
Machine Learning-Based Prediction of Drug-Drug Interactions for Histamine Antagonist Using Hybrid Chemical Features

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
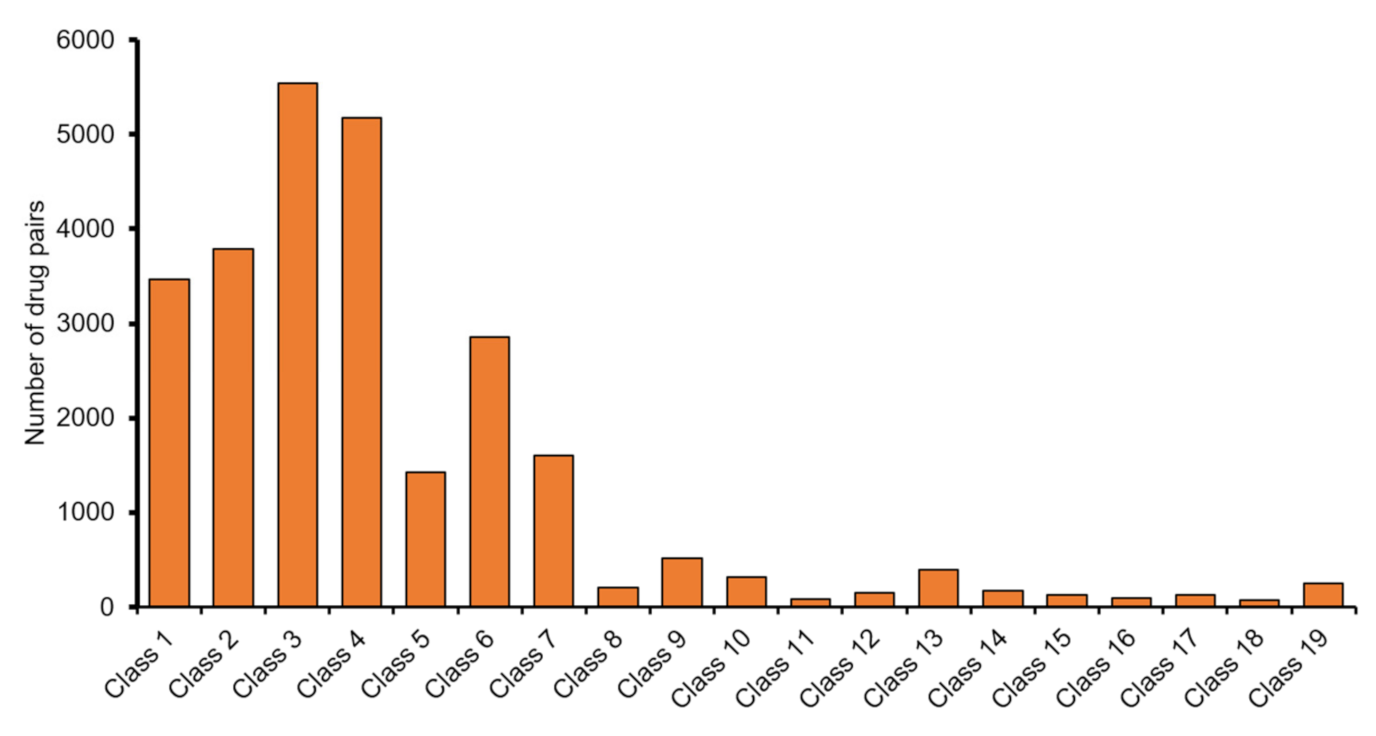
2.1. Data Preparation
2.2. Data Preprocessing
2.2.1. Chemical Interaction Feature Extraction
2.2.2. CYP450-Related Interaction Feature Extraction
2.3. Development of the Predictive HAINI Model
- ▪
- Naive Bayes (NB)
- -
- is the posterior probability of a class (c, target) of a given predictor (x, attributes).
- -
- is the prior probability of a class.
- -
- is the likelihood, which is the probability of a predictor of a given class.
- -
- is the prior probability of the predictor.
- -
- Vector represents some n features.
- ▪
- Decision Tree (DT)
- ▪
- Random Forest (RF)
- ▪
- Logistic Regression (LR)
- ▪
- XGBoost (XGB)
3. Results
3.1. Evaluation of HAINI Performance
3.2. Improvement of HAINI Performance
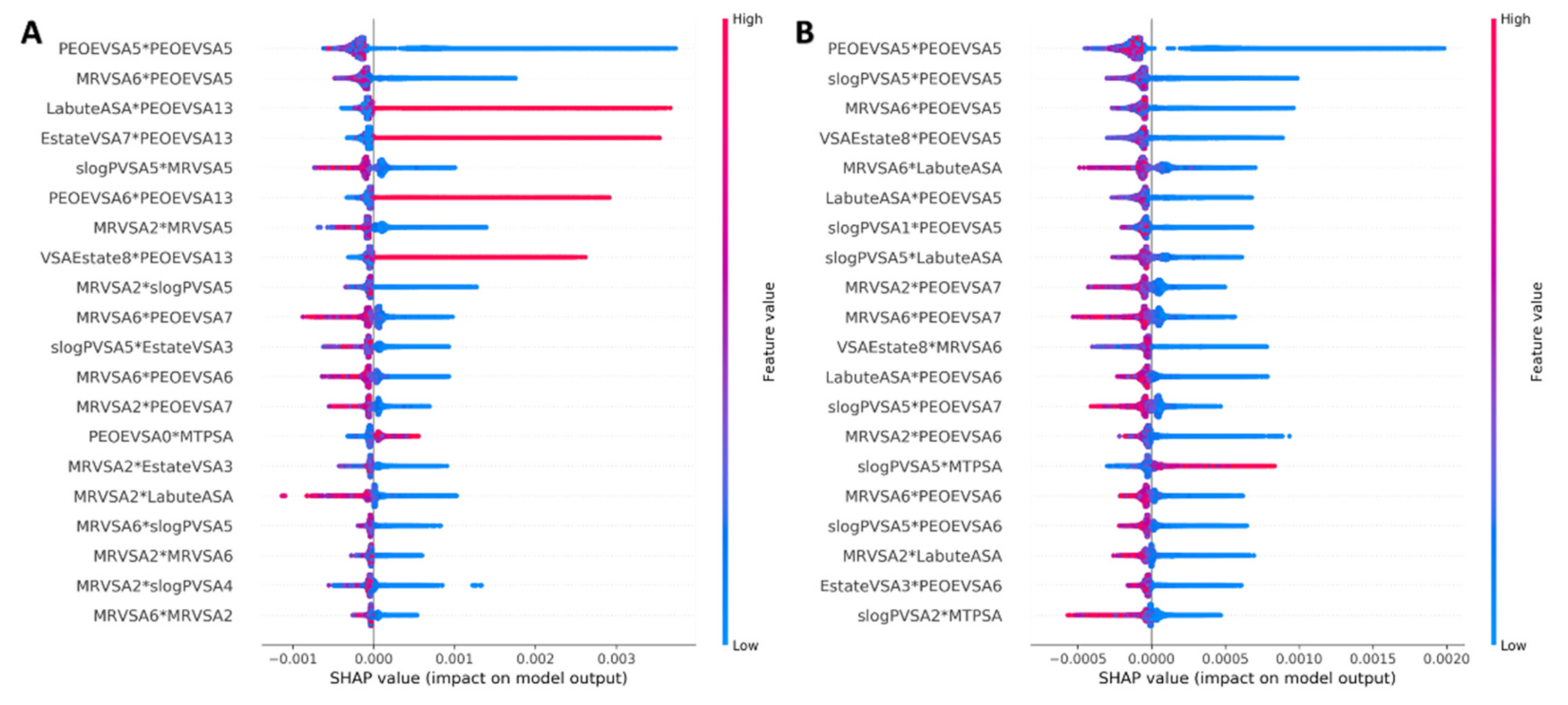
3.2.1. Feature Selection
3.2.2. Applying Synthetic Minority Oversampling Technique (SMOTE)
3.3. HAINI Performance on the Validation Dataset
3.4. Comparison of HAINI Performance on Previous Studies Using Chemical Similarity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ogu, C.C.; Maxa, J.L. Drug interactions due to cytochrome P450. Bayl. Univ. Med. Cent. Proc. 2000, 13, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A.L. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]
- Barry, A.R.; Koshman, S.L.; Pearson, G.J. Adverse drug reactions: The importance of maintaining pharmacovigilance. Can. Pharm. J./Rev. Pharm. Can. 2014, 147, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Honig, P.K.; Woosley, R.L.; Zamani, K.; Conner, D.P.; Cantilena, L.R., Jr. Changes in the pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine with concomitant administration of erythromycin. Clin. Pharmacol. Ther. 1992, 52, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Neilan, T. Merck pulls Vioxx painkiller from market, and stock plunges. The New York Times, 30 September 2004; p. 30. [Google Scholar]
- Dougherty, T.S. Antihistamines (H1 Receptor Antagonists). In Side Effects of Drugs Annual; Elsevier: Amsterdam, The Netherlands, 2017; Volume 39, pp. 157–159. [Google Scholar]
- Lawton, G.; Nussbaumer, P. The Evolving Role of the Medicinal Chemist. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; Volume 55, pp. 193–226. [Google Scholar]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: The hunt for new therapeutic targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [Green Version]
- Bystritsky, A.; Khalsa, S.S.; Cameron, M.E.; Schiffman, J. Current diagnosis and treatment of anxiety disorders. Pharm. Ther. 2013, 38, 30. [Google Scholar]
- Nutt, D.J. Overview of diagnosis and drug treatments of anxiety disorders. CNS Spectr. 2005, 10, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tyrer, P.; Baldwin, D. Generalised anxiety disorder. Lancet 2006, 368, 2156–2166. [Google Scholar] [CrossRef]
- Cantú, T.G.; Korek, J.S. Central nervous system reactions to histamine-2 receptor blockers. Ann. Intern. Med. 1991, 114, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the nervous system. Physiol. Rev. 2008. [Google Scholar] [CrossRef]
- Starmer, G. Antihistamines and highway safety. Accid. Anal. Prev. 1985, 17, 311–317. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Ghasemi, F.; Mehridehnavi, A.; Pérez-Garrido, A.; Pérez-Sánchez, H. Neural network and deep-learning algorithms used in QSAR studies: Merits and drawbacks. Drug Discov. Today 2018, 23, 1784–1790. [Google Scholar] [CrossRef]
- Varmuza, K.; Dehmer, M.; Bonchev, D. Statistical Modelling of Molecular Descriptors in QSAR/QSPR; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Deng, Y.; Xu, X.; Qiu, Y.; Xia, J.; Zhang, W.; Liu, S. A multimodal deep learning framework for predicting drug–drug interaction events. Bioinformatics 2020, 36, 4316–4322. [Google Scholar] [CrossRef]
- Shtar, G.; Rokach, L.; Shapira, B. Detecting drug-drug interactions using artificial neural networks and classic graph similarity measures. PLoS ONE 2019, 14, e0219796. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning improves prediction of drug–drug and drug–food interactions. Proc. Natl. Acad. Sci. USA 2018, 115, E4304–E4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, R.; Dallas, S.; Dickmann, L.J.; Fahmi, O.A.; Kenny, J.R.; Kraynov, E.; Nguyen, T.; Patel, A.H.; Slatter, J.G.; Zhang, L. Critical review of preclinical approaches to investigate cytochrome P450–mediated therapeutic protein drug-drug interactions and recommendations for best practices: A white paper. Drug Metab. Dispos. 2013, 41, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Izumi, S.; Nozaki, Y.; Maeda, K.; Komori, T.; Takenaka, O.; Kusuhara, H.; Sugiyama, Y. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab. Dispos. 2015, 43, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Shah, Y.; Cheung, C.; Guo, G.L.; Feigenbaum, L.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. The PREgnane X receptor gene-humanized mouse: A model for investigating drug-drug interactions mediated by cytochromes P450 3A. Drug Metab. Dispos. 2007, 35, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Ferdousi, R.; Safdari, R.; Omidi, Y. Computational prediction of drug-drug interactions based on drugs functional similarities. J. Biomed. Inform. 2017, 70, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Uriarte, E.; Santana, L.; Tatonetti, N.P.; Friedman, C. Detection of drug-drug interactions by modeling interaction profile fingerprints. PLoS ONE 2013, 8, e58321. [Google Scholar]
- Gottlieb, A.; Stein, G.Y.; Oron, Y.; Ruppin, E.; Sharan, R. INDI: A computational framework for inferring drug interactions and their associated recommendations. Mol. Syst. Biol. 2012, 8, 592. [Google Scholar] [CrossRef]
- You, J.; McLeod, R.D.; Hu, P. Predicting drug-target interaction network using deep learning model. Comput. Biol. Chem. 2019, 80, 90–101. [Google Scholar] [CrossRef]
- Bougiatiotis, K.; Aisopos, F.; Nentidis, A.; Krithara, A.; Paliouras, G. Drug-Drug Interaction Prediction on a Biomedical Literature Knowledge Graph. In Artificial Intelligence in Medicine, Proceedings of the 18th International Conference on Artificial Intelligence in Medicine, Minneapolis, MN, USA, 25–28 August 2020; Springer: Cham, Switzerland, 2020; Volume 12299, pp. 122–132. [Google Scholar]
- Dong, J.; Yao, Z.-J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rish, I. An empirical study of the naive Bayes classifier. In Proceedings of the IJCAI 2001 Workshop on Empirical Methods in Artificial Intelligence, Seattle, MA, USA, 4–6 August 2001; pp. 41–46. [Google Scholar]
- Farid, D.M.; Zhang, L.; Rahman, C.M.; Hossain, M.A.; Strachan, R. Hybrid decision tree and naïve Bayes classifiers for multi-class classification tasks. Expert Syst. Appl. 2014, 41, 1937–1946. [Google Scholar] [CrossRef]
- Ho, T.K. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; pp. 278–282. [Google Scholar]
- Bagley, S.C.; White, H.; Golomb, B.A. Logistic regression in the medical literature: Standards for use and reporting, with particular attention to one medical domain. J. Clin. Epidemiol. 2001, 54, 979–985. [Google Scholar] [CrossRef]
- Kha, Q.-H.; Le, V.-H.; Hung, T.N.K.; Le, N.Q.K. Development and Validation of an Efficient MRI Radiomics Signature for Improving the Predictive Performance of 1p/19q Co-Deletion in Lower-Grade Gliomas. Cancers 2021, 13, 5398. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, F.; Kang, J.; Chen, W. mRNALocater: Enhance the prediction accuracy of eukaryotic mRNA subcellular localization by using model fusion strategy. Mol. Ther. 2021, 29, 2617–2623. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Kha, Q.H.; Nguyen, V.H.; Chen, Y.-C.; Cheng, S.-J.; Chen, C.-Y. Machine Learning-Based Radiomics Signatures for EGFR and KRAS Mutations Prediction in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 9254. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, Y.; Chen, Y.; Sun, J.; Zhao, Z.; Chen, X.-w.; Matheny, M.E.; Xu, H. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J. Am. Med. Inform. Assoc. 2012, 19, e28–e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohani, N.; Eslahchi, C. Drug-Drug interaction predicting by neural network Using integrated Similarity. Sci. Rep. 2019, 9, 1–11. [Google Scholar]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinform. 2017, 18, 18. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VectorA | VectorB | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A2 | Subtrate | 1 | VectorA = | 1 | 1A2 | No interaction | 0 | VectorB = | 0 |
| 2A6 | Non-subtrate | 0 | 0 | 2A6 | No interaction | 0 | 0 | ||
| 2B6 | Non-subtrate | 0 | 0 | 2B6 | Inhibitor | 1 | 1 | ||
| 2C18 | Non-subtrate | 0 | 0 | 2C18 | No interaction | 0 | 0 | ||
| 2C19 | Non-subtrate | 0 | 0 | 2C19 | No interaction | 0 | 0 | ||
| 2C8 | Non-subtrate | 0 | 0 | 2C8 | Inhibitor | 1 | 1 | ||
| 2C9 | Non-subtrate | 0 | 0 | 2C9 | No interaction | 0 | 0 | ||
| 2D6 | Non-subtrate | 0 | 0 | 2D6 | Inducer | −1 | −1 | ||
| 2E1 | Non-subtrate | 0 | 0 | 2E1 | No interaction | 0 | 0 | ||
| 3A4 | Subtrate | 1 | 1 | 3A4 | Inducer | −1 | −1 | ||
| 3A5 | Non-subtrate | 0 | 0 | 3A5 | No interaction | 0 | 0 | ||
| 3A7 | Non-subtrate | 0 | 0 | 3A7 | No interaction | 0 | 0 | ||
| Algorithms | Hyperparameter Grid | Optimal Parameter | |
|---|---|---|---|
| Naïve Bayes | C: | 0.001, 0.01, 0.1, 1, 10, 100, 1000 | C: 100 |
| gamma: | 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0 | gamma: 0.5 | |
| kernel: | rbf, linear | kernel: rbf | |
| Logistic Regression | Penalty: | 11, 12 | Penalty: 12 |
| C: | 0.001, 0.01, 0.1, 1, 10, 100, 1000 | C: 100 | |
| Decision Tree | criterion: | gini, entropy | criterion: Gini |
| max_depth: | 10, 20, 30, 40, 50, None | max_depth: 10 | |
| min_samples_leaf: | 1, 2, 4 | min_samples_leaf: 2 | |
| min_samples_split: | 2, 5, 10 | min_samples_split: 5 | |
| Random Forest | bootstrap: | True, False | bootstrap: False |
| max_depth: | 10, 20, 30, 40, 50, None | max_depth: 10 | |
| max_features: | auto, sqrt | max_features: sqrt | |
| min_samples_leaf: | 1, 2, 4 | min_samples_leaf: 2 | |
| min_samples_split: | 2, 5, 10 | min_samples_split: 5 | |
| n_estimators: | 20, 40, 60, 80, 100, 200, 500, 1000, 1500 | n_estimators: 1500 | |
| XGBoost | max_depth: | 10, 20, 30, 40, 50, None | max_depth: 10 |
| max_features: | auto, sqrt | max_features: sqrt | |
| min_samples_leaf: | 1, 2, 4 | min_samples_leaf: 2 | |
| min_samples_split: | 2, 5, 10 | min_samples_split: 5 | |
| n_estimators: | 20, 40, 60, 80, 100, 200, 500, 1000, 1500 | n_estimators: 1500 | |
| Type of Interactions * | Recall | Precision | F1-Score | Rank |
|---|---|---|---|---|
| Class 13 | 0.906 | 0.904 | 0.999 | 1 |
| Class 15 | 0.839 | 0.838 | 1.000 | 2 |
| Class 6 | 0.837 | 0.818 | 0.984 | 3 |
| Class 3 | 0.799 | 0.745 | 0.966 | 4 |
| Class 17 | 0.769 | 0.777 | 0.981 | 5 |
| Class 4 | 0.749 | 0.685 | 0.939 | 6 |
| Class 7 | 0.742 | 0.729 | 0.959 | 7 |
| Class 2 | 0.703 | 0.65 | 0.941 | 8 |
| Class 1 | 0.681 | 0.63 | 0.946 | 9 |
| Class 8 | 0.68 | 0.681 | 0.995 | 10 |
| Type of Interaction * | Recall | Precision | F1 | Number of Drug-Drug Pairs |
|---|---|---|---|---|
| 15 | 0.68 | 0.75 | 0.65 | 7 |
| 6 | 0.71 | 0.73 | 0.60 | 165 |
| 3 | 0.73 | 0.57 | 0.64 | 570 |
| 17 | 0.46 | 0.45 | 0.38 | 10 |
| 4 | 0.65 | 0.67 | 0.59 | 670 |
| Recall | Precision | F1 | |
|---|---|---|---|
| Average performance * | 0.734 | 0.783 | 0.758 |
| Best performance ** | 0.921 | 0.778 | 0.838 |
| Narjes Rohani et al. | 0.899 | 0.373 | 0.527 |
| Mei Liu et al. | 0.493 | 0.434 | N/A |
| Wen Zhang et al. | 0.765 | 0.617 | 0.683 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, L.H.; Dung, N.T.; Quang, L.X.; Hung, L.Q.; Le, N.H.; Le, N.T.N.; Diem, N.T.; Nga, N.T.T.; Hung, S.-H.; Le, N.Q.K. Machine Learning-Based Prediction of Drug-Drug Interactions for Histamine Antagonist Using Hybrid Chemical Features. Cells 2021, 10, 3092. https://doi.org/10.3390/cells10113092
Dang LH, Dung NT, Quang LX, Hung LQ, Le NH, Le NTN, Diem NT, Nga NTT, Hung S-H, Le NQK. Machine Learning-Based Prediction of Drug-Drug Interactions for Histamine Antagonist Using Hybrid Chemical Features. Cells. 2021; 10(11):3092. https://doi.org/10.3390/cells10113092
Chicago/Turabian StyleDang, Luong Huu, Nguyen Tan Dung, Ly Xuan Quang, Le Quang Hung, Ngoc Hoang Le, Nhi Thao Ngoc Le, Nguyen Thi Diem, Nguyen Thi Thuy Nga, Shih-Han Hung, and Nguyen Quoc Khanh Le. 2021. "Machine Learning-Based Prediction of Drug-Drug Interactions for Histamine Antagonist Using Hybrid Chemical Features" Cells 10, no. 11: 3092. https://doi.org/10.3390/cells10113092