Induction of the Endoplasmic-Reticulum-Stress Response: MicroRNA-34a Targeting of the IRE1α-Branch

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
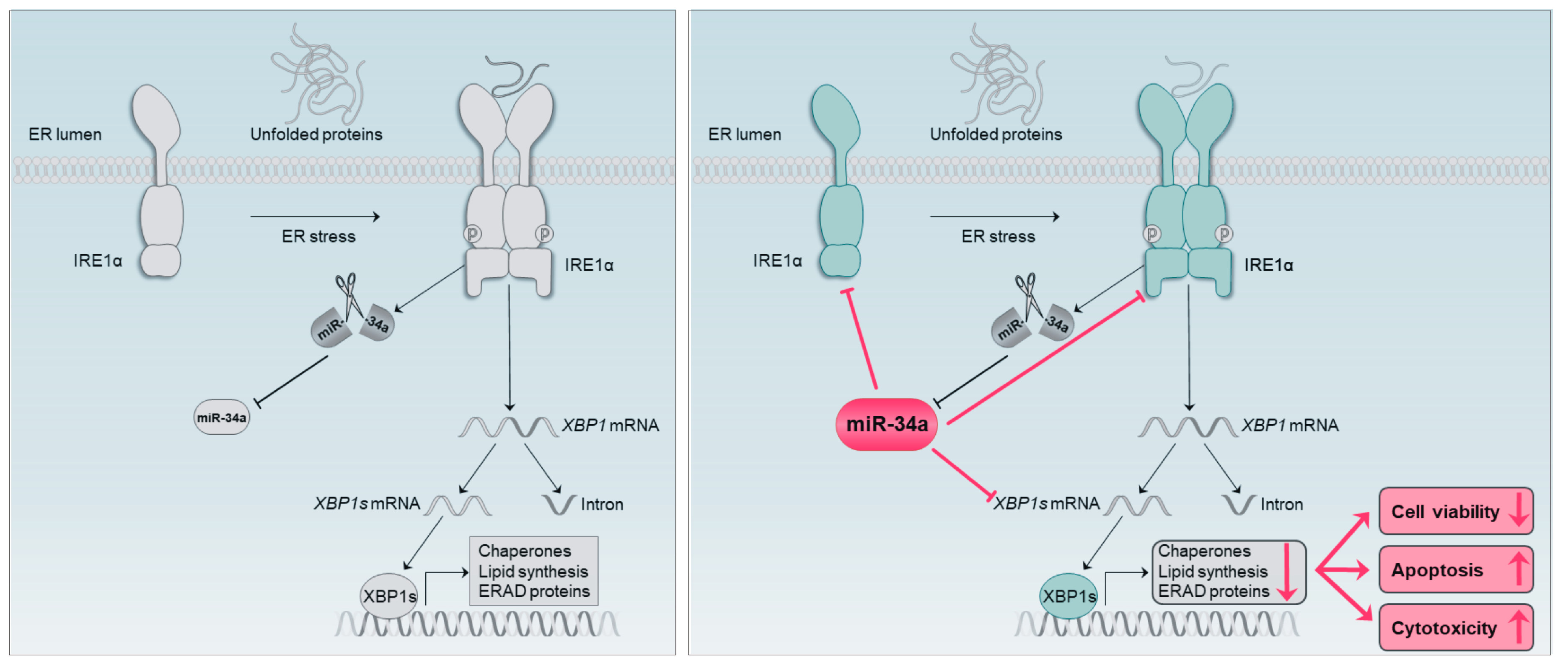
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
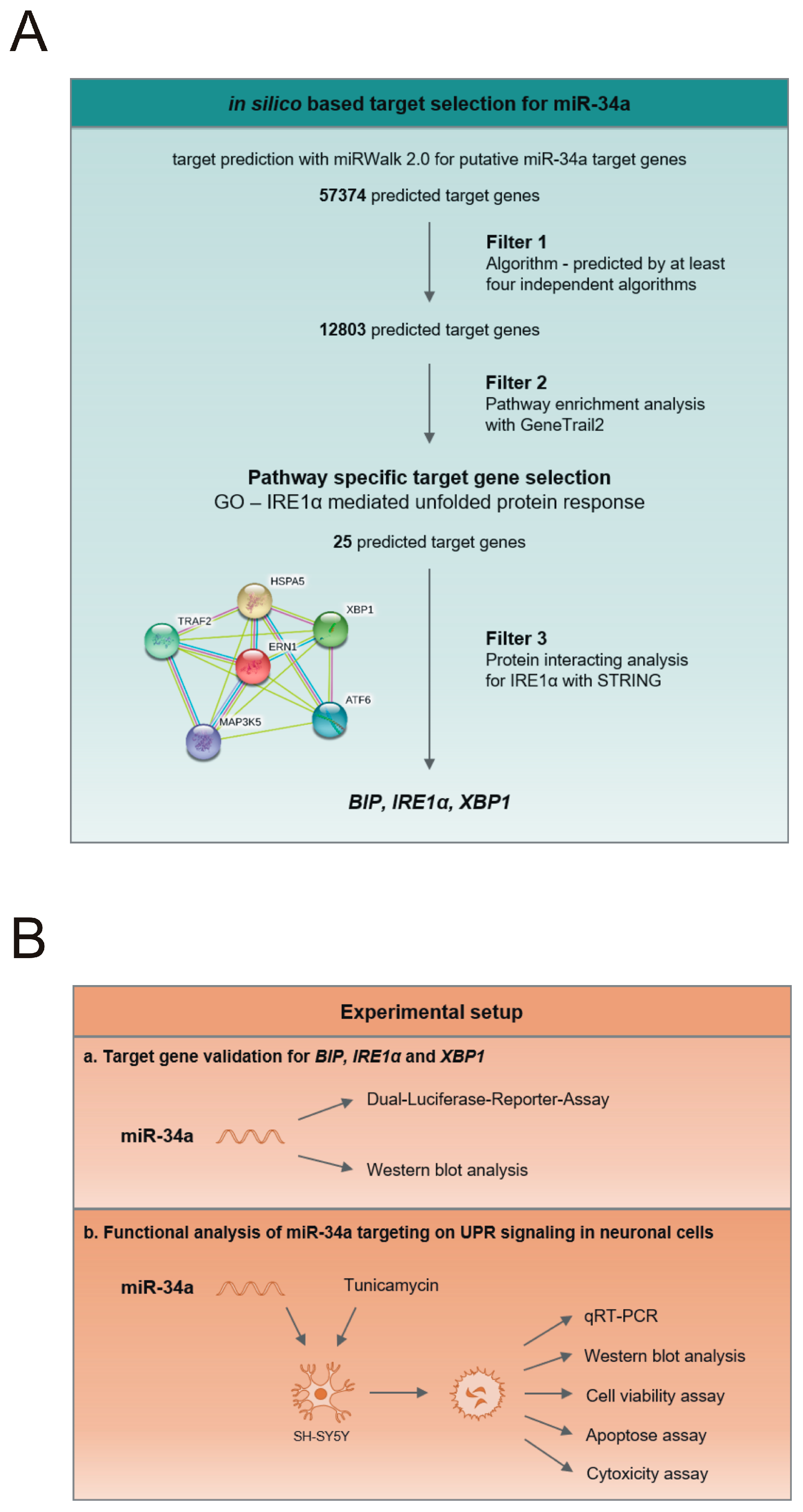
2.2. In-Silico-Based Target Selection for miR-34a
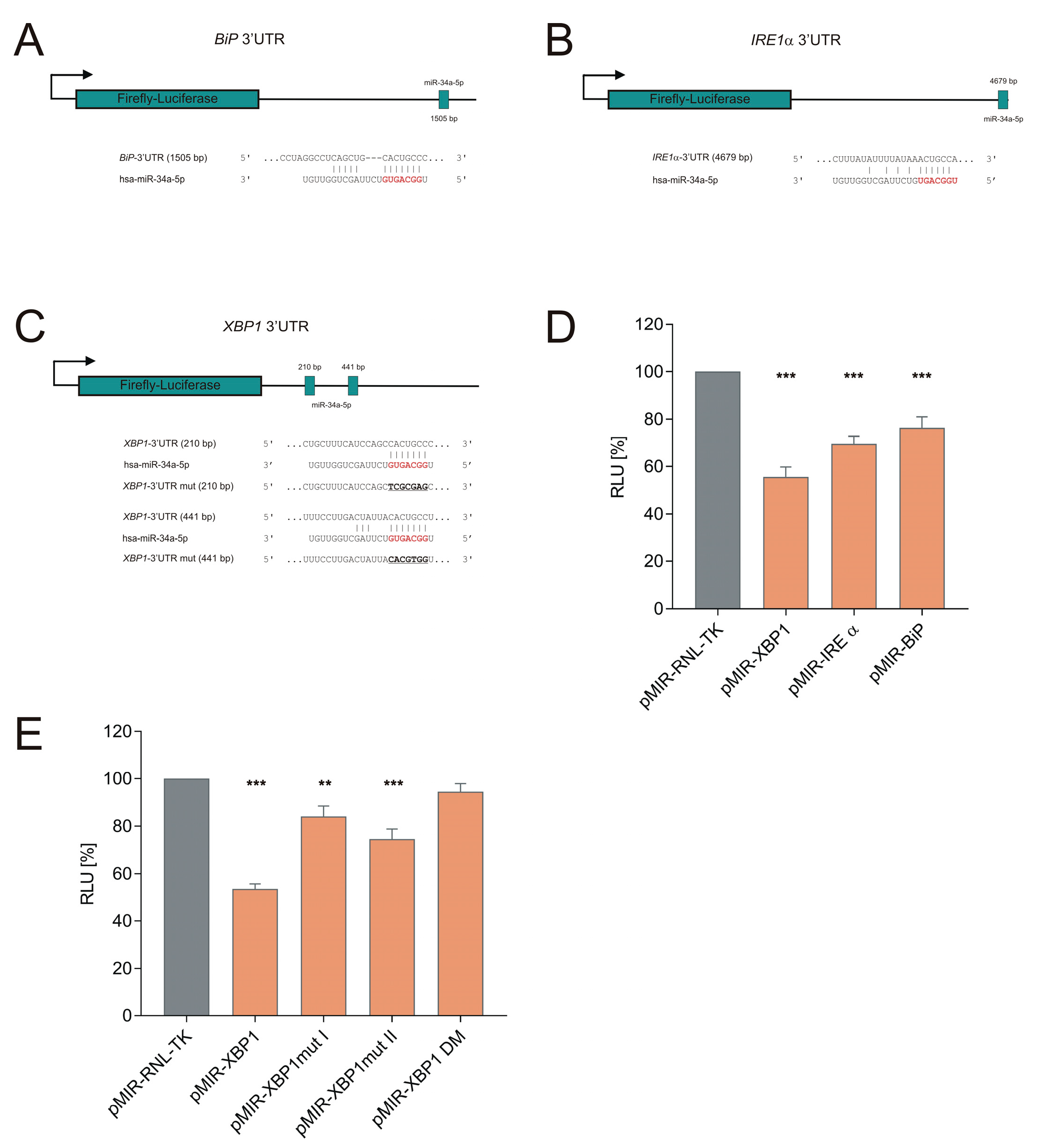
2.3. Generation of 3′UTR Reporter Constructs
2.4. Dual-Luciferase Reporter Assay
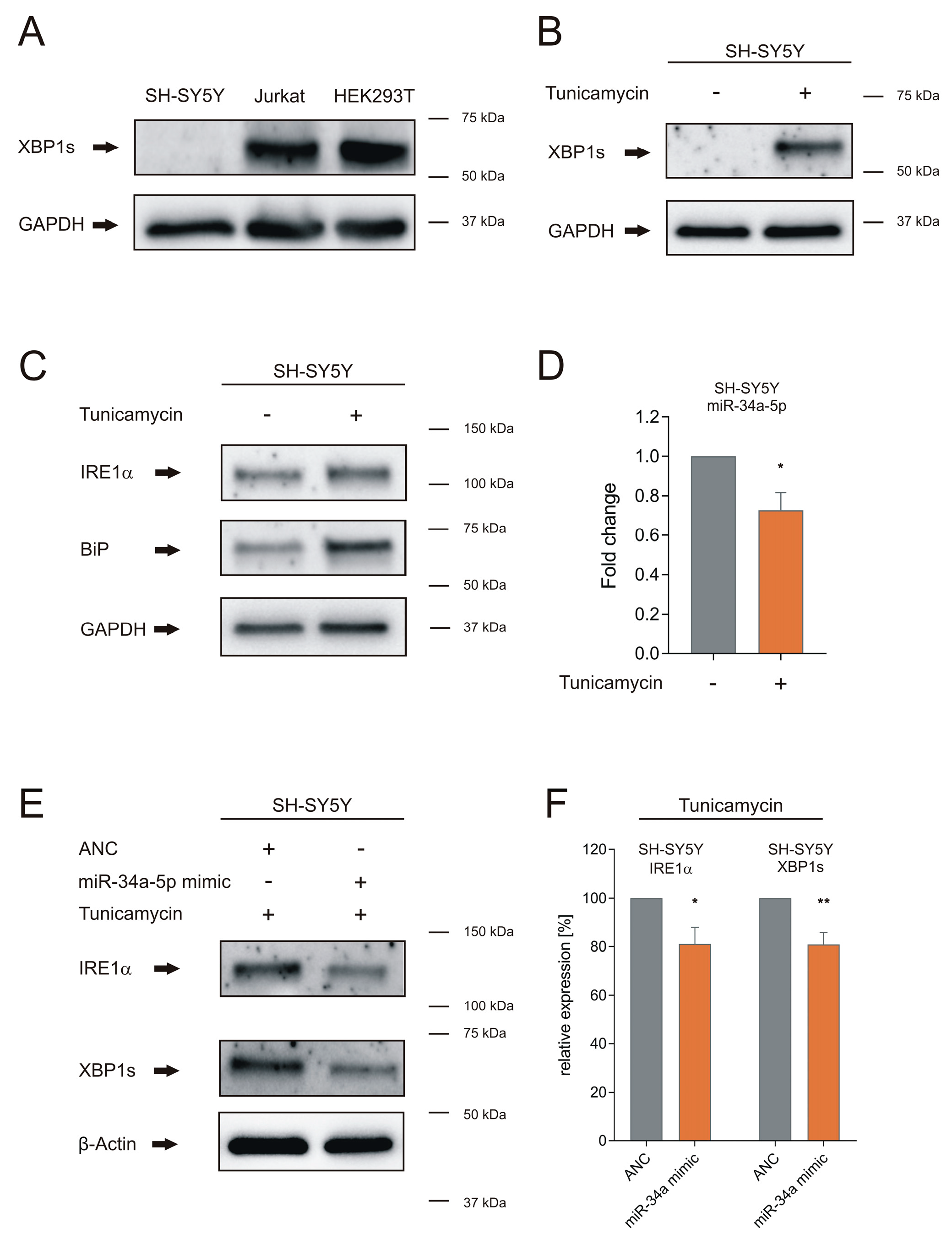
2.5. Tunicamycin Treatment
2.6. Western Blot
2.7. RNA-Isolation, Quantitative Real-Time PCR and Northern Blot
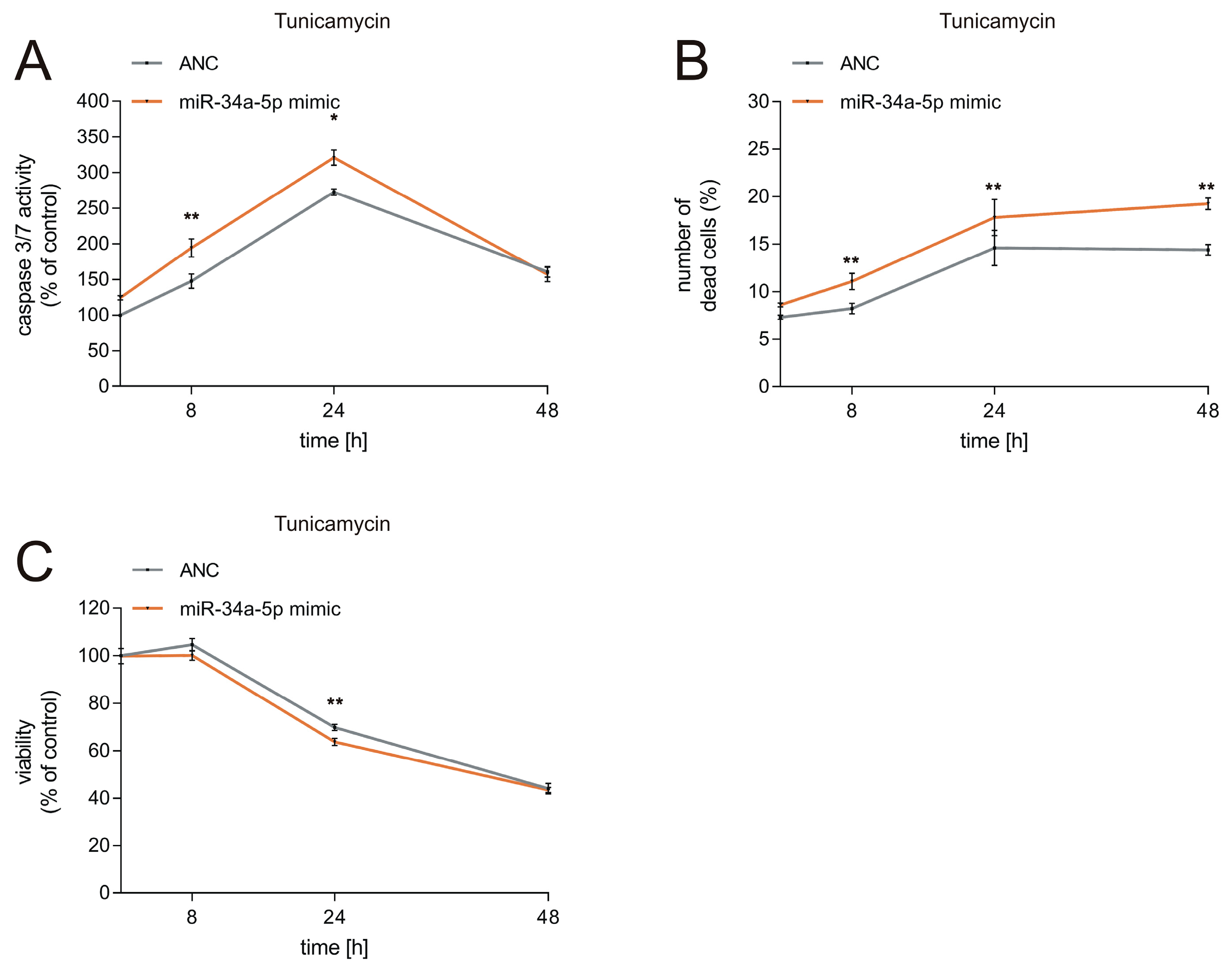
2.8. Cell Viability and Cytotoxicity Assays
2.9. Quantification and Statistical Analysis
3. Results
3.1. miR-34a-5p Directly Targets IRE1α Branch of the UPR
3.2. miR-34a-5p Overexpression Overwrites the Effect of ER Stress on IRE1α and XBP1s Proteins in Neuronal Cells
3.3. Effects of miR-34a-5p Overexpression on Viability of Neuronal Cells during ER Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Kalathur, R.K.; Giner-Lamia, J.; Machado, S.; Barata, T.; Ayasolla, K.R.; Futschik, M.E. The unfolded protein response and its potential role in Huntington’s disease elucidated by a systems biology approach. F1000Research 2015, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Hacki, J.; Egger, L.; Monney, L.; Conus, S.; Rosse, T.; Fellay, I.; Borner, C. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 2000, 19, 2286–2295. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders-therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef]
- Jiang, D.; Niwa, M.; Koong, A.C. Targeting the IRE1alpha-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Engels, B.M.; Hutvagner, G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 2006, 25, 6163–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, F.; Thermann, R.; Hentze, M.W. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA 2010, 16, 2493–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Maciotta, S.; Meregalli, M.; Torrente, Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell. Neurosci. 2013, 7, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backes, C.; Meese, E.; Keller, A. Specific miRNA disease biomarkers in blood, serum and plasma: Challenges and prospects. Mol. Diagn. Ther. 2016, 20, 509–518. [Google Scholar] [CrossRef]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stahler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14, R78. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Jun, S.; Rellick, S.; Quintana, D.D.; Cavendish, J.Z.; Simpkins, J.W. Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 2016, 1646, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Briggs, C.E.; Wang, Y.; Kong, B.; Woo, T.U.; Iyer, L.K.; Sonntag, K.C. Midbrain dopamine neurons in Parkinson’s disease exhibit a dysregulated miRNA and target-gene network. Brain Res. 2015, 1618, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.; Rheinheimer, S.; Leidinger, P.; Backes, C.; Menegatti, J.; Fehlmann, T.; Grasser, F.; Keller, A.; Meese, E. Identification of miR-34a-target interactions by a combined network based and experimental approach. Oncotarget 2016, 7, 34288–34299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Stockel, D.; Kehl, T.; Trampert, P.; Schneider, L.; Backes, C.; Ludwig, N.; Gerasch, A.; Kaufmann, M.; Gessler, M.; Graf, N.; et al. Multi-omics enrichment analysis using the GeneTrail2 web service. Bioinformatics 2016, 32, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Cabaj, A.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. miR-34c-5p modulates X-box-binding protein 1 (XBP1) expression during the adaptive phase of the unfolded protein response. FASEB J. 2019, 33, 11541–11554. [Google Scholar] [CrossRef]
- Byrd, A.E.; Aragon, I.V.; Brewer, J.W. MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J. Cell Biol. 2012, 196, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola-Segovia, V.; Cavarsan, C.; Maia, F.G.; Ferraz, A.C.; Nakao, L.S.; Lima, M.M.; Zanata, S.M. ER Stress Induced by Tunicamycin Triggers alpha-Synuclein Oligomerization, Dopaminergic Neurons Death and Locomotor Impairment: A New Model of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 5798–5806. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzym. 2011, 490, 71–92. [Google Scholar] [CrossRef] [Green Version]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi Masouleh, B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Valdes, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [Green Version]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krammes, L.; Hart, M.; Rheinheimer, S.; Diener, C.; Menegatti, J.; Grässer, F.; Keller, A.; Meese, E. Induction of the Endoplasmic-Reticulum-Stress Response: MicroRNA-34a Targeting of the IRE1α-Branch. Cells 2020, 9, 1442. https://doi.org/10.3390/cells9061442
Krammes L, Hart M, Rheinheimer S, Diener C, Menegatti J, Grässer F, Keller A, Meese E. Induction of the Endoplasmic-Reticulum-Stress Response: MicroRNA-34a Targeting of the IRE1α-Branch. Cells. 2020; 9(6):1442. https://doi.org/10.3390/cells9061442
Chicago/Turabian StyleKrammes, Lena, Martin Hart, Stefanie Rheinheimer, Caroline Diener, Jennifer Menegatti, Friedrich Grässer, Andreas Keller, and Eckart Meese. 2020. "Induction of the Endoplasmic-Reticulum-Stress Response: MicroRNA-34a Targeting of the IRE1α-Branch" Cells 9, no. 6: 1442. https://doi.org/10.3390/cells9061442