Dual CXCR4 and E-Selectin Inhibitor, GMI-1359, Shows Anti-Bone Metastatic Effects and Synergizes with Docetaxel in Prostate Cancer Cell Intraosseous Growth

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Drug Preparation
2.2. Cell Lines
2.3. Generation of Docetaxel Resistant Cells
2.4. Facs Analysis
2.5. Growth Assays
2.6. Western Blot
2.7. Dot Blot
2.8. Subcutaneous Xenograft Model
2.9. Evaluation of Treatment Response In Vivo
2.10. Intracardiac (IC) Tumour Model
2.11. Intratibial (IT) Tumour Model
2.12. Assessment of Treatment Response in Bone Tumour Models
2.13. Treatments for In Vivo Experiments
Immunohistochemical Analyses
2.14. Statistical Analysis
3. Results
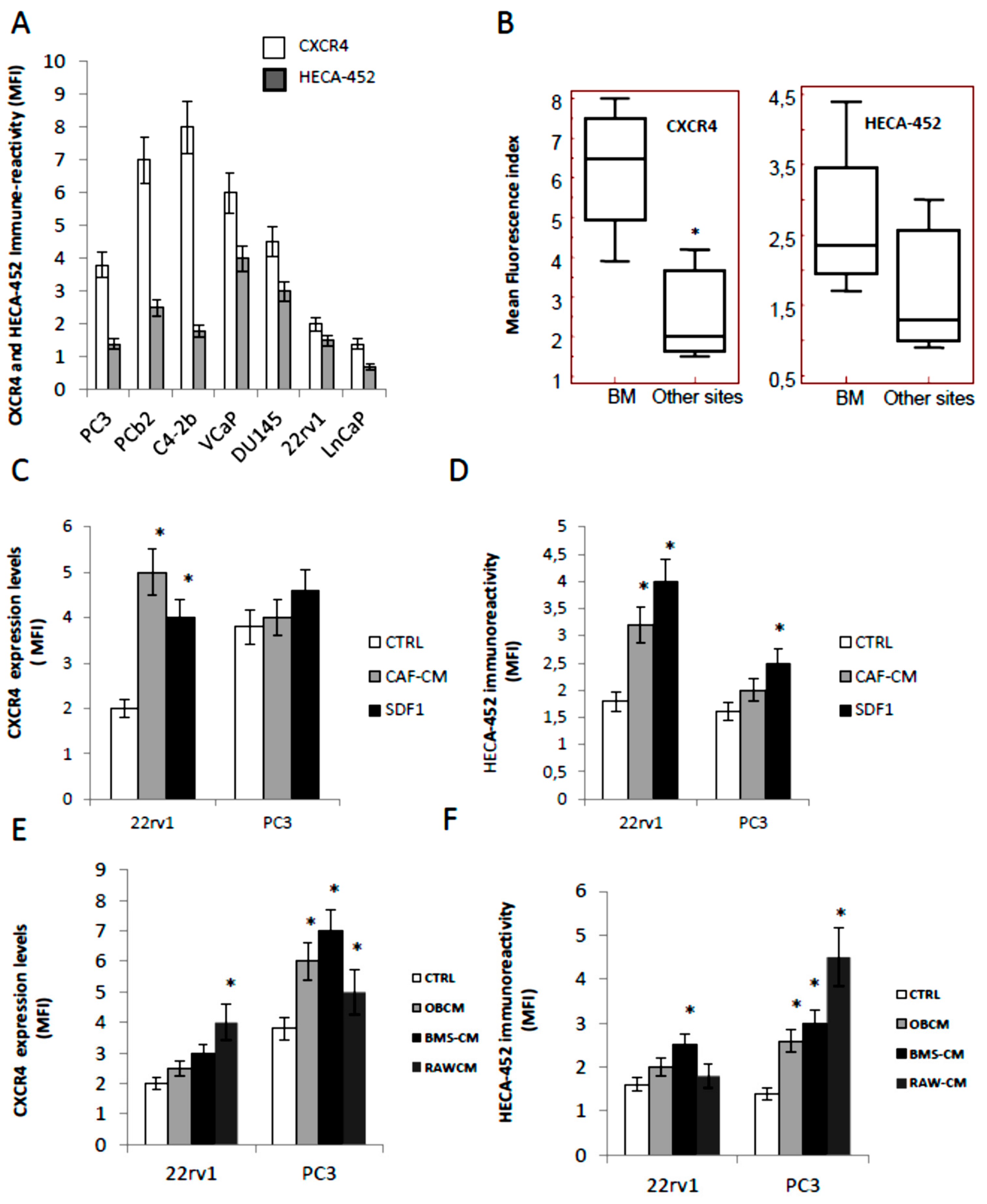
3.1. CXCR4 and HECA-452 Immune-Reactivity in Prostate Cancer Cells
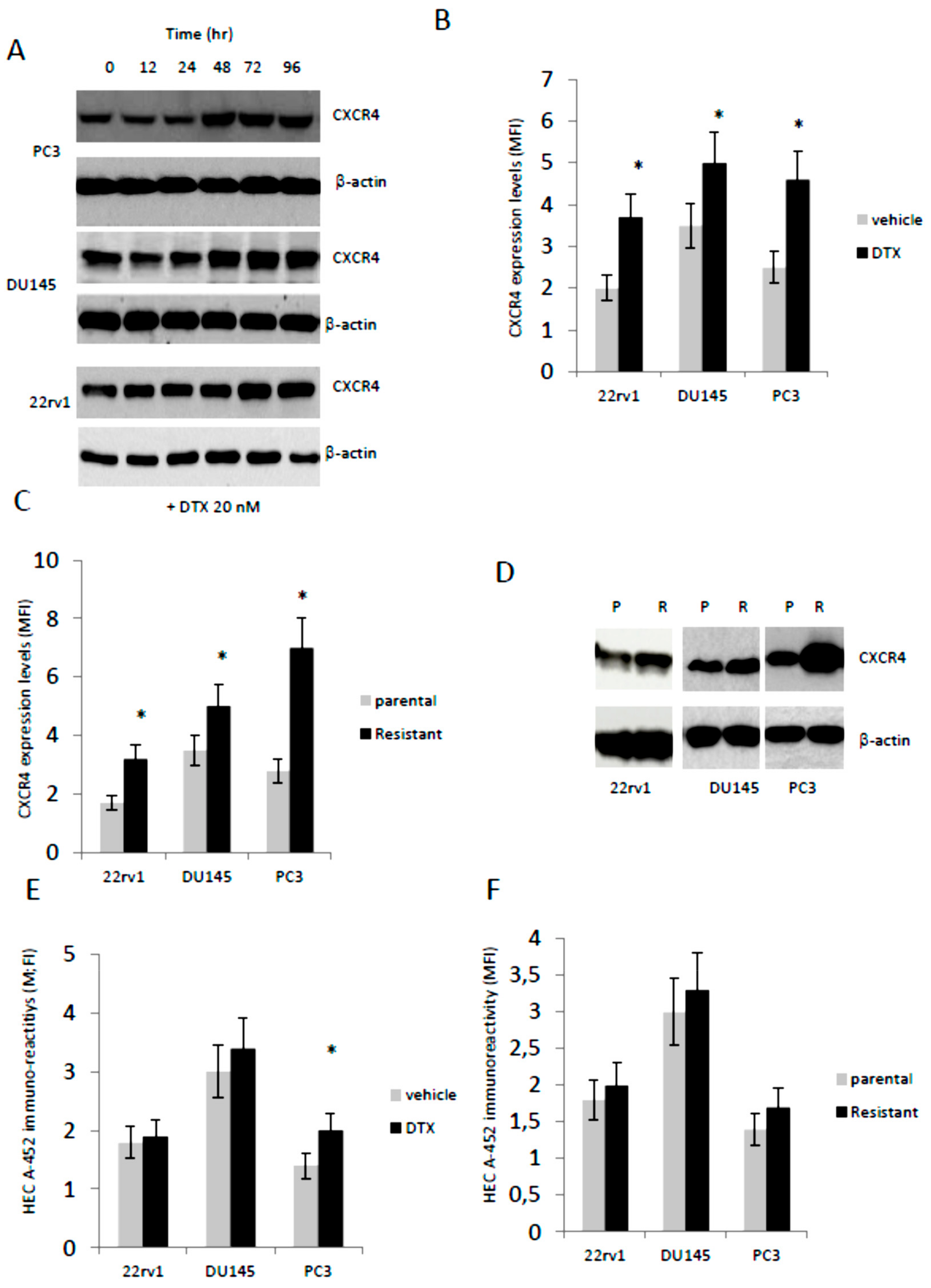
3.2. Docetaxel (DTX) Increases CXCR4 Expression in Docetaxel Sensitive and Resistant Cells In Vitro
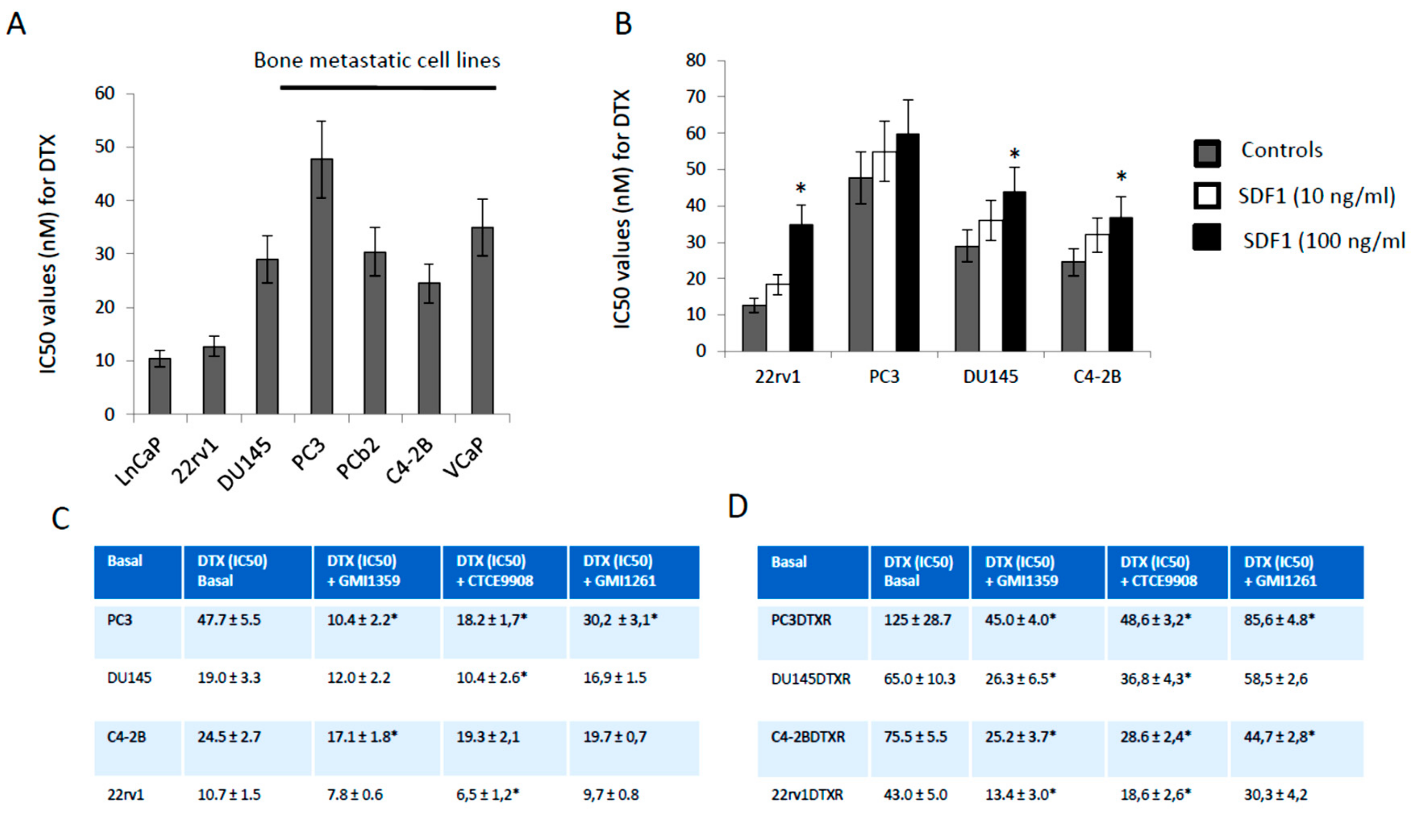
3.3. GMI-1359 Potentiates the Cytotoxicity of DTX in Drug Sensitive Cells and Sensitizes to DTX in Drug-Resistant Cells In Vitro: Comparison with CTCE-9908 and GMI1272
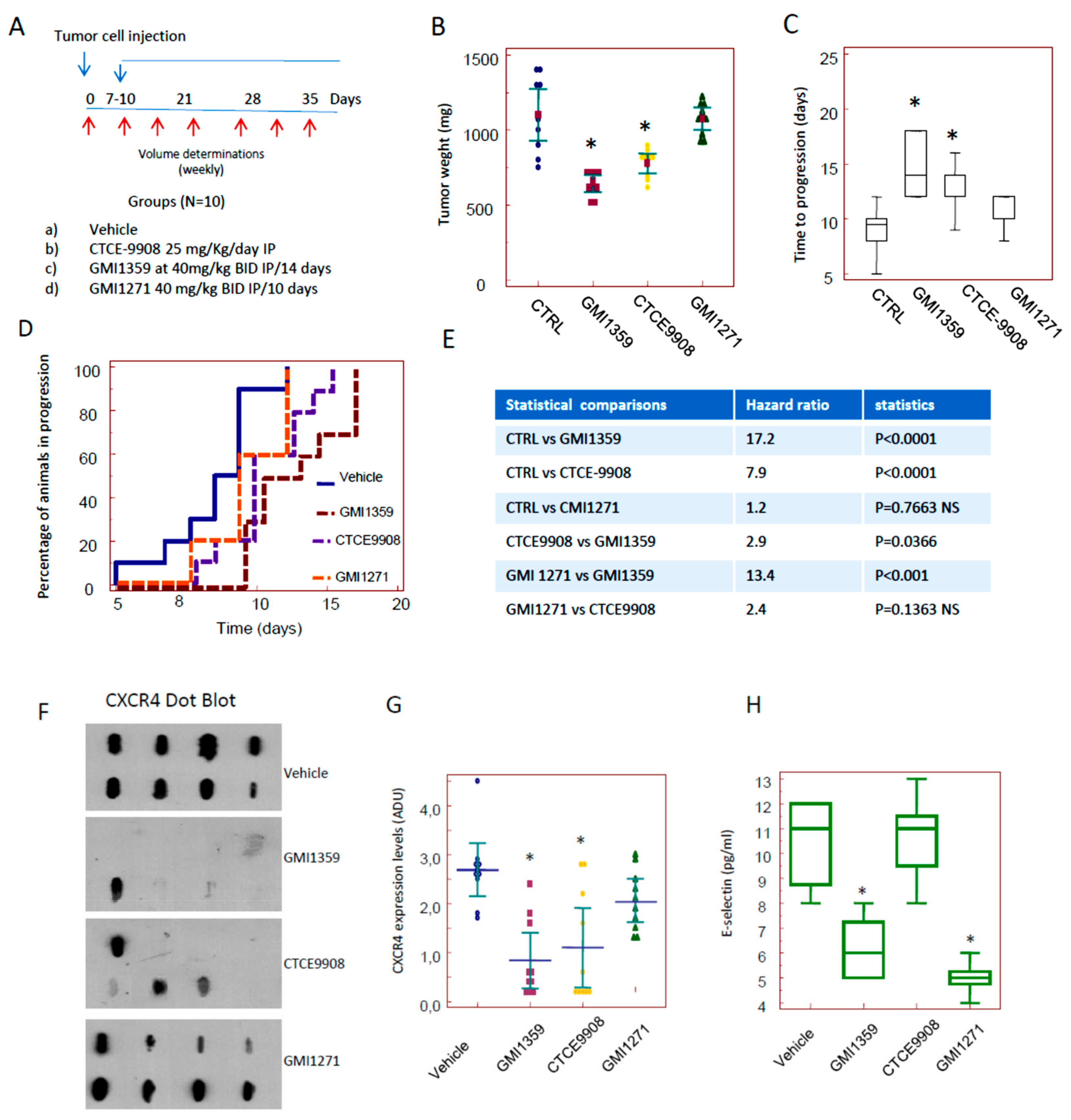
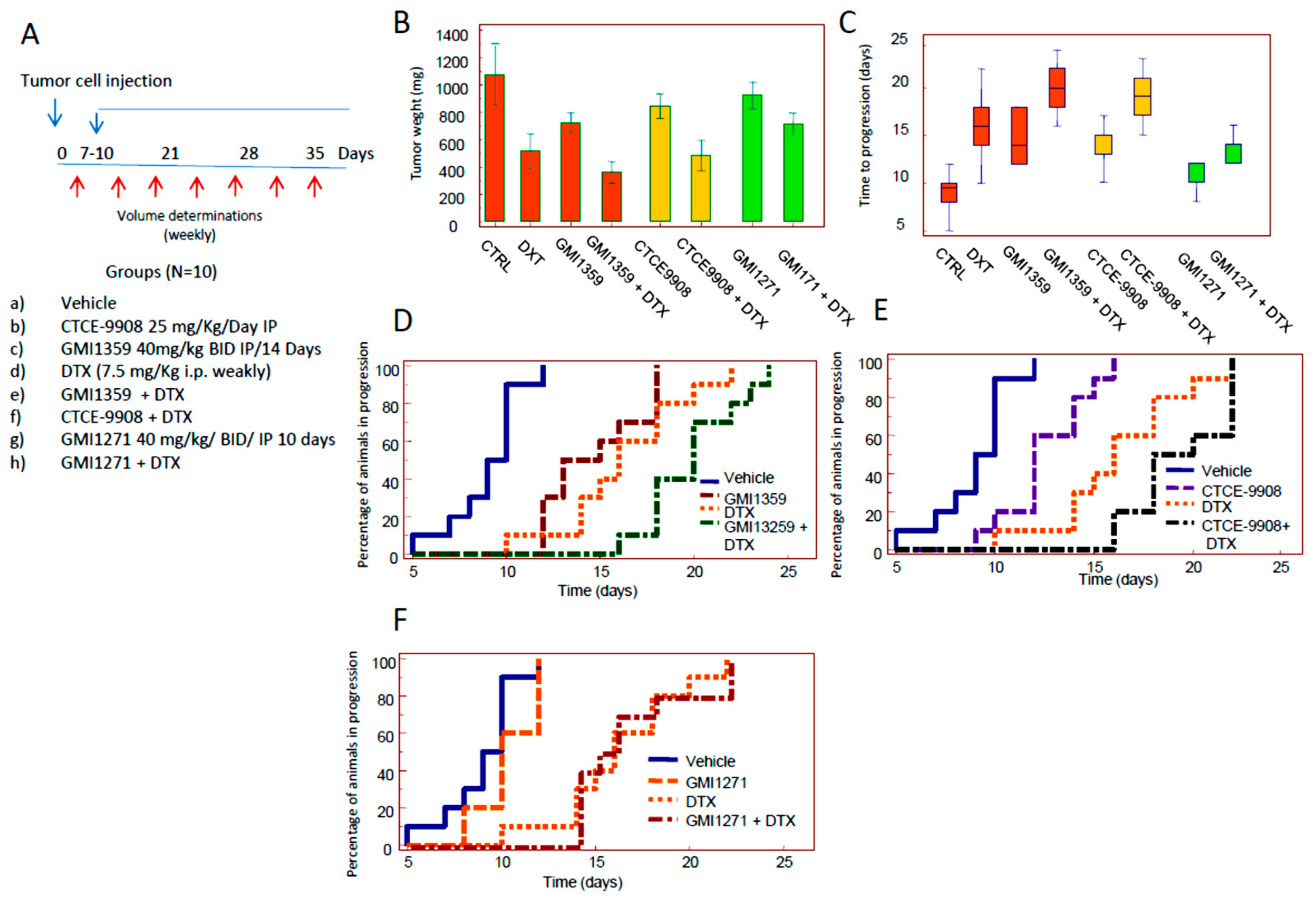
3.4. Compared effects of GMI-1359, CTCE-9908 and GMI-1271 Single Treatments in Locally Aggressive/Non Metastatic 22rv1 Xenografts
3.5. Compared Effects of GMI-1359, CTCE-9908 and GMI-1271 as DTX Chemo-Sensitizing Agents in 22rv1 Xenografts
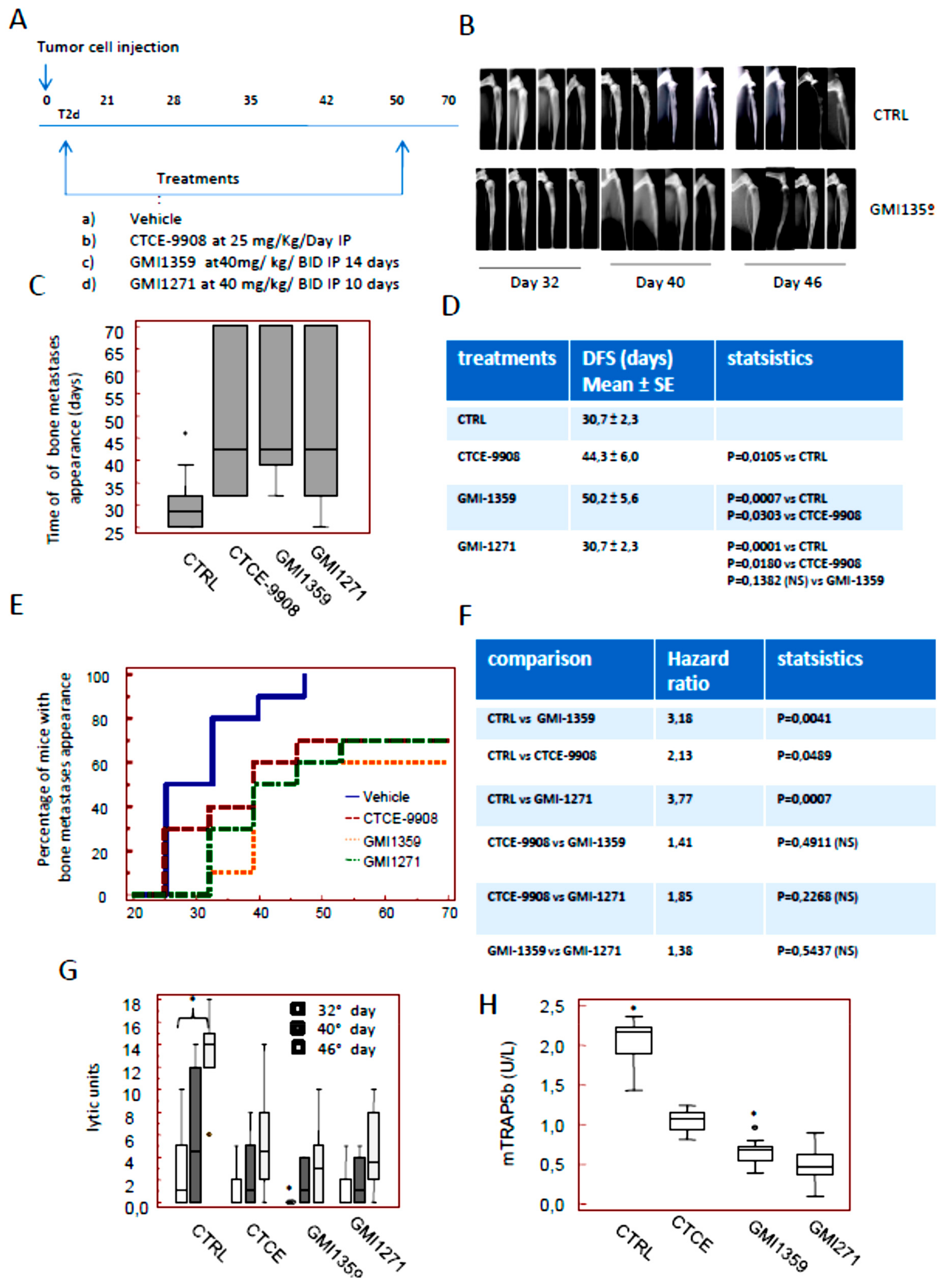
3.6. Intraventricular Tumour Cell Injection: Reduction of Bone Marrow Colonization (Anti-Bone Metastatic Activities) from GMI-1359, GMI-1271 and CTCE-9908
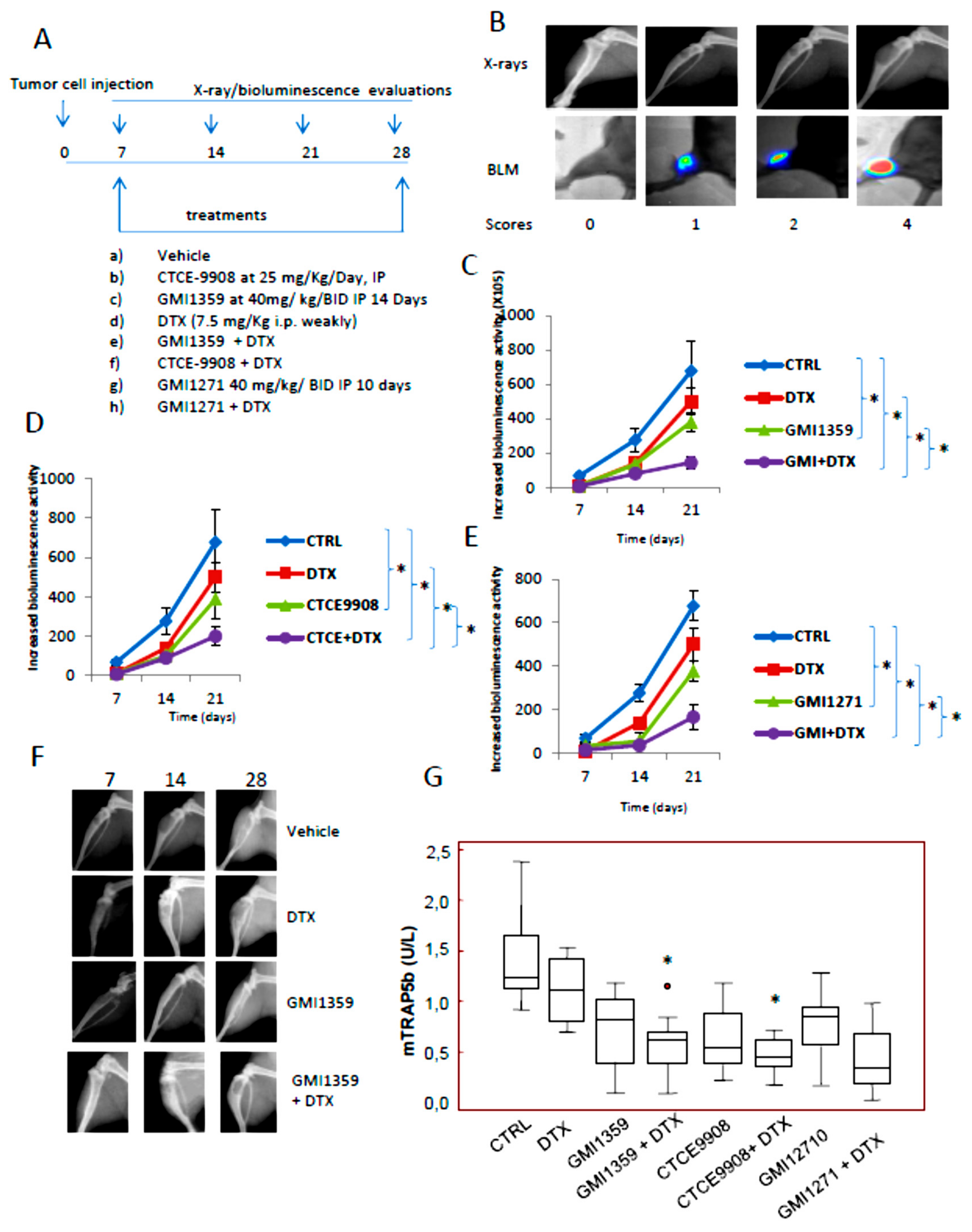
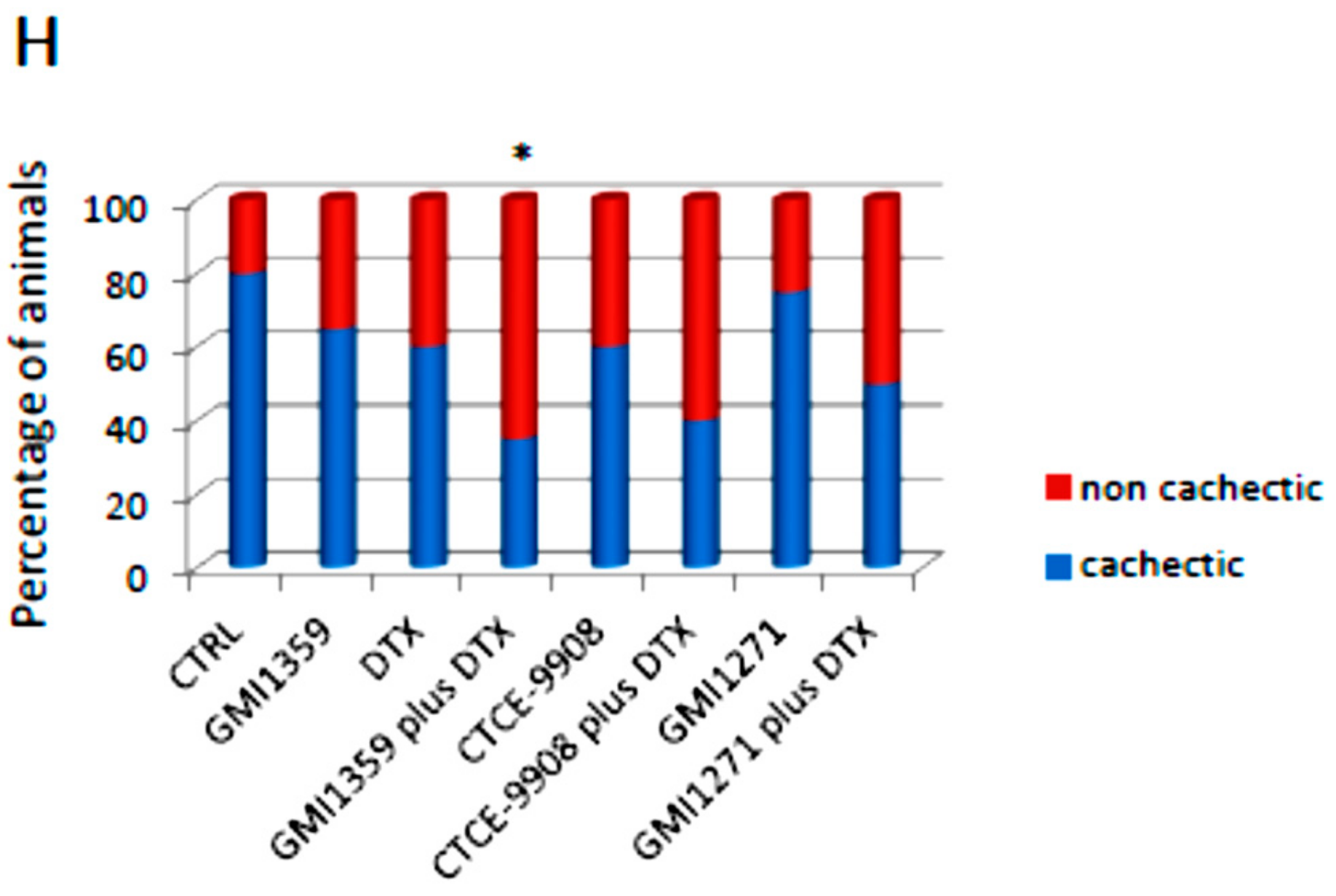
3.7. GMI-1359 Affects Intra-Osseous Tumour Growth and Increases the Efficacy of Docetaxel
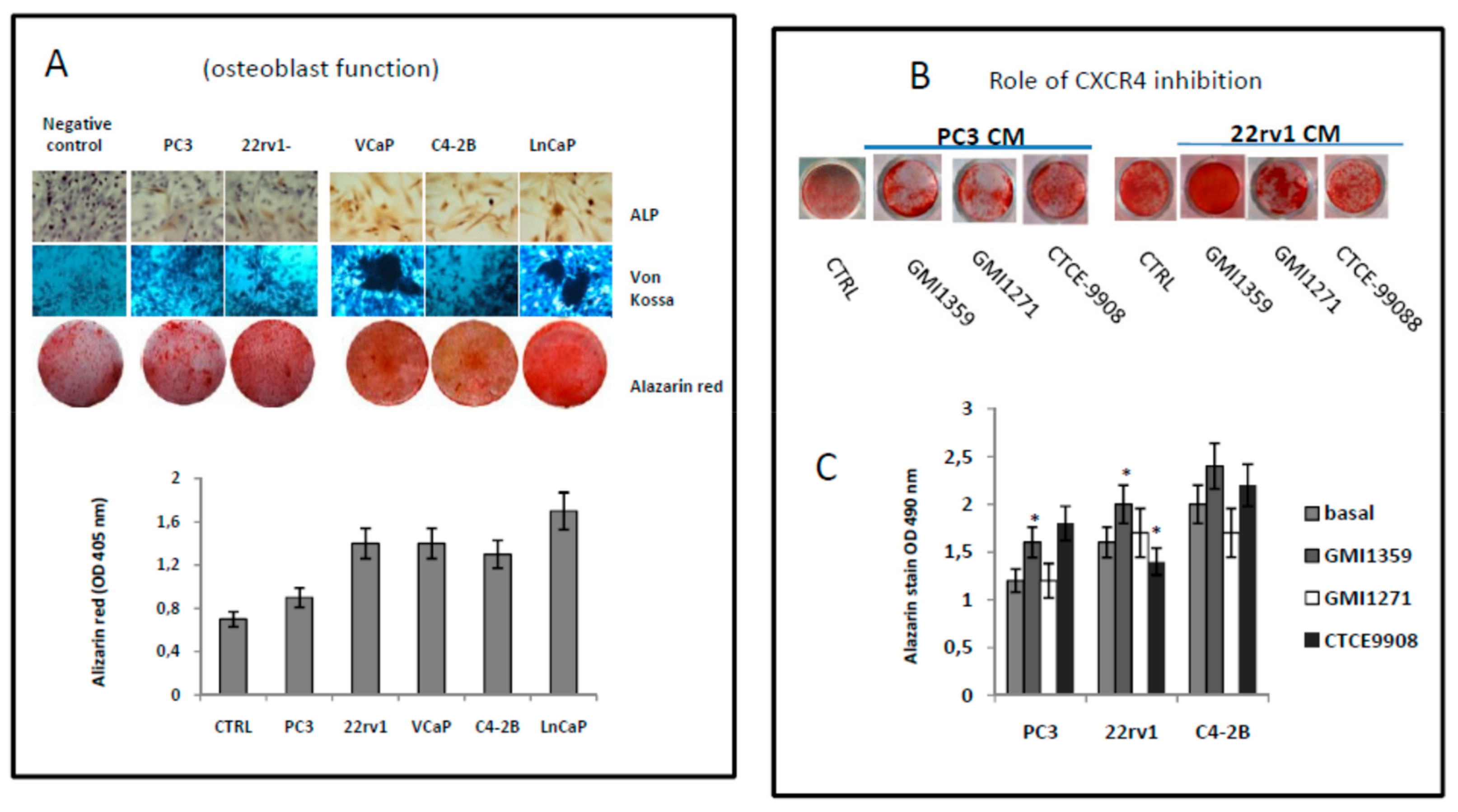
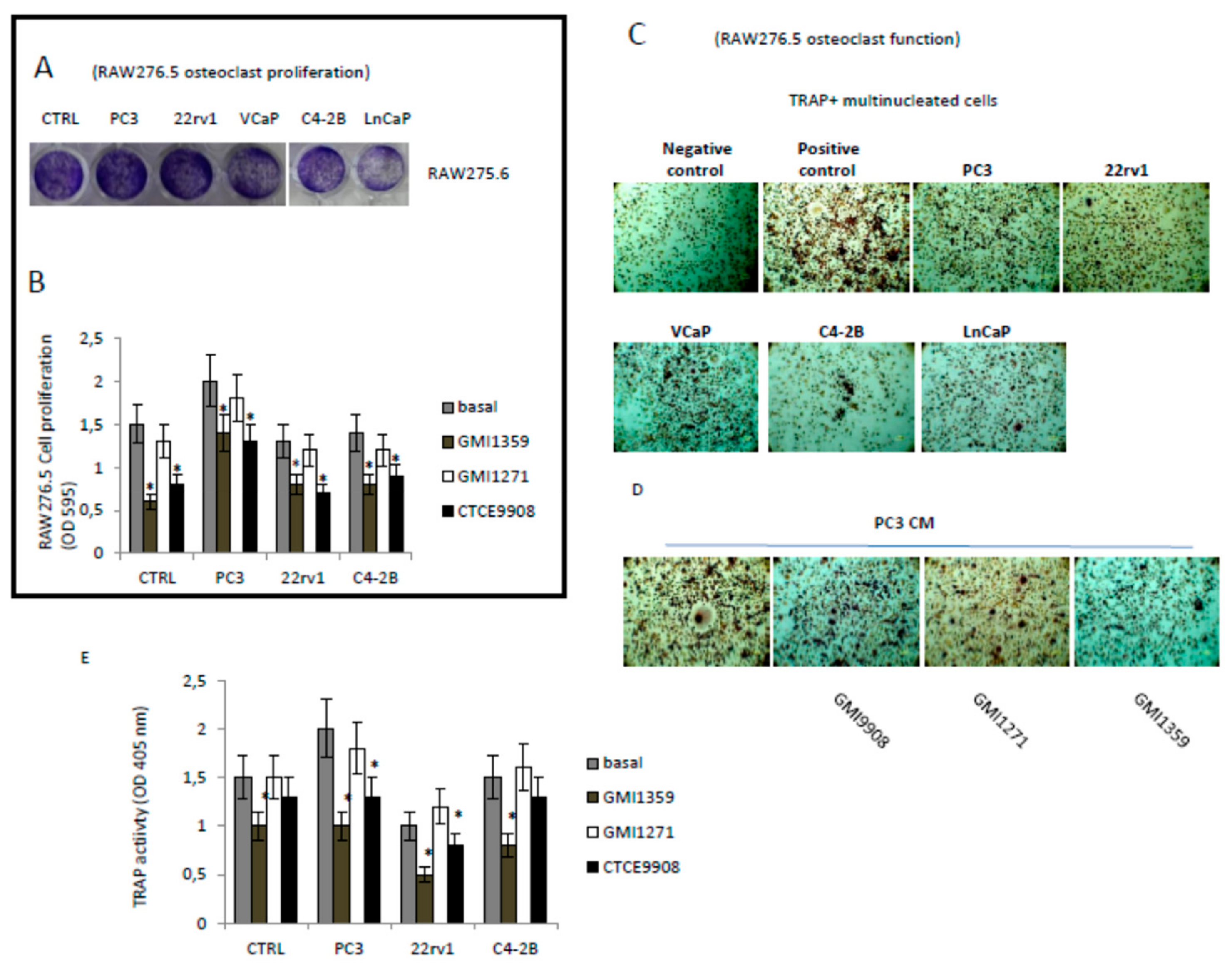
3.8. Effects of CXCR4 and E-Selectin Antagonism on Osteoblast and Osteoclast Growth and Function In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hashim, D.; Boffetta, P.; La Vecchia, C.; Rota, M.; Bertuccio, P.; Malvezzi, M.; Negri, E. The global decrease in cancer mortality: Trends and disparities. Ann. Oncol. 2016, 27, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, M.S.; Lowrance, W.T.; Murad, M.H.; Kibel, A.S.; American Urological Association. Castration-resistant prostate cancer: AUA guideline amendment. J. Urol. 2015, 193, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Joniau, S.; Mason, M.; Matveev, V.; Schmid, H.P.; Van der Kwast, T.; Wiegel, T.; Zattoni, F.; et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 2011, 59, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.M.; Shiri, S.; Farsinejad, S. Metastasis review: From bench to bedside. Tumor Biol. 2014, 35, 8483–8523. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Festuccia, C.; Teti, A.; Bianco, P.; Guerra, F.; Vicentini, C.; Tennina, R.; Villanova, I.; Sciortino, G.; Bologna, M. Human prostatic tumour cells in culture produce growth and differentiation factors active on osteoblasts: A new biological and clinical parameter for prostatic carcinoma. Oncol. Res. 1997, 9, 419–431. [Google Scholar]
- Coupland, L.A.; Parish, C.R. Platelets, selectins, and the control of tumour metastasis. Semin. Oncol. 2014, 41, 422–434. [Google Scholar] [CrossRef]
- Barthel, S.R.; Gavino, J.D.; Wiese, G.K.; Jaynes, J.M.; Siddiqui, J.; Dimitroff, C.J. Analysis of glycosyltransferase expression in metastatic prostate cancer cells capable of rolling activity on microvascular endothelial (E)-selectin. Glycobiology 2008, 18, 806–817. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Borentain, P.; Carmona, S.; Mathieu, S.; Jouve, E.; El-Battari, A.; Gérolami, R. Inhibition of E-selectin expression on the surface of endothelial cells inhibits hepatocellular carcinoma growth by preventing tumour angiogenesis. Cancer Chemother. Pharmacol. 2016, 77, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.; Meissner, T.; Moehler, T. A Furan-Based Lewis-Y-(CD174)-Saccharide Mimetic Inhibits Endothelial Functions and In Vitro Angiogenesis. Adv. Clin. Exp. Med. 2015, 24, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kang, D.H.; Chung, D.Y.; Kwon, J.K.; Lee, H.; Cho, N.H.; Choi, Y.D.; Hong, S.J.; Cho, K.S. Meta-Analysis of the Relationship between CXCR4 Expression and Metastasis in Prostate Cancer. World J. Men’s Health 2014, 32, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Işman, F.K.; Kucukgergin, C.; Daşdemir, S.; Cakmakoglu, B.; Sanli, O.; Seckin, S. Association between SDF1-3’A or CXCR4 gene polymorphisms with predisposition to and clinicopathological characteristics of prostate cancer with or without metastases. Mol. Biol. Rep. 2012, 39, 11073–11079. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Muzi, P.; Ventura, L.; Biordi, L.; Ricevuto, E.; Pompili, S.; Mattei, C.; Di Cesare, E.; Jannini, E.A.; et al. CXCR4 pharmacogical inhibition reduces bone and soft tissue metastatic burden by affecting tumour growth and tumourigenic potential in prostate cancer preclinical models. Prostate 2015, 75, 1227–1246. [Google Scholar] [CrossRef]
- Zhu, W.B.; Zhao, Z.F.; Zhou, X. AMD3100 inhibits epithelial-mesenchymal transition, cell invasion, and metastasis in the liver and the lung through blocking the SDF-1α/CXCR4 signaling pathway in prostate cancer. J. Cell Physiol. 2019, 234, 11746–11759. [Google Scholar] [CrossRef]
- Chen, Q.; Zhong, T. The association of CXCR4 expression with clinicopathological significance and potential drug target in prostate cancer: A meta-analysis and literature review. Drug Des. Devel. Ther. 2015, 9, 5115–5122. [Google Scholar] [CrossRef] [Green Version]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 68. [Google Scholar] [CrossRef]
- Wong, D.; Kandagatla, P.; Korz, W.; Chinni, S.R. Targeting CXCR4 with CTCE-9908 inhibits prostate tumour metastasis. BMC Urol. 2014, 14, 12. [Google Scholar] [CrossRef] [Green Version]
- Rettig, M.P.; Ramirez, P.; Nervi, B.; DiPersio, J.F. CXCR4 and mobilization of hematopoietic precursors. Methods Enzymol. 2009, 460, 57–90. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. Hematopoietic stem cell niche is a potential therapeutic target for bone metastatic tumours. Clin. Cancer Res. 2011, 17, 5553–5558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C.; Vincent, L.; Cook, K.R.; Lucci, A. A CXCR4 antagonist CTCE-9908 inhibits primary tumour growth and metastasis of breast cancer. J. Surg. Res. 2009, 155, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Engl, T.; Relja, B.; Marian, D.; Blumenberg, C.; Müller, I.; Beecken, W.D.; Jones, J.; Ringel, E.M.; Bereiter-Hahn, J.; Jonas, D.; et al. CXCR4 chemokine receptor mediates prostate tumour cell adhesion through α5 and β3 integrins. Neoplasia 2006, 8, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Li, Y.; Graziani, G.M.; Filion, L.; Allan, D.S. E-selectin mediated adhesion and migration of endothelial colony forming cells is enhanced by SDF-1α/CXCR4. PLoS ONE 2013, 8, e60890. [Google Scholar] [CrossRef] [Green Version]
- Katkoori, V.R.; Basson, M.D.; Bond, V.C.; Manne, U.; Bumpers, H.L. Nef-M1, a peptide antagonist of CXCR4, inhibits tumour angiogenesis and epithelial-to-mesenchymal transition in colon and breast cancers. Oncotarget 2015, 6, 27763–27777. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Wang, L.; Xu, H.; Xu, Z.; Feng, C.; Ding, Q.; Fang, Z.; Wu, Z.; Jiang, H.; Xu, J.; et al. Mesenchymal stem cells in prostate cancer have higher expressions of SDF-1, CXCR4 and VEGF. Gen. Physiol. Biophys. 2013, 32, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Jiang, Q.W.; Wu, J.Y.; Qiu, J.G.; Zhang, W.J.; Mei, X.L.; Shi, Z.; Di, J.M. Regulation of migration and invasion by Toll-like receptor-9 signaling network in prostate cancer. Oncotarget 2015, 6, 22564–22574. [Google Scholar] [CrossRef] [Green Version]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar]
- Wu, H.C.; Hsieh, J.T.; Gleave, M.E.; Brown, N.M.; Pathak, S.; Chung, L.W. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. [Google Scholar] [CrossRef]
- Korenchuk, S.; Lehr, J.E.; MClean, L.; Lee, Y.G.; Whitney, S.; Vessella, R.; Lin, D.L.; Pienta, K.J. VCaP, a cell-based model system of human prostate cancer. In Vivo 2001, 15, 163–168. [Google Scholar] [PubMed]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Sramkoski, R.M.; Pretlow, T.G., II; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell. Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Koeneman, K.S.; Yeung, F.; Chung, L.W. Osteomimetic properties of prostate cancer cells: A hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate 1999, 39, 246–261. [Google Scholar] [CrossRef]
- Lin, D.L.; Tarnowski, C.P.; Zhang, J.; Dai, J.; Rohn, E.; Patel, A.H.; Morris, M.D.; Keller, E.T. Bone metastatic LNCaP-derivative C4–2B prostate cancer cell line mineralizes in vitro (PDF). Prostate 2001, 47, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Hensel, J.; Wang, N.; Kruithof-de Julio, M.; Shiozawaa, Y. Mouse models for studying prostate cancer bone metastasis. Bonekey Rep. 2016, 5, 777. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, A.; Gravina, G.L.; Rucci, N.; Festuccia, C.; Muzi, P.; Vicentini, C.; Teti, A.; Bologna, M. Evaluation of metastatic potential in prostate carcinoma: An in vivo model. Int. J. Oncol. 2004, 25, 1713–1720. [Google Scholar] [CrossRef]
- Fujio, M.; Yamamoto, A.; Ando, Y.; Shohara, R.; Kinoshita, K.; Kaneko, T.; Hibi, H.; Ueda, M. Stromal cell-derived factor-1 enhances distraction osteogenesis-mediated skeletal tissue regeneration through the recruitment of endothelial precursors. Bone 2011, 49, 693–700. [Google Scholar] [CrossRef]
- Morikis, V.A.; Chase, S.; Wun, T.; Chaikof, E.L.; Magnani, J.L.; Simon, S.I. Selectin catch-bonds mechanotransduce integrin activation and neutrophil arrest on inflamed endothelium under shear flow. Blood 2017, 130, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, Z.; Pan, X.; Xia, L.; Chen, P.; Yang, Y.; Hu, H.; Zhang, J.; Li, K.; Ge, J.; et al. Inhibition of angiogenesis, fibrosis and thrombosis by tetramethylpyrazine: Mechanisms contributing to the SDF-1/CXCR4 axis. PLoS ONE 2014, 9, e88176. [Google Scholar] [CrossRef] [PubMed]
- Geary, L.A.; Nash, K.A.; Adisetiyo, H.; Liang, M.; Liao, C.P.; Jeong, J.H.; Zandi, E.; Roy-Burman, P. CAF-secreted annexin A1 induces prostate cancer cells to gain stem cell-like features. Mol. Cancer Res. 2014, 12, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festuccia, C.; Bologna, M.; Gravina, G.L.; Guerra, F.; Angelucci, A.; Villanova, I.; Millimaggi, D.; Teti, A. Osteoblast conditioned media contain TGF-beta1 and modulate the migration of prostate tumor cells and their interactions with extracellular matrix components. Int. J. Cancer 1999, 81, 395–403. [Google Scholar] [CrossRef]
- Mancini, A.; Colapietro, A.; Pompili, S.; Del Fattore, A.; Delle Monache, S.; Biordi, L.A.; Angelucci, A.; Mattei, V.; Liang, C.; Gravina, G.L.; et al. Dual PI3 K/mTOR inhibition reduces prostate cancer bone engraftment altering tumor-induced bone remodeling. Tumor Biol. 2018, 40, 1010428318771773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanika, S.; Karantanos, T.; Kurosaka, S.; Wang, J.; Hirayama, T.; Yang, G.; Park, S.; Golstov, A.A.; Tanimoto, R.; Li, L.; et al. GLIPR1-ΔTM synergizes with docetaxel in cell death and suppresses resistance to docetaxel in prostate cancer cells. Mol. Cancer 2015, 14, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Marampon, F.; Sferra, R.; Pompili, S.; Biordi, L.A.; Iorio, R.; Flati, V.; Argueta, C.; et al. Pharmacological treatment with inhibitors of nuclear export enhances the antitumor activity of docetaxel in human prostate cancer. Oncotarget 2017, 8, 111225–111245. [Google Scholar] [CrossRef] [Green Version]
- Spinello, I.; Quaranta, M.T.; Riccioni, R.; Riti, V.; Pasquini, L.; Boe, A.; Pelosi, E.; Vitale, A.; Foà, R.; Testa, U.; et al. MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J. 2011, 1, e26. [Google Scholar] [CrossRef]
- Available online: https://www.sciencegateway.org/protocols/cellbio/drug/hcic50.htm (accessed on 7 November 2011).
- Gravina, G.L.; Tortoreto, M.; Mancini, A.; Addis, A.; Di Cesare, E.; Lenzi, A.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. XPO1/CRM1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J. Hematol. Oncol. 2014, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Kalilani, L.; Atashili, J. Measuring additive interaction using odds ratios. Epidemiol. Perspect. Innov. 2006, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Kemper, E.M.; Leenders, W.; Kusters, B.; Lyons, S.; Buckle, T.; Heerschap, A.; Boogerd, W.; Beijnen, J.H.; van Tellingen, O. Development of luciferase tagged brain tumour models in mice for chemotherapy intervention studies. Eur. J. Cancer 2006, 42, 3294–3303. [Google Scholar] [CrossRef] [PubMed]
- Cosette, J.; Ben Abdelwahed, R.; Donnou-Triffault, S.; Sautès-Fridman, C.; Flaud, P.; Fisson, S. Bioluminescence-Based Tumor Quantification Method for Monitoring Tumor Progression and Treatment Effects in Mouse Lymphoma Models. J. Vis. Exp. 2016, 113, e53609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Monache, S.D.; Angelucci, A.; Calgani, A.; Fogler, W.E.; Magnani, J.L.; Festuccia, C. Dual E-selectin and CXCR4 inhibition reduces tumor growth and increases the sensitivity to docetaxel in experimental bone metastases of prostate cancer. In Proceedings of the 105th AACR Meeting, Philadelphia, PA, USA, 5–22 April 2015. [Google Scholar] [CrossRef]
- Richert, M.M.; Vaidya, K.S.; Mills, C.N.; Wong, D.; Korz, W.; Hurst, D.R.; Welch, D.R. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol. Rep. 2009, 21, 761–767. [Google Scholar] [PubMed]
- Natoni, A.; Smith, T.A.G.; Keane, N.; McEllistrim, C.; Connolly, C.; Jha, A.; Andrulis, M.; Ellert, E.; Raab, M.S.; Glavey, S.V.; et al. E-selectin ligands recognised by HECA452 induce drug resistance in myeloma, which is overcome by the E-selectin antagonist, GMI-1271. Leukemia 2017, 31, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, S.; Ansa-Addo, E.A.; Kholia, S.; Stratton, D.; Valley, S.; Lange, S.; Inal, J. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci. Rep. 2015, 5, 13006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festuccia, C.; Gravina, G.L.; D’Alessandro, A.M.; Muzi, P.; Millimaggi, D.; Dolo, V.; Ricevuto, E.; Vicentini, C.; Bologna, M. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr. Relat. Cancer 2009, 16, 401–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Wei, J.; Qian, X.; Chen, G.; Yu, L.; Ding, Y.; Liu, B. CXCR4, a potential predictive marker for docetaxel sensitivity in gastric cancer. Anticancer Res. 2010, 30, 2209–2216. [Google Scholar]
- Liu, Y.; Ren, C.C.; Yang, L.; Xu, Y.M.; Chen, Y.N. Role of CXCL12-CXCR4 axis in ovarian cancer metastasis and CXCL12-CXCR4 blockade with AMD3100 suppresses tumor cell migration and invasion in vitro. J. Cell. Physiol. 2019, 234, 3897–3909. [Google Scholar] [CrossRef]
- Hirsch, H.R.; Engelberg, J. Determination of the cell doubling-time distribution from culture growth-rate data. J. Theor. Biol. 1965, 9, 297–302. [Google Scholar] [CrossRef]
- Yang, M.; Burton, D.W.; Geller, J.; Hillegonds, D.J.; Hastings, R.H.; Deftos, L.J.; Hoffman, R.M. The bisphosphonate olpadronate inhibits skeletal prostate cancer progression in a green fluorescent protein nude mouse model. Clin. Cancer Res. 2006, 12, 2602–2606. [Google Scholar] [CrossRef] [Green Version]
- Allocca, G.; Hughes, R.; Wang, N.; Brown, H.K.; Ottewell, P.D.; Brown, N.J.; Holen, I. The bone metastasis niche in breast cancer-potential overlap with the haematopoietic stem cell niche in vivo. J. Bone Oncol. 2019, 17, 100244. [Google Scholar] [CrossRef] [PubMed]
- Meeson, R.; Sanghani-Keri, A.; Coathup, M.; Blunn, G. VEGF with AMD3100 endogenously mobilizes mesenchymal stem cells and improves fracture healing. J. Orthop. Res. 2019, 37, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Oude Munnink, T.H.; Kruizinga, R.C.; Ananias, H.J.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.; de Jong, I.J.; et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Kitagawa, Y.; Zhang, J.; Yao, Z.; Mizokami, A.; Cheng, S.; Nör, J.; McCauley, L.K.; Taichman, R.S.; Keller, E.T. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004, 64, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Cackowski, F.C.; Yumoto, K.; Decker, A.M.; Wang, J.; Kim, J.K.; Lee, E.; Wang, Y.; Chung, J.S.; Gursky, A.M.; et al. CXCL12γ Promotes Metastatic Castration-Resistant Prostate Cancer by Inducing Cancer Stem Cell and Neuroendocrine Phenotypes. Cancer Res. 2018, 78, 2026–2039. [Google Scholar] [CrossRef] [Green Version]
- Hatano, K.; Yamaguchi, S.; Nimura, K.; Murakami, K.; Nagahara, A.; Fujita, K.; Uemura, M.; Nakai, Y.; Tsuchiya, M.; Nakayama, M.; et al. Residual prostate cancer cells after docetaxel therapy increase the tumourigenic potential via constitutive signaling of CXCR4, ERK1/2 and c-Myc. Mol. Cancer Res. 2013, 11, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Srivastava, S.K.; Singh, S.; Arora, S.; Tyagi, N.; Andrews, J.; McClellan, S.; Carter, J.E.; Singh, A.P. CXCL12/CXCR4 signaling counteracts docetaxel-induced microtubule stabilization via p21-activated kinase 4-dependent activation of LIM domain kinase 1. Oncotarget 2014, 5, 11490–11500. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.D.; Silva, M.D.; Wen, S.; Siebert, E.; Solin, E.; Chandra, S.; Worland, P.J. Spiculated periosteal response induced by intraosseous injection of 22Rv1 prostate cancer cells resembles subset of bone metastases in prostate cancer patients. Prostate 2005, 65, 347–354. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Liesveld, J.L.; Jonas, B.A.; O’Dwyer, M.E.; Bixby, D.L.; Magnani, J.L.; Thackray, H.M.; Becker, P.S. A Phase I/II Study of GMI-1271, a Novel E-Selectin Antagonist, in Combination with Induction Chemotherapy in Relapsed/Refractory and Elderly Previously Untreated Acute Myeloid Leukemia; Results to Date. Blood 2016, 128, 4049. [Google Scholar] [CrossRef]
- Muz, B.; Azab, F.; Fiala, M.; King, J.; Kohnen, D.; Fogler, W.E.; Smith, T.; Magnani, J.L.; Vij, R.; Azab, A.K. Inhibition of E-Selectin (GMI-1271) or E-selectin together with CXCR4 (GMI-1359) re-sensitizes multiple myeloma to therapy. Blood Cancer J. 2019, 9, 68. [Google Scholar] [CrossRef]
- Zhang, W.; Mu, H.; Zhang, Q.; Patel, N.B.; Fogler, W.E.; Magnani, J.L.; Andreeff, M. The E-selectin/CXCR4 Inhibitor GMI-1359 Effectively Mobilizes Bone Marrow Leukemia Cells and Enhances FLT3 Inhibitor Efficacy in a Murine AML Model. 107th AACR Annual Meeting New Orleans. Blood 2016, 128, 3519. [Google Scholar] [CrossRef]
- Li, J.; Guillebon, A.D.; Hsu, J.W.; Barthel, S.R.; Dimitroff, C.J.; Lee, Y.F.; King, M.R. Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br. J. Cancer 2013, 109, 3014–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, S.; Ivetic, A.; Grigoriadis, A.; QiZe, D.; Burford, B.; Sproviero, D.; Picco, G.; Gillett, C.; Papp, S.L.; Schaffer, L.; et al. Selectin ligand sialyl-Lewis x antigen drives metastasis of hormone-dependent breast cancers. Cancer Res. 2011, 71, 7683–7693. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | 7° day | 14° day | 21° day | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive Tibiae | Lytic Scores | Bio Signal Mean ± SE | Lytic Units Mean ± SE | Positive Tibiae | Lytic Scores | Bio Signal Mean ± SE | Lytic Units Mean ± SE | Positive Tibiae | Lytic Scores | Bio Signal Mean ± SE | Lytic Units Mean ± SE | |
| Vehicle | 13/16 81.25% | 0 (3/16); 1 (8/16); 2 (5/16) | 40.2 × 104 ± 7.3 | 8.5 ± 3.7 | 13/16 81.25% | 0 (3/16); 1 (5/16); 2 (5/16); 3 (3/16) | 101.0 × 104 ± 16.3 | 16.8 ± 2.2 | 15/16 93.75% | 0 (1/16); 1 (2/16); 2 (3/16); 3 (4/16); 4 (6/16) | 850.3 × 104 ± 85.0 | 32.3 ± 5.8 |
| Docetaxel | 6/16 37.5% | 0(10/16); 1 (2/16); 2 (4/16) | 31.9 × 104 ± 11.8 | 3.2 ± 1.4 | 7/16 43.75% | 0 (9/16); 1 (2/16); 2 (5/16) | 45.3 × 104 ± 17.7 | 3.9 ± 1.1 | 10/16 62.5% | 0 (6/16); 1 (3/16); 2 (3/16); 3 (1/16); 4 (3/16) | 348.8 × 104 ± 76.5 | 19.4 ± 3.7 |
| GMI1359 | 4/16 25.0 % | 0(12/16); 1 (2/16); 2 (2/16) | 25.5 × 104 ± 12.4 | 2.4 ± 1.0 | 8/16 50.0% | 0 (8/16); 1 (3/16); 2 (2/16); 3 (3/16); | 98.1 × 104 ± 31.6 | 3.4 ± 1.8 | 8/16 50.0 % | 0 (8/16); 1 (1/16); 2 (3/16); 3 (3/16); 4 (1/16) | 218.5 × 104 ± 49.3 | 12.9 ± 4.0 |
| GMI1359 + DTX | 2/16 12.5 % | 0 (14/16); 1 (2/16); | 8.75 × 104 ± 6.05 | 1.7 ± 0.5 | 4/16 25.0 % | 0 (12/16); 1 (2/16); 2 (2/16) | 37.5 × 104 ± 19.05 | 2.0 ± 0.9 | 4/16 25.0 % | 0 (10/16); 1 (2/16); 3 (2/16) | 67.1 × 103 ± 30.6 | 6.3 ± 1.7 |
| GMI1271 | 8/16 50.0 % | 0 (8/16); 1 (3/16); 2 (4/16); 3 (1/16) | 23.4 × 104 ± 7.3 | 6.7 ± 2.9 | 10/16 62.5 % | 0 (6/16); 1 (2/16); 2 (3/16) 3 (2/16); 4 (3/16) | 122.9 × 104 ± 78.3 | 11.8 ± 3.5 | 10/16 62.5 % | 0 (6/16); 1 (1/16); 2 (1/16); 3 (4/16); 4 (4/16) | 385.0 × 104 ± 96.6 | 22.4 ± 5.0 |
| GMI1271 + DTX | 4/16 25.0 % | 0 (12/16); 1 (4/16) | 8.8 × 104 ± 8.2 | 1.7 ± 0.3 | 7/16 43.75% | 0 (9/16); 1 (4/16); 2 (3/16) | 55.8 × 104 ± 22.7 | 2.5 ± 1.4 | 8/16 50.0 % | 0 (8/16); 1 (2/16); 2 (2/16); 3 (3/16); 4 (1/16) | 116.2 × 105 ± 45.6 | 14.6 ± 2.7 |
| CTCE9908 | 4/16 25.0 % | 0 (12/16); 1 (2/16); 2 (2/16) | 33.7 × 104 ± 18.0 | 2.7 ± 0.8 | 6/16 37.5 % | 0 (10/16); 1 (2/16); 2 (2/16); 3 (2/16) | 105.8 × 104 ± 35.6 | 4.0 ± 1.5 | 8/16 50.0 % | 0 (8/16); 1 (2/16); 2 (2/16); 3 (2/16) 4 (2/16) | 257.6 × 104 ± 67.6 | 15.4 ± 4.2 |
| CTCE9908 + DTX | 3/16 18.25% | 0 (12/16); 1 (4/16); | 17.5 × 104 ± 12.4 | 1.2 ± 0.4 | 5/16 31.25 % | 0 (11/16); 1 (2/16); 2 (2/16); 3 (1/16) | 46.3 × 104 ± 18.2 | 1.8 ± 0.9 | 6/16 37.5 % | 0 (10/16); 2 (2/16); 3 (2/16); 4 (4/16) | 85.5 × 104 ± 30.9 | 9.4 ± 2.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Festuccia, C.; Mancini, A.; Gravina, G.L.; Colapietro, A.; Vetuschi, A.; Pompili, S.; Ventura, L.; Delle Monache, S.; Iorio, R.; Del Fattore, A.; et al. Dual CXCR4 and E-Selectin Inhibitor, GMI-1359, Shows Anti-Bone Metastatic Effects and Synergizes with Docetaxel in Prostate Cancer Cell Intraosseous Growth. Cells 2020, 9, 32. https://doi.org/10.3390/cells9010032
Festuccia C, Mancini A, Gravina GL, Colapietro A, Vetuschi A, Pompili S, Ventura L, Delle Monache S, Iorio R, Del Fattore A, et al. Dual CXCR4 and E-Selectin Inhibitor, GMI-1359, Shows Anti-Bone Metastatic Effects and Synergizes with Docetaxel in Prostate Cancer Cell Intraosseous Growth. Cells. 2020; 9(1):32. https://doi.org/10.3390/cells9010032
Chicago/Turabian StyleFestuccia, Claudio, Andrea Mancini, Giovanni Luca Gravina, Alessandro Colapietro, Antonella Vetuschi, Simona Pompili, Luca Ventura, Simona Delle Monache, Roberto Iorio, Andrea Del Fattore, and et al. 2020. "Dual CXCR4 and E-Selectin Inhibitor, GMI-1359, Shows Anti-Bone Metastatic Effects and Synergizes with Docetaxel in Prostate Cancer Cell Intraosseous Growth" Cells 9, no. 1: 32. https://doi.org/10.3390/cells9010032