. In StatPearls; StatPearls Publishing LLC: Treaure Island, FL, USA, 2020. [Google Scholar]
- Linder, C.H.; Englund, U.H.; Narisawa, S.; Millán, J.L.; Magnusson, P. Isozyme profile and tissue-origin of alkaline phosphatases in mouse serum. Bone 2013, 53, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Economopoulos, K.P.; Ward, N.L.; Phillips, C.D.; Teshager, A.; Patel, P.; Mohamed, M.M.; Hakimian, S.; Cox, S.B.; Ahmed, R.; Moaven, O.; et al. Prevention of antibiotic-associated metabolic syndrome in mice by intestinal alkaline phosphatase. Diabetes Obes. Metab. 2016, 18, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Hamarneh, S.R.; Mohamed, M.M.; Economopoulos, K.P.; Morrison, S.A.; Phupitakphol, T.; Tantillo, T.J.; Gul, S.S.; Gharedaghi, M.H.; Tao, Q.; Kaliannan, K.; et al. A novel approach to maintain gut mucosal integrity using an oral enzyme supplement. Ann. Surg. 2014, 260, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Malo, M.S.; Moaven, O.; Muhammad, N.; Biswas, B.; Alam, S.N.; Economopoulos, K.P.; Gul, S.S.; Hamarneh, S.R.; Malo, N.S.; Teshager, A.; et al. Intestinal alkaline phosphatase promotes gut bacterial growth by reducing the concentration of luminal nucleotide triphosphates. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G826–G838. [Google Scholar] [CrossRef]
- Caussy, C.; Hsu, C.; Lo, M.-T.; Liu, A.; Bettencourt, R.; Ajmera, V.H.; Bassirian, S.; Hooker, J.; Sy, E.; Richards, L.; et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018, 68, 918–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.T.; Malo, M.S.; Beasley-Topliffe, L.K.; Poelstra, K.; Millan, J.L.; Mostafa, G.; Alam, S.N.; Ramasamy, S.; Warren, H.S.; Hohmann, E.L.; et al. A role for intestinal alkaline phosphatase in the maintenance of local gut immunity. Dig. Dis. Sci. 2011, 56, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.S.; Alam, S.N.; Mostafa, G.; Zeller, S.J.; Johnson, P.V.; Mohammad, N.; Chen, K.T.; Moss, A.K.; Ramasamy, S.; Faruqui, A.; et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut 2010, 59, 1476–1484. [Google Scholar] [CrossRef]
- Kaliannan, K.; Hamarneh, S.R.; Economopoulos, K.P.; Alam, S.N.; Moaven, O.; Patel, P.; Malo, N.S.; Ray, M.; Abtahi, S.M.; Muhammad, N.; et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 7003–7008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, F.; Adiliaghdam, F.; Cavallaro, P.M.; Hamarneh, S.R.; Tsurumi, A.; Hoda, R.S.; Munoz, A.R.; Dhole, Y.; Ramirez, J.M.; Liu, E.; et al. Intestinal alkaline phosphatase targets the gut barrier to prevent aging. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Liu, Y.; Cavallaro, P.M.; Kim, B.M.; Liu, T.; Wang, H.; Kuehn, F.; Adiliaghdam, F.; Liu, E.; Vasan, R.; Samarbafzadeh, E.; et al. A role for intestinal alkaline phosphatase in preventing liver fibrosis. Theranostics 2020, 11, 14–26. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse primers: | ||
| Gene | Forward Primer | Reverse Primer |
| β-actin | 5′ ATC GTG CGT GAC ATC AAA GA 3′ | 3′ ATG CCA CAG GAT TCC ATA CC 5′ |
| TNF-α | 5′ CAT CTT CTCA AAA TTC GAG TGA CAA 3′ | 3′ GAG TAG ACA AGG TAC AAC CC 5′ |
| IL-1β | 5′ GCC AAG ACA GGT CGC TCA GGG 3′ | 3′ CCC CCA CAC GTT GAC AGC TAG G 5′ |
| IL-6 | 5′ TGA TGC TGG TGA CAA CCA CGG C 3′ | 3′ TAA GCC TCC GAC TTG TGA AGT GGT A 5′ |
| Col1a1 | 5′ TGA CTG GAA GAG CGG AGA GT 3′ | 3′ ATC CAT CGG TCA TGC TCT CT 5′ |
| αSMA | 5′ ACT ACT GCC GAG CGT GAG AT 3′ | 3′ CCA ATG AAA GAT GGC TGG AA 5′ |
| Rat primers: | ||
| Gene | Forward Primer | Reverse Primer |
| β-actin | 5′ GGC ATC CTG ACC CTG AAG TA 3′ | 5′ GGG GTG TTG AAG GTC TCA AA 3′ |
| IL-6 | 5′ CCG GAG AGG AGA CTT CAC AG 3′ | 5 ‘ACA GTG CAT CAT CGC TGT TC 3′ |
| IL-8 | 5′ GGC AGG GAT TCA CTT CAA GA 3′ | 5′ GCC ATC GGT GCA ATC TAT CT 3′ |
| TLR4 | 5′ AAC TTC CTG GGG AAA AAC TCT TG 3′ | 5′ TGC CAC CAT TTA CAG TTC GTC AT 3′ |
| CD14 | 5′ GTT ACA CAA CAG GCT GGA TAG G 3′ | 5′ ACT ACG CCA GAG TTA TAC GC 3′ |
| MMP-1 | 5′ CTT GCG GGA ATC CTG AAG AAG TCT A 3′ | 5′ GCC AAG CTC ATG GGC AGC AAC AAT 3′ |
| MMP-13 | 5′ GGA AGA CCC TCT TCT TCT CA 3′ | 5′ TCA TAG ACA GCA TCT ACT TTG TC 3′ |
| TGFβ | 5′ ATA CGC CTG AGT GGC TGT CT 3′ | 5′ TGG GAC TGA TCC CAT TGA TT 3′ |
| Col1a1 | 5′ AGC CTG AGC CAG CAG ATT GA 3′ | 5′ CCA GGT TGC AGC CTT GGT TA 3′ |
| αSMA | 5′ GAC ACC AGG GAG TGA TGG TT 3′ | 5′ GTT AGC AAG GTC GGA TGC TC 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schippers, M.; Post, E.; Eichhorn, I.; Langeland, J.; Beljaars, L.; Malo, M.S.; Hodin, R.A.; Millán, J.L.; Popov, Y.; Schuppan, D.; et al. Phosphate Groups in the Lipid A Moiety Determine the Effects of LPS on Hepatic Stellate Cells: A Role for LPS-Dephosphorylating Activity in Liver Fibrosis. Cells 2020, 9, 2708. https://doi.org/10.3390/cells9122708
Schippers M, Post E, Eichhorn I, Langeland J, Beljaars L, Malo MS, Hodin RA, Millán JL, Popov Y, Schuppan D, et al. Phosphate Groups in the Lipid A Moiety Determine the Effects of LPS on Hepatic Stellate Cells: A Role for LPS-Dephosphorylating Activity in Liver Fibrosis. Cells. 2020; 9(12):2708. https://doi.org/10.3390/cells9122708
Chicago/Turabian StyleSchippers, Marlies, Eduard Post, Ilse Eichhorn, Jitske Langeland, Leonie Beljaars, Madhu S. Malo, Richard A. Hodin, José Luis Millán, Yury Popov, Detlef Schuppan, and et al. 2020. "Phosphate Groups in the Lipid A Moiety Determine the Effects of LPS on Hepatic Stellate Cells: A Role for LPS-Dephosphorylating Activity in Liver Fibrosis" Cells 9, no. 12: 2708. https://doi.org/10.3390/cells9122708