Insights into Early Recovery from Influenza Pneumonia by Spatial and Temporal Quantification of Putative Lung Regenerating Cells and by Lung Proteomics

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Influenza Virus and Infection of Animals
2.2. Tissue Processing and Histologic Staining
2.3. Immunofluorescence Staining
2.4. Imaging and Quantification of H&E and Immunofluorescence Slides
2.5. Global Lung Protein Expression by Proteomic Analysis
3. Results
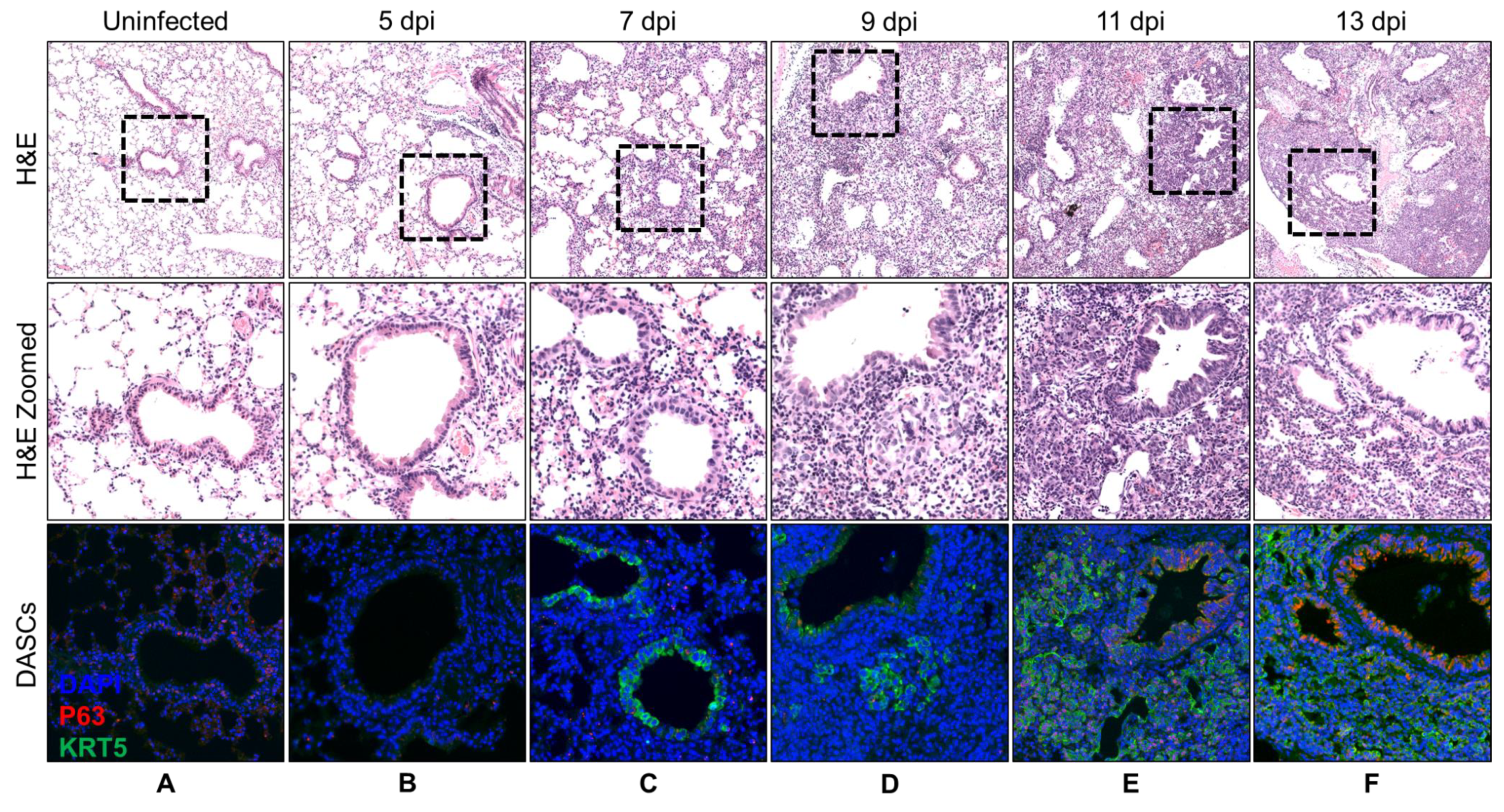
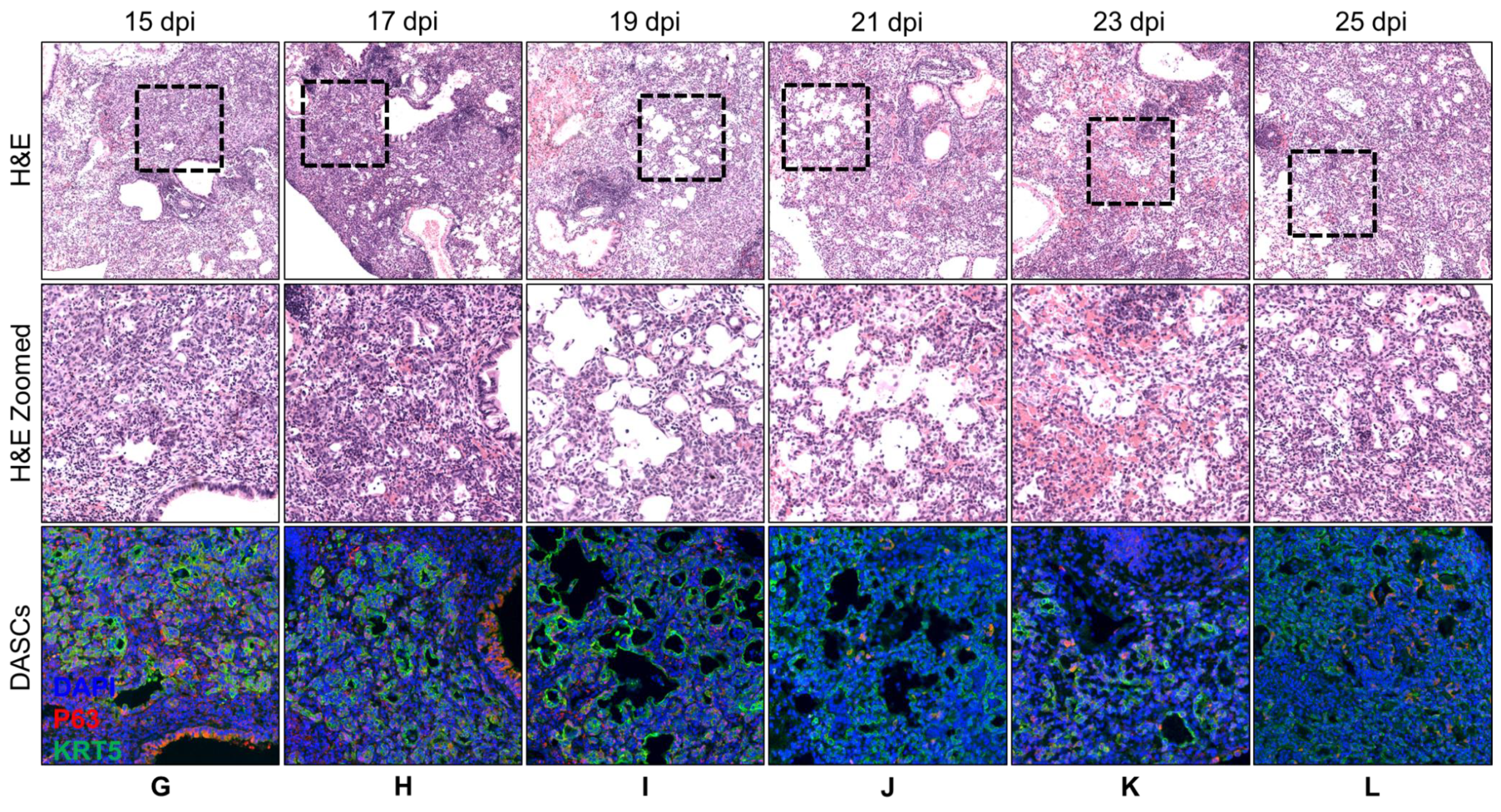
3.1. Spatial and Temporal Distribution of DASCs Following Infection to Early Recovery
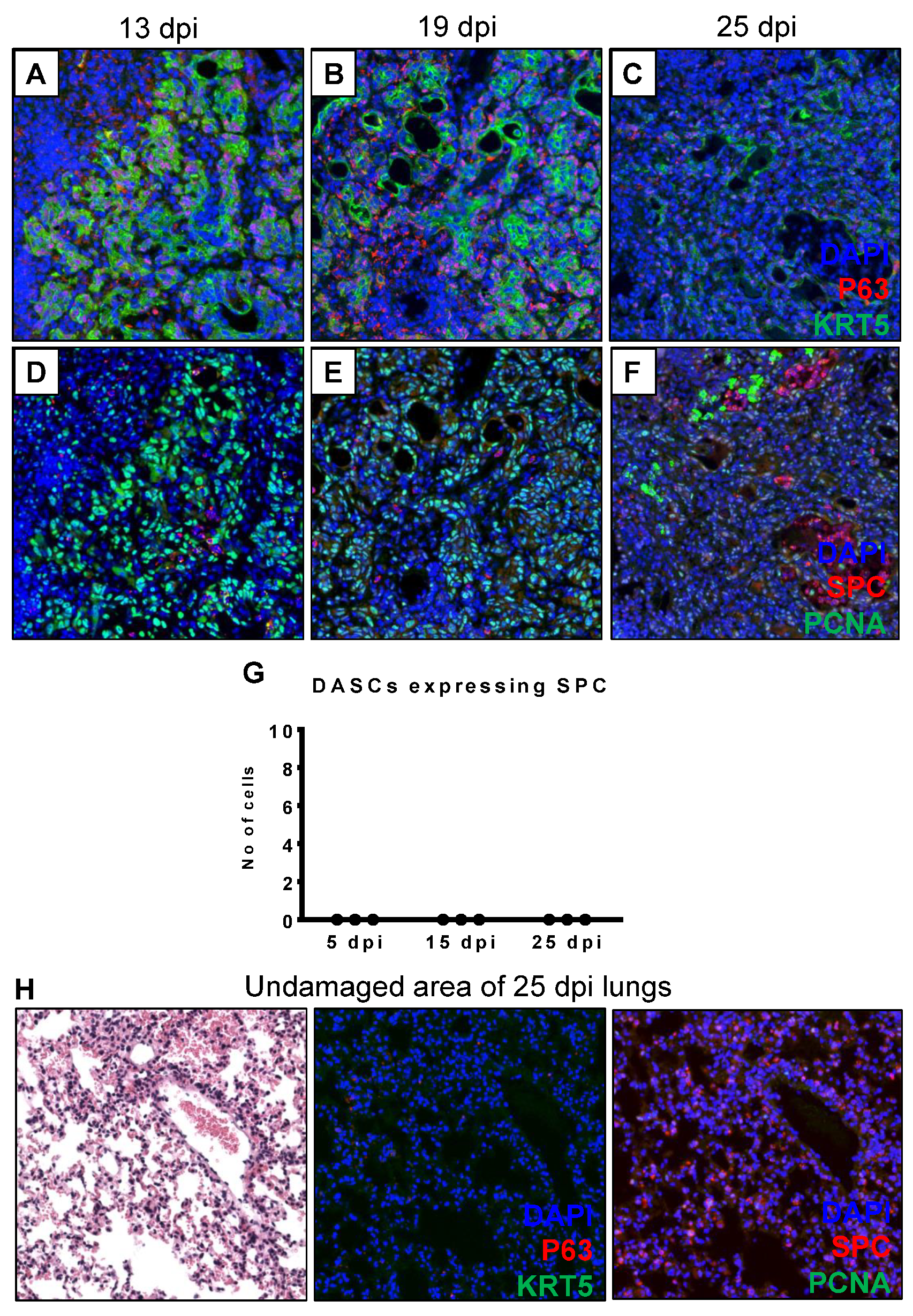
3.2. DASCs Do Not Conclusively Differentiate to Alveolar Epithelial Type II cells by 25 dpi
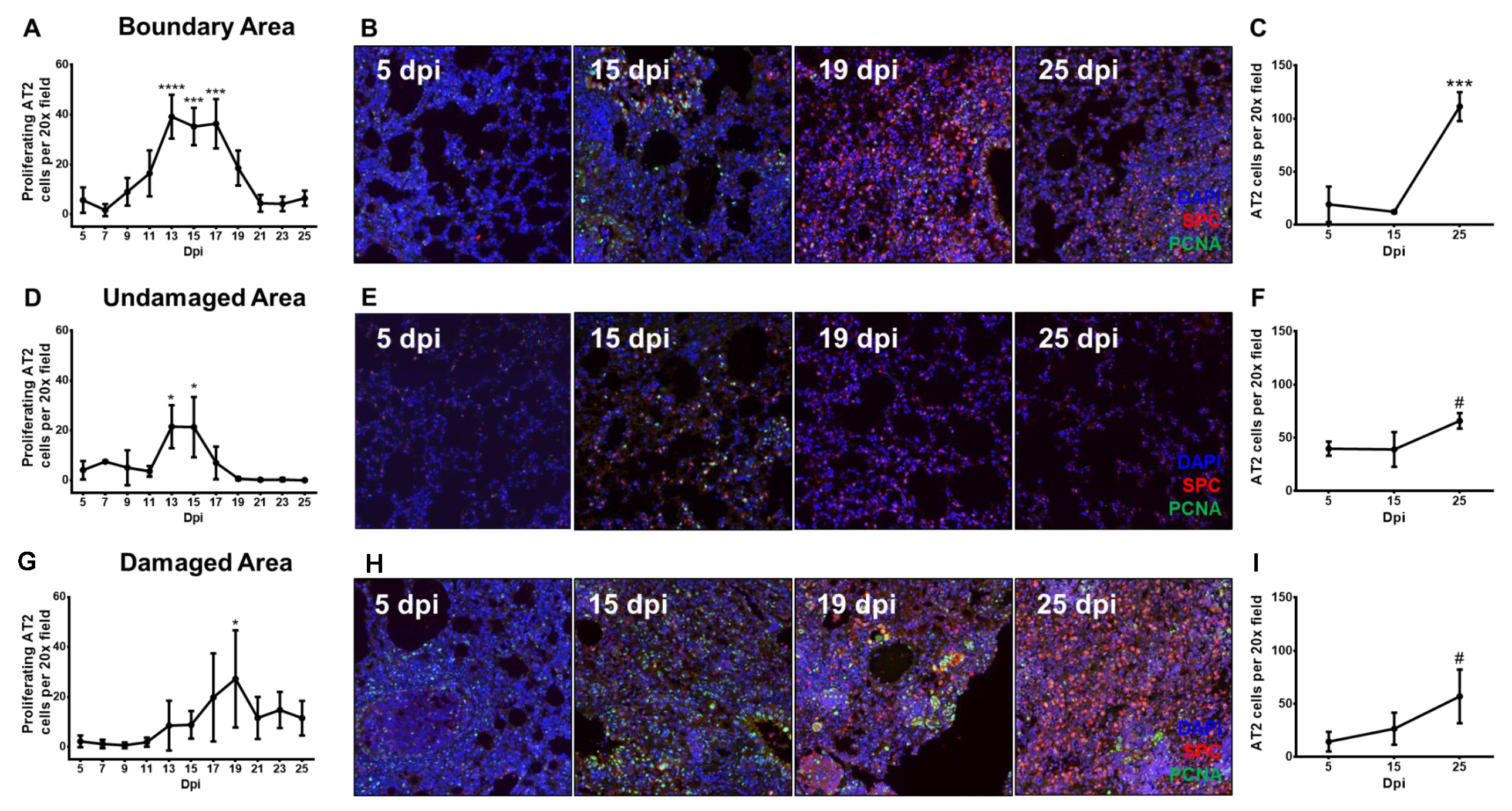
3.3. Proliferating Alveolar Type II Cells Increase During Recovery From Influenza Pneumonia
3.4. Global Protein Expression of Mouse Lungs at 7 and 15 dpi
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van der Vliet, A.; Cross, C.E. Oxidants, nitrosants, and the lung. Am. J. Med. 2000, 109, 398–421. [Google Scholar] [CrossRef]
- Rolfes, M.A.; Foppa, I.M.; Garg, S.; Flannery, B.; Brammer, L.; Singleton, J.A.; Burns, E.; Jernigan, D.; Olsen, S.J.; Bresee, J.; et al. Annual estimates of the burden of seasonal influenza in the United States: A tool for strengthening influenza surveillance and preparedness. Influenza Other Respir. Viruses 2018, 12, 132–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riel, D.; den Bakker, M.A.; Leijten, L.M.E.; Chutinimitkul, S.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Kuiken, T. Seasonal and pandemic human influenza viruses attach better to human upper respiratory tract epithelium than avian influenza viruses. Am. J. Pathol. 2010, 176, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.C.L.; Chan, R.W.Y.; Wang, J.; Travanty, E.A.; Nicholls, J.M.; Peiris, J.S.M.; Mason, R.J.; Chan, M.C.W. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J. Virol. 2011, 85, 6844–6855. [Google Scholar] [CrossRef] [PubMed]
- Ghebrehewet, S.; MacPherson, P.; Ho, A. Influenza. Br. Med. J. 2016, 355, i6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauad, T.; Hajjar, L.A.; Callegari, G.D.; da Silva, L.F.F.; Schout, D.; Galas, F.R.B.G.; Alves, V.A.F.; Malheiros, D.M.A.C.; Auler, J.O.C.; Ferreira, A.F.; et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am. J. Respir. Crit. Care Med. 2010, 181, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayan, S.; Stanford, R.E.; Petty, T.L. Prognosis after recovery from adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1976, 113, 7–16. [Google Scholar] [PubMed]
- Yahav, J.; Lieberman, P.; Molho, M. Pulmonary function following the adult respiratory distress syndrome. Chest 1978, 74, 247–250. [Google Scholar] [CrossRef]
- Elliott, C.G.; Morris, A.H.; Cengiz, M. Pulmonary function and exercise gas exchange in survivors of adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1981, 123, 492–495. [Google Scholar]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.E. Pulmonary epithelial stem cells. Cell Prolif. 2004, 37, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Zhang, T.; Wu, D.Z.; Guan, S.P.; Liew, A.-A.; Yamamoto, Y.; Wang, X.; Lim, S.J.; Vincent, M.; Lessard, M.; et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 2015, 517, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef]
- Kanegai, C.M.; Xi, Y.; Donne, M.L.; Gotts, J.E.; Driver, I.H.; Amidzic, G.; Lechner, A.J.; Jones, K.D.; Vaughan, A.E.; Chapman, H.A.; et al. Persistent pathology in influenza-infected mouse lungs. Am. J. Respir. Cell Mol. Biol. 2016, 55, 613–615. [Google Scholar] [CrossRef]
- Oliveira, V.C.; Carrara, R.C.V.; Simoes, D.L.C.; Saggioro, F.P.; Carlotti, C.G.; Covas, D.T.; Neder, L. Sudan Black B treatment reduces autofluorescence and improves resolution of in situ hybridization specific fluorescent signals of brain sections. Histol. Histopathol. 2010, 25, 1017–1024. [Google Scholar] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Teo, G.; Kim, S.; Tsou, C.-C.; Collins, B.; Gingras, A.-C.; Nesvizhskii, A.I.; Choi, H. mapDIA: Preprocessing and statistical analysis of quantitative proteomics data from data independent acquisition mass spectrometry. J. Proteom. 2015, 129, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [PubMed]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB—A database for integrating human functional interaction networks. Nucleic Acids Res. 2009, 37, D623–D628. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Stephens, R.J.; Freeman, G. Effects of nitrogen dioxide on cell renewal in the rat lung. Arch. Intern. Med. 1971, 128, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Adamson, I.Y.; Bowden, D.H. The type 2 cell as progenitor of alveolar epithelial regeneration. A cytodynamic study in mice after exposure to oxygen. Lab. Investig. 1974, 30, 35–42. [Google Scholar] [PubMed]
- Zheng, D.; Limmon, G.V.; Yin, L.; Leung, N.H.N.; Yu, H.; Chow, V.T.K.; Chen, J. Regeneration of alveolar type I and II cells from Scgb1a1-expressing cells following severe pulmonary damage induced by bleomycin and influenza. PLoS ONE 2012, 7, e48451. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef]
- Ortmann, B.; Copeman, J.; Lehner, P.J.; Sadasivan, B.; Herberg, J.A.; Grandea, A.G.; Riddell, S.R.; Tampé, R.; Spies, T.; Trowsdale, J.; et al. A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science 1997, 277, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Lampton, P.W.; Crooker, R.J.; Newmark, J.A.; Warner, C.M. Expression of major histocompatibility complex class I proteins and their antigen processing chaperones in mouse embryonic stem cells from fertilized and parthenogenetic embryos. Tissue Antigens 2008, 72, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Kim, D.; Choi, I.; Yang, J.S.; Lee, D.-S.; Lee, J.R.; Kang, K.; Kim, S.; Hwang, W.S.; Lee, J.S.; et al. MHC expression in a human adult stem cell line and its down-regulation by hCMV US gene transfection. Int. J. Biochem. Cell Biol. 2005, 37, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Y.; Ye, K.; Wu, N.; Li, J.; Liu, N.; He, M.; Lu, B.; Zhou, W.; Hu, R. Knockdown of tubulin polymerization promoting protein family member 3 suppresses proliferation and induces apoptosis in non-small-cell lung cancer. J. Cancer 2016, 7, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Boopathy, G.T.K.; Kulkarni, M.; Ho, S.Y.; Boey, A.; Chua, E.W.M.; Barathi, V.A.; Carney, T.J.; Wang, X.; Hong, W. Cavin-2 regulates the activity and stability of endothelial nitric-oxide synthase (eNOS) in angiogenesis. J. Biol. Chem. 2017, 292, 17760–17776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallen, L.C.; Burki, Y.; Ebeling, M.; Broger, C.; Siegrist, F.; Oroszlan-Szovik, K.; Bohrmann, B.; Certa, U.; Foser, S. Antiproliferative activity of the human IFN-alpha-inducible protein IFI44. J. Interferon Cytokine Res. 2007, 27, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Uthaiah, R.C.; Praefcke, G.J.K.; Howard, J.C.; Herrmann, C. IIGP1, an interferon-gamma-inducible 47-kDa GTPase of the mouse, showing cooperative enzymatic activity and GTP-dependent multimerization. J. Biol. Chem. 2003, 278, 29336–29343. [Google Scholar] [CrossRef]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Pepys, M.B.; Baltz, M.; Gomer, K.; Davies, A.J.; Doenhoff, M. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature 1979, 278, 259–261. [Google Scholar] [CrossRef]
- Steel, D.M.; Whitehead, A.S. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol. Today 1994, 15, 81–88. [Google Scholar] [CrossRef]
- Wassell, J. Haptoglobin: Function and polymorphism. Clin. Lab. 2000, 46, 547–552. [Google Scholar] [PubMed]
- Matsuura, K.; Bunai, Y.; Ohya, I.; Hara, A.; Nakanishi, M.; Sawada, H. Ultrastructural localization of carbonyl reductase in mouse lung. Histochem. J. 1994, 26, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Oehrle, B.; Gerckens, M.; White, E.S.; Schiller, H.B.; Eickelberg, O. The instructive extracellular matrix of the lung: Basic composition and alterations in chronic lung disease. Eur. Respir. J. 2017, 50, 27. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adh. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotton, D.N.; Morrisey, E.E. Lung regeneration: Mechanisms, applications and emerging stem cell populations. Nat. Med. 2014, 20, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, L.K.; Rims, C.R.; Gill, S.E.; McGuire, J.K.; Manicone, A.M. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 417–426. [Google Scholar] [CrossRef]
- Yamada, M.; Fujino, N.; Ichinose, M. Inflammatory responses in the initiation of lung repair and regeneration: Their role in stimulating lung resident stem cells. Inflamm. Regen. 2016, 36, 15. [Google Scholar] [CrossRef]
- Tan, K.S.; Choi, H.; Jiang, X.; Yin, L.; Seet, J.E.; Patzel, V.; Engelward, B.P.; Chow, V.T. Micro-RNAs in regenerating lungs: An integrative systems biology analysis of murine influenza pneumonia. BMC Genom. 2014, 15, 587. [Google Scholar] [CrossRef]
- Yanagita, K.; Matsumoto, K.; Sekiguchi, K.; Ishibashi, H.; Niho, Y.; Nakamura, T. Hepatocyte growth factor may act as a pulmotrophic factor on lung regeneration after acute lung injury. J. Biol. Chem. 1993, 268, 21212–21217. [Google Scholar]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Panganiban, R.A.M.; Day, R.M. Hepatocyte growth factor in lung repair and pulmonary fibrosis. Acta Pharmacol. Sin. 2011, 32, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Ng, H.H.; Phoon, M.C.; Chow, V.T.K. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Tan, K.S.; Moorthy, A.N.; Phoon, M.C.; Rooijen, N.; Choi, H.W.; Chow, V.T. Combination therapy with hepatocyte growth factor and oseltamivir confers enhanced protection against influenza viral pneumonia. Curr. Mol. Med. 2014, 14, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Kaza, A.K.; Kron, I.L.; Leuwerke, S.M.; Tribble, C.G.; Laubach, V.E. Keratinocyte growth factor enhances post-pneumonectomy lung growth by alveolar proliferation. Circulation 2002, 106, 120–124. [Google Scholar]
- Desai, T.J.; Cardoso, W.V. Growth factors in lung development and disease: Friends or foe? Respir. Res. 2002, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.F.; Schamberger, A.C.; Nayakanti, S.; Hatz, R.; Behr, J.; Eickelberg, O. Detection and quantification of epithelial progenitor cell populations in human healthy and IPF lungs. Respir. Res. 2016, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Xu, S.; Cheng, J.; Zheng, D.; Limmon, G.V.; Leung, N.H.N.; Rajapakse, J.C.; Chow, V.T.K.; Chen, J.; Yu, H. Spatiotemporal quantification of cell dynamics in the lung following influenza virus infection. J. Biomed. Opt. 2013, 18, 046001. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.J.; Driver, I.H.; Lee, J.; Conroy, C.M.; Nagle, A.; Locksley, R.M.; Rock, J.R. Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy. Cell Stem Cell 2017, 21, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Chamoto, K.; Gibney, B.C.; Ackermann, M.; Lee, G.S.; Lin, M.; Konerding, M.A.; Tsuda, A.; Mentzer, S.J. Alveolar macrophage dynamics in murine lung regeneration. J. Cell. Physiol. 2012, 227, 3208–3215. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, M.; Ito, T.; Oka, T.; Kitazawa, T.; Miyoshi-Akiyama, T.; Kirikae, T.; Yamashita, M.; Kudo, K. Serial histopathological examination of the lungs of mice infected with influenza A virus PR8 strain. PLoS ONE 2011, 6, e21207. [Google Scholar] [CrossRef] [PubMed]
- Rutigliano, J.A.; Sharma, S.; Morris, M.Y.; Oguin, T.H.; McClaren, J.L.; Doherty, P.C.; Thomas, P.G. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J. Virol. 2014, 88, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zheng, D.; Limmon, G.V.; Leung, N.H.N.; Xu, S.; Rajapakse, J.C.; Yu, H.; Chow, V.T.K.; Chen, J. Aging exacerbates damage and delays repair of alveolar epithelia following influenza viral pneumonia. Respir. Res. 2014, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.K.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell. Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Day | Histology Description | KRT5 Expression |
|---|---|---|
| 5 | No recovery. | No KRT5+ cells. |
| 7 | No recovery. | KRT5+ cells at bronchioles. |
| 9 | Small pods 1 seen adjacent to bronchioles. | Small KRT5+ pods near bronchioles. |
| 11 | Epithelial regeneration seen as obvious pods as well as single cells radiating out from airway. | Obvious KRT5+ pods. |
| 13 | Epithelial regeneration seen as mainly pods with formation of a few lumens. | Obvious KRT5+ pods. |
| 15 | Epithelial regeneration seen in all damaged alveoli. Most of the pods were beginning to show lumens. | Obvious KRT5+ pods. |
| 17 | Similar to 15 dpi. More pods showing lumens. | KRT5 expression still strong. |
| 19 | Lumens began to flatten out and to line alveolar spaces. | KRT5 expression still strong. |
| 21 | Progression from 19 dpi. | KRT5 staining intensity reducing. |
| 23 | Progression from 21 dpi. | KRT5 staining faint. |
| 25 | Progression from 23 dpi. | KRT5 staining even fainter. |
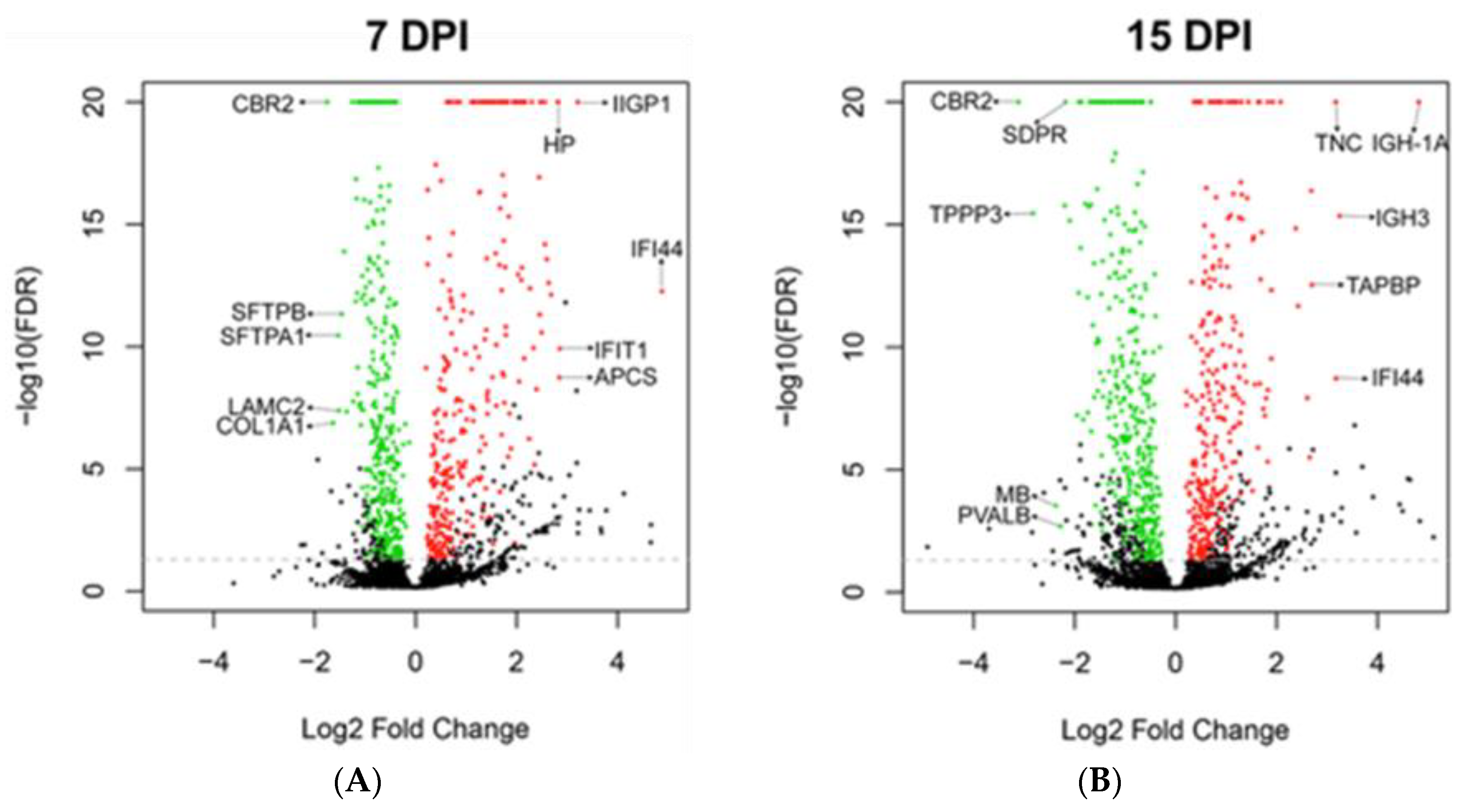
| Day | Regulation | Gene | Function | 7 dpi | 15 dpi |
|---|---|---|---|---|---|
| 7 | Up | IFI44 | Interferon alpha induced protein. Anti-proliferative activity [36]. | +29.4 | +9 |
| IIGP1 | Interferon inducible GTPase 1. Contributes to resistance against intracellular pathogen [37]. | +9.3 | +6.4 | ||
| IFIT1 | Interferon inducible antiviral RNA-binding protein. Antiviral activity against influenza virus [38]. | +7.2 | +2.9 | ||
| APCS | Serum amyloid P-component. Acute phase protein [39,40]. | +7.2 | +3.5 | ||
| HP | Haptoglobin. Acute phase protein [41]. | +7.1 | N.D. | ||
| 7 | Down | CBR2 | Carbonyl reductase. Involved in pulmonary metabolism and highly expressed in alveolar type II cells [42]. | −3.4 | −8.7 |
| COL1A1 | Collagen alpha-1(I) chain, part of type I collagen. Part of lung extracellular matrix [43]. | −3.1 | −1.9 | ||
| SFTPA1 | Surfactant protein A1. Marker of alveolar type II cells. | −2.9 | −2.9 | ||
| LAMC2 | Laminin subunit gamma-2. Subunit of laminin, essential for adherence of epithelial cells to basal membrane [44]. | −2.9 | N.D. | ||
| SFTPB | Surfactant protein B. Marker of alveolar type II cells. | −2.8 | −1.6 | ||
| 15 | Up | IGH-1A | Immunoglobulin gamma 2A chain C region. | +4.7 | +28.2 |
| IGH-3 | Immunoglobulin gamma 2B chain C region. | +3.5 | +9.5 | ||
| IFI44 | Interferon alpha induced protein. Anti-proliferative activity [36]. | +29.4 | +9.0 | ||
| TNC | Tenascin C. Implicated in fibrosis persistence [30]. | +2.2 | +9.0 | ||
| TAPBP | Tapasin. Essential for MHC Class I-TAP complex synthesis [31]. Lowly expressed in stem cells [32,33]. | +6.4 | +6.5 | ||
| 15 | Down | CBR2 | Carbonyl reductase. Involved in pulmonary metabolism and highly expressed in alveolar type II cells [42]. | −3.4 | −8.7 |
| TPPP3 | Tubulin polymerization promoting protein. Associated with proliferation [34]. | −2.7 | −7.1 | ||
| MB | Myoglobin and parvalbumin. Associated with muscles. | N.D. | −5.2 | ||
| PVALB | N.D. | −4.8 | |||
| SDPR | Caveolae-associated protein 2. Associated with angiogenesis by regulating eNOS activity [35]. | −2.4 | −4.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, J.W.J.; Tan, K.S.; Ler, S.G.; Gunaratne, J.; Choi, H.; Seet, J.E.; Chow, V.T.-K. Insights into Early Recovery from Influenza Pneumonia by Spatial and Temporal Quantification of Putative Lung Regenerating Cells and by Lung Proteomics. Cells 2019, 8, 975. https://doi.org/10.3390/cells8090975
Ong JWJ, Tan KS, Ler SG, Gunaratne J, Choi H, Seet JE, Chow VT-K. Insights into Early Recovery from Influenza Pneumonia by Spatial and Temporal Quantification of Putative Lung Regenerating Cells and by Lung Proteomics. Cells. 2019; 8(9):975. https://doi.org/10.3390/cells8090975
Chicago/Turabian StyleOng, Joe Wee Jian, Kai Sen Tan, Siok Ghee Ler, Jayantha Gunaratne, Hyungwon Choi, Ju Ee Seet, and Vincent Tak-Kwong Chow. 2019. "Insights into Early Recovery from Influenza Pneumonia by Spatial and Temporal Quantification of Putative Lung Regenerating Cells and by Lung Proteomics" Cells 8, no. 9: 975. https://doi.org/10.3390/cells8090975