Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease?



Department of Physiology, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Cardiovascular Sciences, 1081 HV Amsterdam, The Netherlands
*
Authors to whom correspondence should be addressed.
Cells 2019, 8(12), 1617; https://doi.org/10.3390/cells8121617
Submission received: 5 November 2019
/
Revised: 6 December 2019
/
Accepted: 10 December 2019
/
Published: 12 December 2019
(This article belongs to the Special Issue The Role of Proteostasis Derailment in Cardiac Diseases)
{kind=link}
{kind=link}
Abstract
:Cardiac disease is still the leading cause of morbidity and mortality worldwide, despite some exciting and innovative improvements in clinical management. In particular, atrial fibrillation (AF) and heart failure show a steep increase in incidence and healthcare costs due to the ageing population. Although research revealed novel insights in pathways driving cardiac disease, the exact underlying mechanisms have not been uncovered so far. Emerging evidence indicates that derailed proteostasis (i.e., the homeostasis of protein expression, function and clearance) is a central component driving cardiac disease. Within proteostasis derailment, key roles for endoplasmic reticulum (ER) and mitochondrial stress have been uncovered. Here, we describe the concept of ER and mitochondrial stress and the role of interactions between the ER and mitochondria, discuss how imbalance in the interactions fuels cardiac ageing and cardiac disease (including AF), and finally assess the potential of drugs directed at conserving the interaction as an innovative therapeutic target to improve cardiac function.
1. Introduction
Cardiac disease is still the leading cause of morbidity and mortality worldwide, even though the clinical management of cardiac diseases has been improved over the last years [1]. In particular, due to the ageing population, atrial fibrillation (AF), the most common cardiac tachyarrhythmia, shows a steep increase in incidence and, therefore, healthcare costs. Its prevalence in the developed world is estimated to be 1.5%–2% of the general population and steadily rises in the population between 75 and 85 years of age [2]. In Europe, more than 6 million people suffer from AF and in the next 50 years its incidence is estimated to at least double due to the ageing population [3]. In addition, heart failure (HF), the most common and chronic stage of cardiac disease, is also approaching a higher prevalence all over the world. HF constitutes a medical and economical problem worldwide, as treatment costs reach as much as 1%–2% of the total healthcare budget [4]. Unfortunately, no specific therapy exists for AF and HF, and, therefore, treatment is severely hampered. Reason for treatment failure is that the exact molecular mechanisms underlying the pathophysiology are still unknown. However, accumulating evidence shows that derailment of proteostasis (i.e., the homeostasis of protein expression, function, and clearance) is a central component driving cardiac ageing and cardiac disease [5,6,7].
A healthy proteostasis network safeguards normal cellular (metabolic) function through proper protein biosynthesis, folding, trafficking, and clearance, thereby maintaining normal heart function [5,8]. Proteostasis functioning is monitored by the protein quality control (PQC) system, which consists of the heat shock response (chaperones), unfolded protein responses, and protein degradation systems, including the ubiquitin–proteasomal system (UPS) and autophagy [9]. In general, cellular stress, including stress caused by cardiac disease, activates multiple stress pathways, including the cytosolic heat shock response, the endoplasmic reticulum (ER) unfolded protein response (UPRER), and the mitochondrial UPR (UPRmt) [10,11,12].
Within the PQC, the ER and mitochondria play a critical role in the regulation of protein homeostasis and the maintenance of normal cellular function. The ER is an essential organelle involved in protein synthesis, folding, and trafficking. As protein folding is an error-prone process, the PQC system of the ER is specialized in optimizing this process, thereby preserving cardiac protein quality and homeostasis [13]. As approximately 30% of the cardiomyocyte volume is comprised of mitochondria, the maintenance of a healthy and functional mitochondrial PQC system, which includes molecular chaperones (e.g., HSP60 and HSP10) and proteases (e.g., ClpP), is critical to conserve the energy balance and cardiomyocyte function. The PQC system in mitochondria activates, upon stress, protein folding assistance mechanisms and promotes clearance of misfolded proteins to preserve mitochondrial function [14,15].
Thus, a healthy proteostasis in cardiomyocytes safeguards the proper contractile function in the heart. The ER and mitochondria are essential organelles for regulating protein homeostasis, thereby guaranteeing cardiomyocyte function and survival. Therefore, a deeper understanding of ER and mitochondrial function during health and cardiac disease is required to ultimately develop novel therapeutic strategies.
2. Role of the ER in Cardiac Health
The ER is an essential organelle supporting multiple cellular processes such as protein synthesis, protein folding, regulation of Ca2+ homeostasis, and contribution to the generation of autophagosomes and peroxisomes [16]. The ER lumen constitutes of a specialized folding environment, including ER chaperones and oxidoreductases, which processes approximately one-third of newly synthesized proteins, including secretory, cell membrane, and ER proteins, to ensure that they are correctly folded and assembled [17]. One of these ER chaperones is glucose-regulated protein 78 (GRP78), a member of the heat shock protein 70 family, which promotes the folding of hydrophobic stretches in newly synthesized polypeptides [18,19]. The oxidizing folding environment in the ER is vital for the disulfide bond formation catalyzed by protein disulfide isomerase (PDI) and ER oxidoreductin 1 (Ero1) [20,21].
Besides protein-related functions, the ER is also a high-capacity reservoir for intracellular Ca2+, which is important for the regulation of muscle contraction and relaxation [22,23]. For example, in cardiomyocytes, the sarcoplasmic reticulum (SR), a muscle-specialized form of the ER, regulates Ca2+ fluxes and controls the excitation–contraction coupling of the heart [24]. In the ER/SR, several major proteins are involved in Ca2+ uptake and release: Ca2+-ATPases (SERCAs), that transport cytosolic Ca2+ into the ER lumen, and the ryanodine (RyRs) and inositol 1,4,5-triphosphate receptors (IP3Rs), which release Ca2+ from the ER lumen into the cytosol [25,26,27].
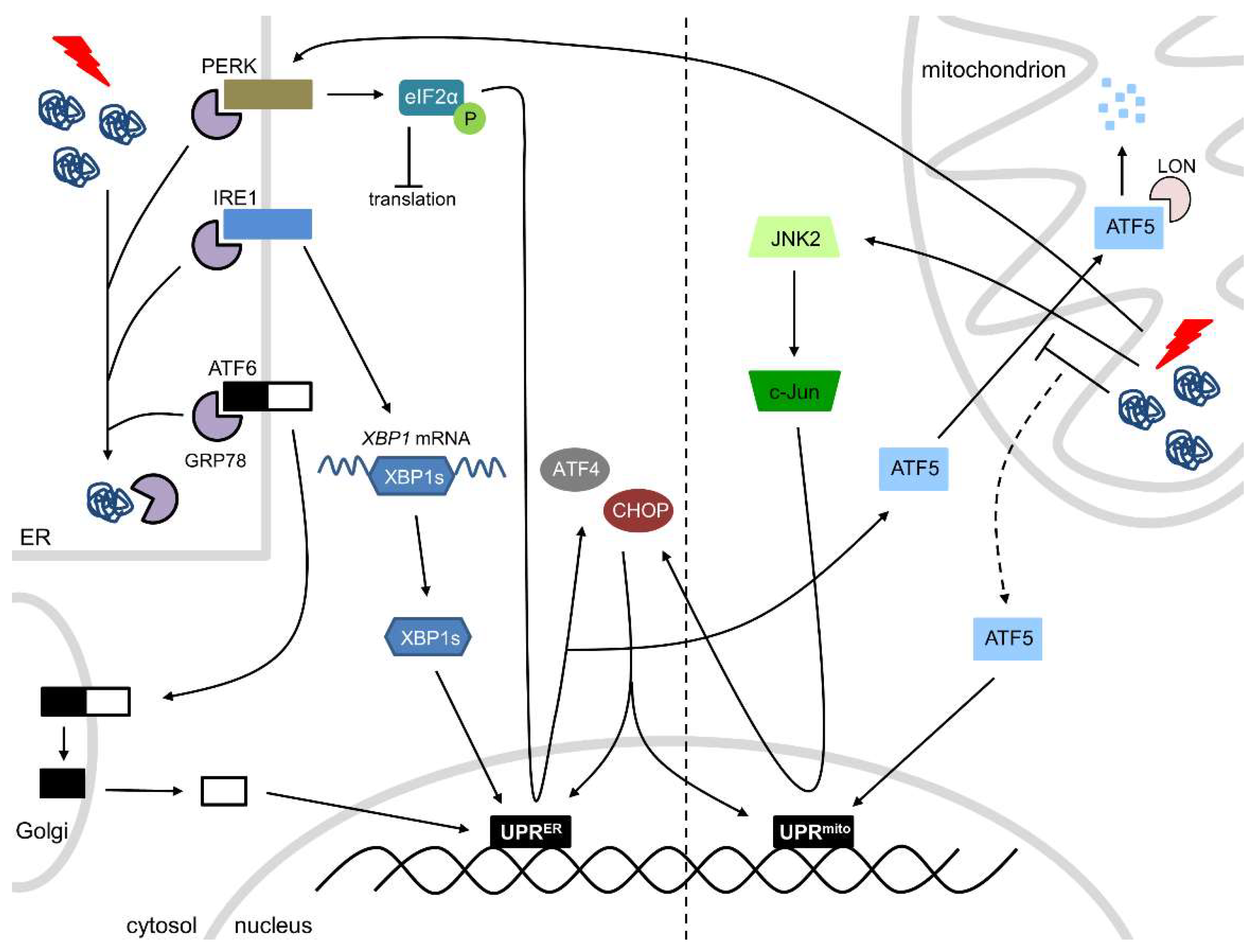
In response to physiological (or pathological) stress, such as proteotoxic stress, oxidative stress, or disturbances in Ca2+ homeostasis, an accumulation of unfolded proteins in the ER triggers the ER stress response. The PQC system in the ER is able to detect the accumulation of misfolded/unfolded proteins and, subsequently, activates the UPRER, a major ER stress-responsive pathway that inhibits protein synthesis and expands ER folding capacity and degradation capacity. The UPRER consists of three stress-responsive arms: activating transcription factor 6 (ATF6), inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1), and protein kinase RNA-like endoplasmic reticulum kinase (PERK) (Figure 1). Under normal circumstances, these three transmembrane proteins are rendered inactive through the binding of GRP78. However, during cellular stress, GRP78 dissociates from ATF6, IRE1, and PERK, thereby activating them. Activated IRE1 splices X-box binding protein 1 (XBP1) mRNA and the spliced XBP1 translocates to the nucleus where it acts as a transcription factor to initiate transcription of molecular chaperones and folding catalysts. ATF6 is cleaved in the Golgi upon ER stress activation and the N-terminal fragment acts as a transcription factor at protective response genes, including those that protect against ER stress and reactive oxygen species (ROS). Activated PERK phosphorylates eukaryotic initiation factor 2 alpha (eIF2α), resulting in a complete blockage of protein translation and initiation, except for the molecular chaperones and catalysts initiated by IRE1 and ATF6, which mRNAs consist of open reading frames in the 5′ untranslated region. In addition, eIF2α phosphorylation induces translation of ATF4, which is able to induce the expression of C/EBP homologous protein (CHOP) (Figure 1). The UPRER is initially an adaptive pathway to restore ER homeostasis, but, if unresolved, chronic/severe activation of the UPRER leads to cell dysfunction and apoptotic cell death, which is initiated by CHOP [6,28,29]. Prolonged ER stress may not be alleviated and cardiomyocytes may undergo apoptosis, which is in any other organ beneficial to clear irreparable damage. However, as cardiomyocytes do not regenerate, apoptosis is detrimental in the heart and prolonged ER stress must be avoided. On the other hand, mild cardiac ER stress may be beneficial to clear unfolded, damaged or old proteins, thereby maintaining proper heart function.
Thus, the ER stress-related signaling pathways activate transcriptional and translational mechanisms that reduce global protein synthesis, increase ER protein-folding capacity, and promote the degradation of misfolded proteins, thereby maintaining normal cardiac health and function.
3. Role of Mitochondria in Cardiac Health
Mitochondria are important organelles, especially in the heart, as they are the source of energy (adenosine triphosphate, ATP) and are required for numerous essential metabolic processes to maintain cardiac contractility and normal heart function [30]. The protein-folding environment in mitochondria is challenged by the complex organelle architecture and the delicate process of assembly of the electron transport chain. In addition, due to their role in energy production, mitochondria undergo continuous additional challenges, such as the management of ROS and the balance in potential mitochondrial DNA mutations [31].
The PQC system of mitochondria ensures mitochondrial integrity and proper mitochondrial function, thereby meeting the metabolic and functional demands of the cell. In response to physiological (or pathological) stress, the accumulation of misfolded/unfolded proteins in the mitochondria activates the UPRmt, initiating the transcription of nuclear-encoded mitochondrial proteases (ClpP), mitochondrial chaperones (HSP60, HSP10) and proteins involved in ROS detoxification and mitochondrial import, thereby ensuring the functional integrity of the mitochondrial proteome [15,32,33]. The exact mechanism of the UPRmt is still somewhat elusive, but two pathways have been described (Figure 1). The first pathway comprises of the transcription factor ATF5, which contains both a nuclear and a mitochondrial targeting sequence. Under physiological conditions, ATF5 is imported into the mitochondria, where it is degraded by the protease LON. However, mitochondrial stress hampers the import of ATF5, which is consequently targeted to the nucleus, where it initiates UPRmt-associated transcription [30,34,35]. The second pathway consists of c-Jun N-terminal kinase 2 (JNK2) and PERK. In accordance with the UPRER, PERK phosphorylates eIF2α, consequently blocking protein translation and initiation. Phosphorylation of eIF2α initiates the translation of ATF4, CHOP, and ATF5. In addition, JNK2 binds to the transcription factor c-Jun, which activates the transcription of CHOP. ATF4, CHOP, and ATF5 all initiate UPRmt-associated transcription (Figure 1) [30,35].
Mitophagy, a specialized form of autophagy, is also activated to safeguard mitochondrial proteostasis in response to mitochondrial stress [36]. It serves to eliminate the most severely defective/damaged mitochondria, while the UPRmt promotes stabilization and recovery of the repairable mitochondria.
Thus, the mitochondrial PQC system monitors protein integrity and prevents accumulation of damaged proteins in the mitochondria to maintain proper protein folding and clearance of misfolded proteins in cells, thereby conserving cardiac contractility and normal heart function.
4. Interactions between the ER and Mitochondria
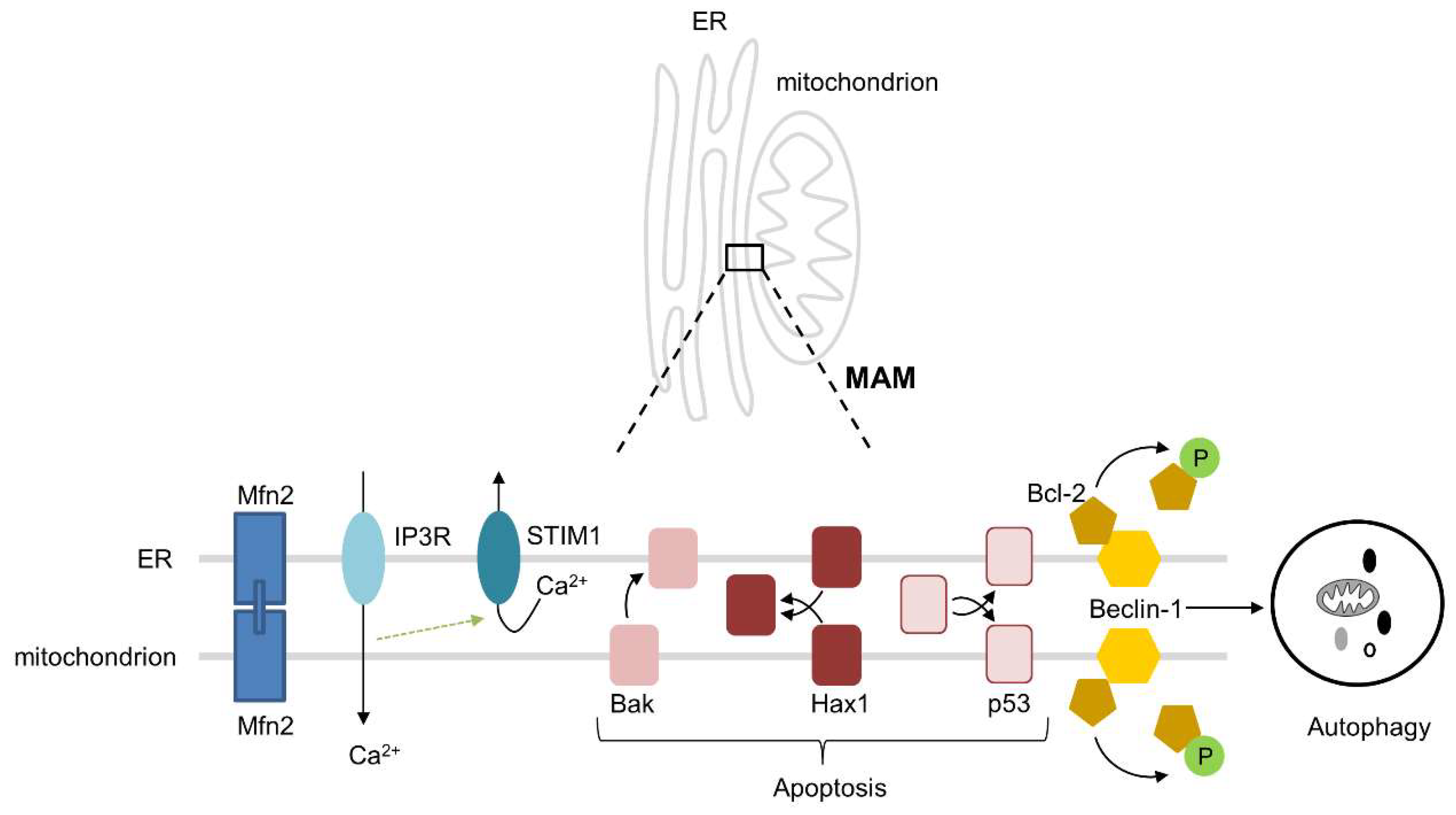
Over the past years, it has been observed that the ER and mitochondria intensively interact with each other and interaction is a prerequisite for healthy cardiac function [37], As a consequence, loss of interaction fuels ageing and cardiac disease. Mitochondria are spatially and functionally organized in close contact with the ER and this contributes to mitochondrial uptake of Ca2+ released from the ER by IP3Rs, thereby providing the mitochondria with Ca2+ that is essential for ATP production [37,38]. During the early phases of ER stress, ER Ca2+ leak and increased ER–mitochondrial coupling lead to an elevated mitochondrial Ca2+ uptake to promote mitochondrial respiration and bioenergetics [39]. Ca2+ signaling between the two organelles is accomplished through mitofusin 2 (Mfn2), an essential physical tether between the ER and mitochondria (Figure 2) [40]. The importance of Mfn2 is exemplified by Chen et al., who showed that cardiac-specific ablation of Mfn2 decreased ER–mitochondrial tethering by 30%, leading to a decreased mitochondrial Ca2+ uptake, which hampers the response to physiological stress [40]. Besides Mfn2, several other proteins, including VAPB, PTPIP51, GRP75, VDAC1, BAP31, FIS1, and Pdzd8, are important for the tethering and interactions, such as Ca2+ exchange, lipid trafficking, apoptosis, autophagy, and mitochondrial fission and fusion, between the ER and mitochondria [41]. Another example to stress the importance of the structural and physiological interactions between the ER and mitochondria consists of stromal interaction molecule 1 (STIM1). STIM1 is an ER Ca2+ sensor, which is essential for normal cardiac homeostasis through the maintenance of ER Ca2+ levels (Figure 2). This has been demonstrated in cardiomyocyte-specific STIM1 knockout mice, which do not only show pronounced ER dilatation, but also smaller mitochondria and a disrupted mitochondrial network. Importantly, these mice have cardiac fibrosis and develop dilated cardiomyopathy [42], suggesting that the structural and physiological interactions between the ER and mitochondria in the heart are of utmost importance for normal and healthy cardiac function.
An important interaction between the ER and mitochondria involves the regulation of autophagy, a fundamental cellular pathway that is activated as a pro-survival pathway under physiological ER stress [43,44]. The IRE1 arm of the UPRER activates JNK and phosphorylates Bcl-2, leading to the dissociation of Bcl-2 from the autophagy-related protein Beclin-1 (Atg6), which is localized at both the ER and mitochondria. This results in the promotion of autophagy as a pro-survival mechanism in the early stages of ER stress (Figure 2) [45,46]. A specialized form of autophagy—mitophagy—is initiated at the mitochondria-associated ER membranes (MAMs), where PINK1 and Beclin-1 re-localize to promote ER-mitochondria tethering and autophagosome formation [47]. The other way around, it has been demonstrated that dysfunctional and/or damaged mitochondria are able to induce ER stress [48]. In addition, the importance of the ER–mitochondrial interactions in the autophagic process comes from the notion that disruption of the ER–mitochondrial contacts impairs the formation of the autophagosome [49]. These findings indicate that interactions between the ER and mitochondria are important for the activation of autophagy to eliminate damaged proteins and organelles, thereby maintaining healthy heart function.
Next to their role in autophagy activation, interactions between the ER and mitochondria can regulate apoptosis under physiological conditions [50]. Bak, known for its localization on the outer mitochondrial membrane where it induces apoptosis upon activation, can also be targeted to the ER, where it depletes Ca2+, induces caspase 12 cleavage, and, subsequently, induces apoptosis (Figure 2) [51]. During the early phase of ER stress, the ER and mitochondria induce an increase in mitochondrial metabolism that depends crucially upon ER–mitochondrial coupling and Ca2+ transfer, which, by enhancing cellular bioenergetics, establishes the metabolic basis for the healthy adaptation to ER stress [39]. In addition, the pro-survival protein HCLS1-associated protein X-1 (Hax1), a novel endogenous inhibitor of apoptosis that is located at both the ER and mitochondria, is associated with maintaining a healthy mitochondrial function and protection from apoptotic cell death (Figure 2). Hax1 is significantly downregulated in cardiac cells upon ER stress, which also resulted in a disrupted mitochondrial morphology, Mfn2 downregulation, and ROS production. Importantly, overexpression of Hax1 protected against ER stress-induced apoptosis and mitochondrial changes, suggesting that Hax1 could be a critical modulator in the cross talk between the ER and mitochondria [52]. The tumor suppressor protein p53 is also an inducer of apoptosis (Figure 2). It can either act indirectly, as a transcription factor increasing or decreasing pro-apoptotic and anti-apoptotic proteins, respectively, or directly, by permeation of the outer mitochondrial membrane through interaction with Bcl-2, thereby releasing cytochrome c from the mitochondria [53,54]. However, p53 is also able to localize to the ER, where it contributes to the imbalance of ER–mitochondrial interactions by inducing mitochondrial damage, including the reduction of oxidative phosphorylation and the release of cytochrome c, leading to apoptotic cell death [55].
To summarize, the functional and balanced interactions, including Ca2+ handling, contractile function, autophagy, and apoptosis, between the ER and mitochondria contribute to the maintenance of cardiomyocyte homeostasis and cardiac contractile function.
5. Interactions between the ER and Mitochondria in Cardiac Ageing and Cardiac Disease
There is compelling evidence that alterations in the interactions between the ER and mitochondria play fundamental roles in the development and progression of cardiac ageing and disease, including ischemic heart disease, AF, and cardiomyopathy.
5.1. Alterations of Interactions between the ER and Mitochondria in Cardiac Ageing
Ageing is a primary risk factor for cardiac disease [56]. Cardiac ageing manifests as a decline in structure and function of the heart, leading to cardiac disease [57,58]. At the cellular level, ageing entails a decline in mitochondrial function and dysregulation of cellular processes, such as oxidative stress, autophagy, and metabolic imbalance, in cardiomyocytes [57]. Mitochondrial alterations, including impaired metabolism and metabolic flexibility with a decreased capacity to oxidize fatty acids and enhanced dependence on glucose metabolism, are critically involved in increased sensitivity to stress in the aged heart [59]. Interestingly, mitochondrial Ca2+ uptake is reduced in cardiomyocytes from aged hearts, and this effect is closely linked with decreased NAD(P)H production and increased mitochondrial ROS production upon increased contractile activity [60]. Moreover, a defective communication between the mitochondrial voltage-dependent anion channel (VDAC) and the SR RyR in cardiomyocytes from aged hearts is associated with a dysregulated Ca2+ handling. Age-dependent alterations in SR Ca2+ transfer to mitochondria and in Ca2+ handling in the SR and mitochondria can be reproduced in cardiomyocytes from young hearts after disrupting the connection between the SR and mitochondria, suggesting that defects in physiological interactions between the SR and mitochondria underlie inefficient inter-organelle Ca2+ exchange, and as such contributes to impaired mitochondrial function and energy demand–supply mismatch in the aged heart. In addition, it has been identified that the protective adaptive response of the UPRER is significantly reduced and pro-apoptotic signaling is more robust during ageing in several tissues, including brain, lung, liver, kidney and heart [61,62]. Moreover, the key quality control proteins, chaperones, and enzymes within the ER, such as GRP78, PDI, calnexin, and glucose-regulated protein 94 (GRP94), are impaired during the aging process [63]. Especially chaperones are progressively oxidized with age, contributing to their functional decline, which is in line with the observed impairment of an adequate cellular response to ER stress during cardiac ageing [63]. Impaired contractility and increased ER stress-induced apoptosis were observed in the heart of mice with cardiac-specific knockout of Sirtuin 1 (Sirt1), an NAD+-dependent histone deacetylase, suggesting that a Sirt1 activator could be a potential modulator of the ER stress response to protect the ageing heart through the inhibition of ER stress-induced apoptosis [64].
These findings indirectly indicate that functional interactions between the ER and mitochondria in the aged heart are hampered and contribute to impairment of autophagy, apoptosis, and mitochondrial dysfunction, leading to cardiac dysfunction during ageing. Although accumulating evidence for loss of the interactions between the ER and mitochondria underlies cardiac ageing, our knowledge on the molecular mechanisms involved is still limited and further research is warranted to conclusively elucidate the importance of loss of interactions during ageing.
5.2. Alterations of Interactions between the ER and Mitochondria in Myocardial Ischemia/Reperfusion
In the clinic, acute myocardial ischemia/reperfusion (I/R) underlies coronary heart disease (CHD). CHD typically arises in patients presenting with an acute myocardial infarction (MI), a major cause of death and disability worldwide [65]. Over the last decade, accumulating evidence shows that alterations in the interactions between the ER and mitochondria contribute to the onset and progression of cardiac I/R injury, in particular interactions involving Ca2+ handling, autophagy, and apoptosis [66,67]. In regards to Ca2+ handling, upon I/R stress, IP3R expression on the ER is upregulated, leading to Ca2+ overload in mitochondria, which, subsequently, activates apoptotic signaling in re-perfused hearts [68,69]. However, returning the ER–mitochondrial interaction back to basal conditions, via the reduction of the upregulated IP3R1 Ca2+ channel complex, decreases the Ca2+ transfer from the ER to mitochondria, subsequently attenuating mitochondrial Ca2+ overload in adult mouse cardiomyocytes during hypoxia-reoxygenation (HR). This suggests that balancing the structural and physiological interactions between the ER and mitochondria during reperfusion could protect cardiomyocytes from lethal reperfusion injury [67]. Thioredoxin-related transmembrane protein 1 (TMX1), a novel SERCA-inhibiting protein, is enriched on the MAMs, which is the site of the ER-mitochondria Ca2+ flux. Cancer cells with low TMX1 levels show increased ER Ca2+ levels, accelerated cytosolic Ca2+ clearance, and reduced Ca2+ transfer to mitochondria, indicating that inhibition of TMX1 may reduce the susceptibility of the heart to I/R injury via reducing MAM contacts [70]. In regards to autophagy, Atg14, a pre-autophagosome/autophagosome protein marker, is re-localized to the MAMs after starvation, and is an indispensable factor for mitophagy activation [71]. However, Atg14 is decreased in a cardiac HR model, resulting in impaired mitophagy and increased cardiomyocyte apoptosis [72]. Finally, in regards to apoptosis, the inhibition of ER stress in in vitro and in vivo myocardial I/R models reveals beneficial effects on cardiac injury, myocardial apoptosis, and infarct area. Inhibition of ER stress increases the expression of superoxide dismutase (SOD) and Bcl-2, which, consequently, decreases the expression of Bax, resulting in the inhibition of apoptosis [73].
These findings suggest that the functional imbalance in the interactions between the ER and mitochondria contribute to increased myocardial apoptosis and damage in cardiac I/R injury.
5.3. Alterations of Interactions between the ER and Mitochondria in AF
AF is the most common progressive cardiac tachyarrhythmia, and is associated with high morbidity and mortality worldwide [3]. There are strong indications that defective interactions between the ER and mitochondria may contribute to AF pathogenesis. In experimental AF models and AF patients, ER stress is present, which induces autophagy and, thereby, cardiac remodeling [74]. Importantly, overexpression of the ER chaperone GRP78 or prevention of ER stress by treatment with the chemical chaperone 4-PBA protected against cardiac remodeling and AF in experimental cardiomyocytes, Drosophila melanogaster, and a dog model for AF [74]. Moreover, ER stress may activate mitogen-activated protein kinases (MAPKs), which initiate the mitochondrial apoptotic pathway [75]. In addition, atria of patients with persistent AF show oxidized RyR2, the cardiac SR Ca2+ release channel. Oxidized RyR2 leads to aberrant intracellular Ca2+ release (Ca2+ sparks) and promotes the development of AF, suggesting that defective communication between the SR and mitochondria contributes to the progression of AF [76]. In concordance, experimental AF models and AF patients show mitochondrial stress, exemplified by increased expression of HSP60 and HSP10, decreased ATP expression, loss of the mitochondrial membrane potential, and mitochondrial network fragmentation, resulting in contractile dysfunction and AF progression [77]. These observations suggest that ER stress may be linked with mitochondrial stress, and that an imbalance in the functional interactions between the ER and mitochondria may contribute to AF progression.
In summary, there is proof for a key role of ER stress and mitochondrial stress in AF. Whether this is a result of loss of interactions between the ER and mitochondria is not known.
5.4. Alteractions of Interactions between the ER and Mitochondria in (Inherited) Cardiomyopathy
Accumulating evidence indicates that ER stress and mitochondrial stress play important roles in cardiomyopathy [78,79,80], of which approximately 50% is inherited [81]. Elevated expression of the ER stress markers GRP78, eIF2α, and XBP1 and increased activation of the UPRER is observed in patients with the p.S143P mutation in the intermediate filament gene lamin A/C, the most frequently reported genetic variant in inherited dilated cardiomyopathy (DCM) [82]. Enhanced mitochondrial oxidative stress and mitochondrial dysfunction were observed in the heart of cats with hypertrophic cardiomyopathy (HCM) [83]. Moreover, impaired autophagy was shown in a Mybpc3-targeted knockin HCM mouse model and in DCM patients with mutations in PLEKHM2 [84,85]. Although ER stress and mitochondrial stress are undeniably present in different forms of cardiomyopathy, it is, however, not known whether loss of the functional interactions between the ER and mitochondria affect the pathogenesis of cardiomyopathies. Therefore, further research is needed to unravel how the molecular mechanisms in the regulation of the interactions between the ER and mitochondria contribute to cardiomyopathies.
5.5. Alterations of Interactions between the ER and Mitochondria in Diabetic Cardiomyopathy
Diabetic cardiomyopathy is characterized as dysfunctional remodeling of the myocardial structure and abnormal cardiac performance in patients with diabetes mellitus, which is independent of vascular pathology [86]. Increasing evidence shows that alterations in the interactions between the ER and mitochondria are involved in the pathophysiological process of diabetic cardiomyopathy. Reduced protein levels of RyR2 in streptozotocin (STZ)-induced diabetic rats contributed to a decrease in the SR Ca2+ storage and decreased rates of Ca2+ release in cardiomyocytes [87], suggesting that reduced contacts between the SR and mitochondria and the subsequent aberrant intracellular Ca2+ signaling may contribute to diabetic cardiomyopathy. On the other hand, reduced contacts between the SR and mitochondria may be beneficial, as the downregulation of Mfn2 expression in STZ-induced type I diabetes inhibited the interaction between the ER and mitochondrial apoptotic pathways [88]. More interestingly, the expression of FUN14 domain containing 1 (Funcd1), an outer mitochondrial membrane protein important for mitophagy and MAMs, was increased in cardiac tissue from diabetic patients. In STZ-induced diabetic mice, increased Funcd1 induced MAM formation, increased mitochondrial Ca2+ influx and, subsequently, mitochondrial dysfunction, resulting in diabetic cardiomyopathy [89]. These observations suggest that the structural and functional interactions between the ER and mitochondria have a fine line between being beneficial and being detrimental and should, therefore, be precisely regulated. In experimental diabetic cardiomyopathy, severe ER stress and prolonged UPRER activation contribute to cardiomyocyte apoptotic death [90,91]. In line, markers of ER stress (GRP78 and CHOP) and apoptosis (cleaved caspase-3) were elevated in myocardium of diabetic patients [92].
Taken together, an imbalance in the interactions between the ER and mitochondria may contribute to mitochondrial dysfunction and lead to cardiomyocyte apoptotic death in diabetic cardiomyopathy.
6. ER and Mitochondria Interactions as Therapeutic Targets
As discussed above, alterations in and loss of the interactions between the ER and mitochondria may induce cardiomyocyte dysfunction and cardiac disease. Therefore, therapeutic interventions to improve interactions between the ER and mitochondria may conserve cardiac function and represent possible promising strategies to delay cardiac ageing and to delay or prevent cardiac disease.
6.1. Lifestyle Interventions—Caloric Restriction and Exercise
Lifestyle change by caloric restriction (CR) is one of the most effective nutritional interventions that reproducibly showed protection against cardiac risk factors and age-related cardiac disease in humans [93]. Besides CR, also exercise training is an effective non-pharmacological approach to improve heart function in cardiac ageing and disease [6]. In an aged rat model, a combination of intermittent ladder-climbing exercise training and a reduced caloric intake were found to decrease the levels of ER stress-related proteins, including phosphorylated PERK and CHOP, proteins that contribute to cardiac muscle damage in ageing [94]. Moreover, high-intensity training can improve cardiac function and reduce cardiac infarction by decreasing the expression of GRP78, phosphorylated PERK, phosphorylated eIF2α, ATF4, ATF6, XBP1, CHOP, and cleaved caspase-3 in an intermittent I/R rat model [95,96]. In addition, treadmill exercise has been shown to ameliorate ER stress by down-regulating phosphorylated eIF2α and ATF6 in diabetic mice [97]. As ageing is an important risk factor for cardiac disease, including AF, lifestyle interventions such as CR and proper exercise training could prevent ER stress, mitochondrial stress, and the progression of AF [98]. However, the effect of CR and exercise training on ER–mitochondrial interactions is so far unknown.
6.2. Pharmacological Interventions—Improved Interactions between the ER and Mitochondria
Pharmacological therapies that improve the balance in interactions between the ER and mitochondria attenuate ER and mitochondrial stress, thereby restoring heart function in ageing and cardiac disease.
One option to improve the ER–mitochondrial balance is by means of taurine, a marketed key nutrient for cardiac health. In ischemic rat cardiomyocytes, administration of taurine attenuated ER stress (GRP78, ATF6, PERK), mitochondrial oxidative stress, and mitochondrial-dependent apoptosis (Bax, Bcl-2) [99]. Another option is the utilization of metformin, a traditional anti-diabetic drug, which decreases ER stress-induced cardiac injury through the protection of mitochondria and attenuation of CHOP expression during cardiac I/R [100]. Moreover, sulodexide (SDX), a glycosaminoglycan, decreases cardiac injury, infarct area, and myocardial apoptosis during I/R through increased Bcl-2 and decreased Bax expression in a mouse model of I/R, suggesting that SDX has in vivo a cardioprotective role in the suppression of ER stress and apoptosis [73]. In addition, ER–mitochondrial cross talk can be regulated by exogenous H2S, which inhibits the activation of apoptotic pathways [88]. Finally, the administration of edaravone, an antioxidant, proved to be beneficial in ameliorating oxidative and ER stress-mediated myocardial apoptosis in an experimental DCM model [101].
Taken together, several compounds may improve the interactions between the ER and mitochondria, thereby having a beneficial effect on cardiac ageing and disease. However, further research is needed to elucidate compounds that directly target the ER–mitochondrial interactions. Nevertheless, with the current knowledge, a combination of lifestyle interventions (caloric restriction and/or high intensity training) and a pharmacological intervention, taurine, may be the most beneficial to improve the ER–mitochondrial interactions in cardiac disease.
7. Conclusions
A healthy proteostasis in cardiomyocytes maintains the proper contractile function in the heart. The ER and mitochondria are key players in the regulation of protein homeostasis and are important for the clearance of stress-induced misfolded proteins, thereby guaranteeing cardiomyocyte health. The structural and functional interactions between the ER and mitochondria are essential for normal cardiac function. Loss of these interactions contribute to the progression of ageing and cardiac disease and preservation of the ER–mitochondrial interactions by pharmacological targeting may represent a promising strategy to conserve cardiac function.
Funding
This research was funded by the Dutch Heart Foundation (2013T096, 2013T144, 2017T029 and 2019T029) and the Netherlands Organization for Scientific Research (AFFIP: 14728).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Camm, A.J.; Lip, G.Y.; De Caterina, R.; Savelieva, I.; Atar, D.; Hohnloser, S.H.; Hindricks, G.; Kirchhof, P.; Guidelines, E.S.C.C.f.P. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: An update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur. Heart J. 2012, 33, 2719–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace 2016, 18, 1609–1678. [Google Scholar] [CrossRef] [PubMed]
- Lesyuk, W.; Kriza, C.; Kolominsky-Rabas, P. Cost-of-illness studies in heart failure: A systematic review 2004–2016. Bmc Cardiovasc. Disord. 2018, 18, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; Henning, R.H.; Brundel, B.J. Derailed Proteostasis as a Determinant of Cardiac Aging. Can. J. Cardiol. 2016, 32, 1166.e11-1166.e20. [Google Scholar] [CrossRef]
- Meijering, R.A.; Zhang, D.; Hoogstra-Berends, F.; Henning, R.H.; Brundel, B.J. Loss of proteostatic control as a substrate for atrial fibrillation: A novel target for upstream therapy by heat shock proteins. Front Physiol. 2012, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, H.; Ranek, M.J. Protein quality control and degradation in cardiomyocytes. J. Mol. Cell Cardiol. 2008, 45, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, D.; Wiersma, M.; Brundel, B. Role of Autophagy in Proteostasis: Friend and Foe in Cardiac Diseases. Cells 2018, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glembotski, C.C.; Arrieta, A.; Blackwood, E.A. Unfolding the Roles of Mitochondria as Therapeutic Targets for Heart Disease. J. Am. Coll. Cardiol. 2019, 73, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.M.; Oster, M.E.; Hebert, D.N. Protein Quality Control in the Endoplasmic Reticulum. Protein J. 2019, 38, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. Embo. J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of endoplasmic reticulum stress in the heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef]
- Ouyang, Y.B.; Xu, L.J.; Emery, J.F.; Lee, A.S.; Giffard, R.G. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion 2011, 11, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Simmen, T.; Lynes, E.M.; Gesson, K.; Thomas, G. Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta. 2010, 1798, 1465–1473. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V. Sarcoplasmic reticulum: Structural determinants and protein dynamics. Int. J. Biochem. Cell Biol. 2011, 43, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F.; Mignery, G.A.; Somlyo, A.V. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 1994, 15, 682–700. [Google Scholar] [CrossRef]
- Otsu, K.; Willard, H.F.; Khanna, V.K.; Zorzato, F.; Green, N.M.; MacLennan, D.H. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J. Biol. Chem. 1990, 265, 13472–13483. [Google Scholar]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharm. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. Embo. Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Valera-Alberni, M.; Canto, C. Mitochondrial stress management: A dynamic journey. Cell Stress 2018, 2, 253–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, S.; Chander, H.; Schafer, P.; Meiss, G.; Kruger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response in mammalian physiology. Mamm. Genome. 2014, 25, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, 170–185. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The Emerging Network of Mitochondria-Organelle Contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Gordaliza-Alaguero, I.; Canto, C.; Zorzano, A. Metabolic implications of organelle-mitochondria communication. Embo. Rep. 2019, 20, 47928. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.E.; He, L.; Zou, L.; Qu, J.; Zhou, L.; Litovsky, S.H.; Yang, Q.; Young, M.E.; Marchase, R.B.; Chatham, J.C. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Matsui, Y.; Sadoshima, J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid. Redox Signal 2007, 9, 1373–1381. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef] [Green Version]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, 2271. [Google Scholar] [CrossRef] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, 4424. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelwahid, E.; Li, H.; Wu, J.; Irioda, A.C.; de Carvalho, K.A.; Luo, X. Endoplasmic reticulum (ER) stress triggers Hax1-dependent mitochondrial apoptotic events in cardiac cells. Apoptosis 2016, 21, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regula, K.M.; Kirshenbaum, L.A. p53 activates the mitochondrial death pathway and apoptosis of ventricular myocytes independent of de novo gene transcription. J. Mol. Cell Cardiol. 2001, 33, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Ishikawa, Y. Apoptosis in heart failure. -The role of the beta-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes. Circ. J. 2011, 75, 1811–1818. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage. Front Cardiovasc. Med. 2019, 6, 10. [Google Scholar] [CrossRef]
- Dhingra, R.; Vasan, R.S. Age as a risk factor. Med. Clin. North Am. 2012, 96, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac ageing: Extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Sanz, C.; Ruiz-Meana, M.; Miro-Casas, E.; Nunez, E.; Castellano, J.; Loureiro, M.; Barba, I.; Poncelas, M.; Rodriguez-Sinovas, A.; Vazquez, J.; et al. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014, 5, 1573. [Google Scholar] [CrossRef] [Green Version]
- Paz Gavilan, M.; Vela, J.; Castano, A.; Ramos, B.; del Rio, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neuro. Biol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.G.; Ramaiah, K.V. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 2007, 355, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Nuss, J.E.; Choksi, K.B.; DeFord, J.H.; Papaconstantinou, J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 2008, 365, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.J.; Hsu, S.C.; Hsu, C.P.; Chen, Y.H.; Chang, Y.L.; Sadoshima, J.; Huang, S.M.; Tsai, C.S.; Lin, C.Y. Sirtuin 1 protects the aging heart from contractile dysfunction mediated through the inhibition of endoplasmic reticulum stress-mediated apoptosis in cardiac-specific Sirtuin 1 knockout mouse model. Int. J. Cardiol. 2017, 228, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER-Mitochondria Microdomains in Cardiac Ischemia-Reperfusion Injury: A Fresh Perspective. Front Physiol. 2018, 9, 755. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Zhu, H.; Jin, Q.; Li, Y.; Ma, Q.; Wang, J.; Li, D.; Zhou, H.; Chen, Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 2018, 23, 101–113. [Google Scholar] [CrossRef]
- Raturi, A.; Gutierrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.H.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huan, L.; Yin, J.; Qin, M.; Zhang, Z.; Zhang, Z.; Zhang, J.; Wang, S. Role of microRNA-130a in myocardial hypoxia/reoxygenation injury. Exp. Med. 2017, 13, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Chen, R.; Zhang, L.; Rao, Z.; Ruan, Y.; Li, L.; Chu, M.; Zhang, Y. Sulodexide attenuates endoplasmic reticulum stress induced by myocardial ischaemia/reperfusion by activating the PI3K/Akt pathway. J. Cell Mol. Med. 2019, 23, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6, 6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Jiang, Q.; Ding, X.; Xu, W.; Wang, D.W.; Chen, M. The ER stress-mediated mitochondrial apoptotic pathway and MAPKs modulate tachypacing-induced apoptosis in HL-1 atrial myocytes. PLoS ONE 2015, 10, e0117567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, M.; van Marion, D.M.S.; Wust, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sun, Y.; Zhuang, Q.; Song, Y.; Wu, B.; Jia, Z.; Pan, H.; Zhou, H.; Hu, S.; Zhang, B.; et al. Mitochondrial dysfunction caused by m.2336T>C mutation with hypertrophic cardiomyopathy in cybrid cell lines. Mitochondrion 2019, 46, 313–320. [Google Scholar] [CrossRef]
- Kopajtich, R.; Nicholls, T.J.; Rorbach, J.; Metodiev, M.D.; Freisinger, P.; Mandel, H.; Vanlander, A.; Ghezzi, D.; Carrozzo, R.; Taylor, R.W.; et al. Mutations in GTPBP3 Cause a Mitochondrial Translation Defect Associated with Hypertrophic Cardiomyopathy, Lactic Acidosis, and Encephalopathy. Am. J. Hum. Genet. 2014, 95, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, G.; Gullmets, J.; Virtanen, L.; Li, S.P.; Keinanen, A.; Shimi, T.; Mauermann, M.; Helio, T.; Kaartinen, M.; Ollila, L.; et al. Deleterious assembly of the lamin A/C mutant p.S143P causes ER stress in familial dilated cardiomyopathy. J. Cell Sci. 2016, 129, 2732–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R.; Zech, A.T.L.; Geertz, B.; Reischmann-Dusener, S.; Osinska, H.; Prondzynski, M.; Kramer, E.; Meng, Q.; Redwood, C.; van der Velden, J.; et al. Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice. Circ. Heart Fail. 2017, 10, 4140. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, E.; Levitas, A.; Singh, S.R.; Braiman, A.; Ofir, R.; Etzion, S.; Sheffield, V.C.; Etzion, Y.; Carrier, L.; Parvari, R. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum. Mol. Genet. 2015, 24, 7227–7240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Yaras, N.; Ugur, M.; Ozdemir, S.; Gurdal, H.; Purali, N.; Lacampagne, A.; Vassort, G.; Turan, B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes 2005, 54, 3082–3088. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, X.; Li, T.; Wu, J.; Zhao, Y.; Liu, J.; Sun, A.; Dong, S.; Wu, J.; Zhong, X.; et al. Exogenous H2S regulates endoplasmic reticulum-mitochondria cross-talk to inhibit apoptotic pathways in STZ-induced type I diabetes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, Q.; Ding, Y.; Wu, Y.; Qiu, Y.; Wang, P.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase alpha2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, Q.; Xu, W.; Cai, L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp. Diabetes Res. 2012, 2012, 827971. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, T.; Dai, H.; Liu, G.; Wang, H.; Sun, Y.; Zhang, Y.; Ge, Z. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J. Clin. Biochem. Nutr. 2007, 41, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubkovic, M.; Gressette, M.; Bulat, C.; Cavar, M.; Bakovic, D.; Fabijanic, D.; Grkovic, I.; Lemaire, C.; Marinovic, J. Disturbed Fatty Acid Oxidation, Endoplasmic Reticulum Stress and Apoptosis in Left Ventricle of Patients with Type 2 Diabetes Mellitus. Diabetes 2019, 68, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Ahn, N.; Jung, S.; Park, S. Effects of intermittent ladder-climbing exercise training on mitochondrial biogenesis and endoplasmic reticulum stress of the cardiac muscle in obese middle-aged rats. Korean J. Physiol. Pharm. 2017, 21, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Bozi, L.H.; Jannig, P.R.; Rolim, N.; Voltarelli, V.A.; Dourado, P.M.; Wisloff, U.; Brum, P.C. Aerobic exercise training rescues cardiac protein quality control and blunts endoplasmic reticulum stress in heart failure rats. J. Cell Mol. Med. 2016, 20, 2208–2212. [Google Scholar] [CrossRef] [Green Version]
- Bourdier, G.; Flore, P.; Sanchez, H.; Pepin, J.L.; Belaidi, E.; Arnaud, C. High-intensity training reduces intermittent hypoxia-induced ER stress and myocardial infarct size. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Wong, W.T.; Zhao, L.; Xu, J.; Wang, L.; Lau, C.W.; Chen, Z.Y.; Ma, R.C.; Xu, A.; Wang, N.; et al. PPARdelta Is Required for Exercise to Attenuate Endoplasmic Reticulum Stress and Endothelial Dysfunction in Diabetic Mice. Diabetes 2017, 66, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Staerk, L.; Wang, B.; Preis, S.R.; Larson, M.G.; Lubitz, S.A.; Ellinor, P.T.; McManus, D.D.; Ko, D.; Weng, L.C.; Lunetta, K.L.; et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: Cohort study based on longitudinal data from the Framingham Heart Study. BMJ 2018, 361, 1453. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, Y.; Liu, X.; Zuo, J.; Wang, K.; Liu, W.; Ge, J. Exogenous taurine attenuates mitochondrial oxidative stress and endoplasmic reticulum stress in rat cardiomyocytes. Acta. Biochim. Biophys. Sin. 2013, 45, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl. Res. 2017, 190, 40–50. [Google Scholar] [CrossRef]
- Arumugam, S.; Thandavarayan, R.A.; Veeraveedu, P.T.; Nakamura, T.; Arozal, W.; Sari, F.R.; Giridharan, V.V.; Soetikno, V.; Palaniyandi, S.S.; Harima, M.; et al. Beneficial effects of edaravone, a novel antioxidant, in rats with dilated cardiomyopathy. J. Cell Mol. Med. 2012, 16, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The ER and mitochondrial UPRs. ER stress activates the three arms of the UPRER: PERK, IRE1, and ATF6. PERK activation leads to phosphorylation of eIF2α, resulting in a protein translation block and transcription of ATF4 and CHOP. IRE1 splices XBP1 mRNA and a spliced form translocates to the nucleus. ATF6 is spliced in the Golgi and the N-terminal fragment acts as a transcription factor. All three arms initiate the transcription of ER-related molecular chaperones and/or folding catalysts. Mitochondrial stress activates the UPRmt, which consists of ATF5, PERK, and JNK2. During mitochondrial stress, the import of ATF5 into the mitochondria is blocked, leading to the translocation of ATF5 to the nucleus. PERK activation leads to the transcription of ATF4, CHOP, and ATF5. JNK2 binds to the transcription factor c-Jun, which activates the transcription of CHOP. ATF5, PERK and JNK2 all initiate the transcription of mitochondrial proteases, mitochondrial molecular chaperones, and proteins involved in ROS detoxification and mitochondrial import.
Figure 1.
The ER and mitochondrial UPRs. ER stress activates the three arms of the UPRER: PERK, IRE1, and ATF6. PERK activation leads to phosphorylation of eIF2α, resulting in a protein translation block and transcription of ATF4 and CHOP. IRE1 splices XBP1 mRNA and a spliced form translocates to the nucleus. ATF6 is spliced in the Golgi and the N-terminal fragment acts as a transcription factor. All three arms initiate the transcription of ER-related molecular chaperones and/or folding catalysts. Mitochondrial stress activates the UPRmt, which consists of ATF5, PERK, and JNK2. During mitochondrial stress, the import of ATF5 into the mitochondria is blocked, leading to the translocation of ATF5 to the nucleus. PERK activation leads to the transcription of ATF4, CHOP, and ATF5. JNK2 binds to the transcription factor c-Jun, which activates the transcription of CHOP. ATF5, PERK and JNK2 all initiate the transcription of mitochondrial proteases, mitochondrial molecular chaperones, and proteins involved in ROS detoxification and mitochondrial import.

Figure 2.
The interactions between the ER and mitochondria. Mfn2 is located on both the ER and mitochondrial membranes and is a physical tether between these two organelles. IP3Rs on the ER release Ca2+, which is taken up by the mitochondria, thereby providing the Ca2+ that is necessary for ATP production by the mitochondria. STIM1 is an ER Ca2+ sensor, which restores the Ca2+ storage in the ER upon Ca2+ release. Bak, Hax1, and p53 can all initiate apoptosis. Bax is located on the mitochondrial membrane and upon translocation to the ER membrane initiates apoptosis. Hax1 is located on both the ER and mitochondrial membranes, where it protects against apoptosis initiation. When p53 translocates to either the ER or mitochondrial membrane, apoptosis is activated. Beclin-1 is localized at both the ER and mitochondria. Upon phosphorylation of Bcl-2, this protein dissociation from Beclin-1, thereby activating autophagy.
Figure 2.
The interactions between the ER and mitochondria. Mfn2 is located on both the ER and mitochondrial membranes and is a physical tether between these two organelles. IP3Rs on the ER release Ca2+, which is taken up by the mitochondria, thereby providing the Ca2+ that is necessary for ATP production by the mitochondria. STIM1 is an ER Ca2+ sensor, which restores the Ca2+ storage in the ER upon Ca2+ release. Bak, Hax1, and p53 can all initiate apoptosis. Bax is located on the mitochondrial membrane and upon translocation to the ER membrane initiates apoptosis. Hax1 is located on both the ER and mitochondrial membranes, where it protects against apoptosis initiation. When p53 translocates to either the ER or mitochondrial membrane, apoptosis is activated. Beclin-1 is localized at both the ER and mitochondria. Upon phosphorylation of Bcl-2, this protein dissociation from Beclin-1, thereby activating autophagy.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, J.; Zhang, D.; Brundel, B.J.J.M.; Wiersma, M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells 2019, 8, 1617. https://doi.org/10.3390/cells8121617
AMA Style
Li J, Zhang D, Brundel BJJM, Wiersma M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells. 2019; 8(12):1617. https://doi.org/10.3390/cells8121617
Chicago/Turabian StyleLi, Jin, Deli Zhang, Bianca J. J. M. Brundel, and Marit Wiersma. 2019. "Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease?" Cells 8, no. 12: 1617. https://doi.org/10.3390/cells8121617
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.