
A Role for the Bone Marrow Microenvironment in Drug Resistance of Acute Myeloid Leukemia

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Genomic and Immunophenotypic Characteristics
3. Treatment
4. Resistance
5. The Normal BM Microenvironment

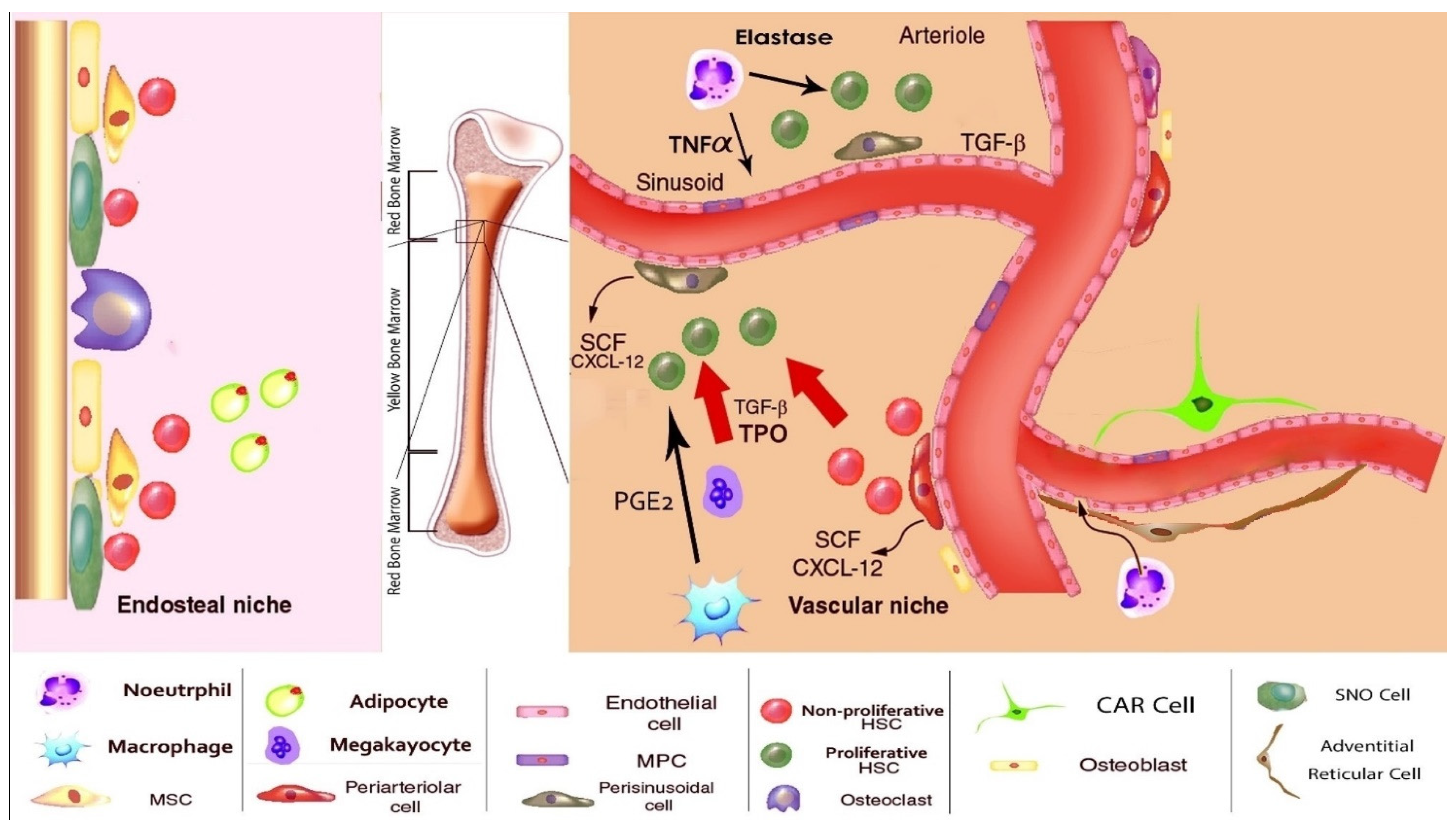{kind=link}
{kind=link}
| Cell | Normal Function and Products | Role in AML | Refs |
|---|---|---|---|
| Adipocyte | 1. Increases in adulthood 2. Adipokine and Adiponectin 3. Hematopoiesis negative regulation | 1. Leukemic cells proliferation 2. Increased adipokinase during leukemia 3. Leukemic cell pro-survival | [44,62,87,89,95] |
| Endothelial cell | 1. Notch L 2. E-selectin, P-selectin 3. Vascular cell adhesion molecule 1 (VCAM 1) 4. Intercellular adhesion molecule 1 (ICAM -1) | 1. Vascular endothelial growth factor (VEGF) production and Granulocyte-macrophage colony-stimulating factor (GM-CSF) (potential mitogen) stimulation 2. AML progression | [87,89,95,96] |
| Osteoblast | 1. N-Cadherin 2. Osteopoietin 3. SCF 4. CXCL12 5. HSC niche establishment | 1. Osteogenesis augmentation 2. AML initiation and progression | [44,86,89,97] |
| CXCL12-abundant reticular cells (CAR cells) | 1. Stromal cell-derived factor 1(SDF-1) 2. VCAM-1 3. E-/P-Selectin 4. CD44 5. Platelet-derived growth factors (PDFG) | Pro-survival | [44,62,87,96] |
| Regulatory T cells (T-reg) | 1. IL-10 2. IL-35 3. Inhibits immune reactions against stem cells | 1. Up-regulated in AML patients 2. AML leukemic cells induce IL-10 secreting T regulatory (iTreg) cells and natural T regulatory (N-Treg) cells through inducible co-stimulator ligand (ICOSL) expression. | |
| Fibroblast | 1. Cancer-associated fibroblasts (CAFs) 2. Growth differentiation factor 15 (GDF15) 3. IL-8 | Chemotherapy resistance | [44,95,98] |
6. Role of the BM Microenvironment in AML and Therapy Resistance
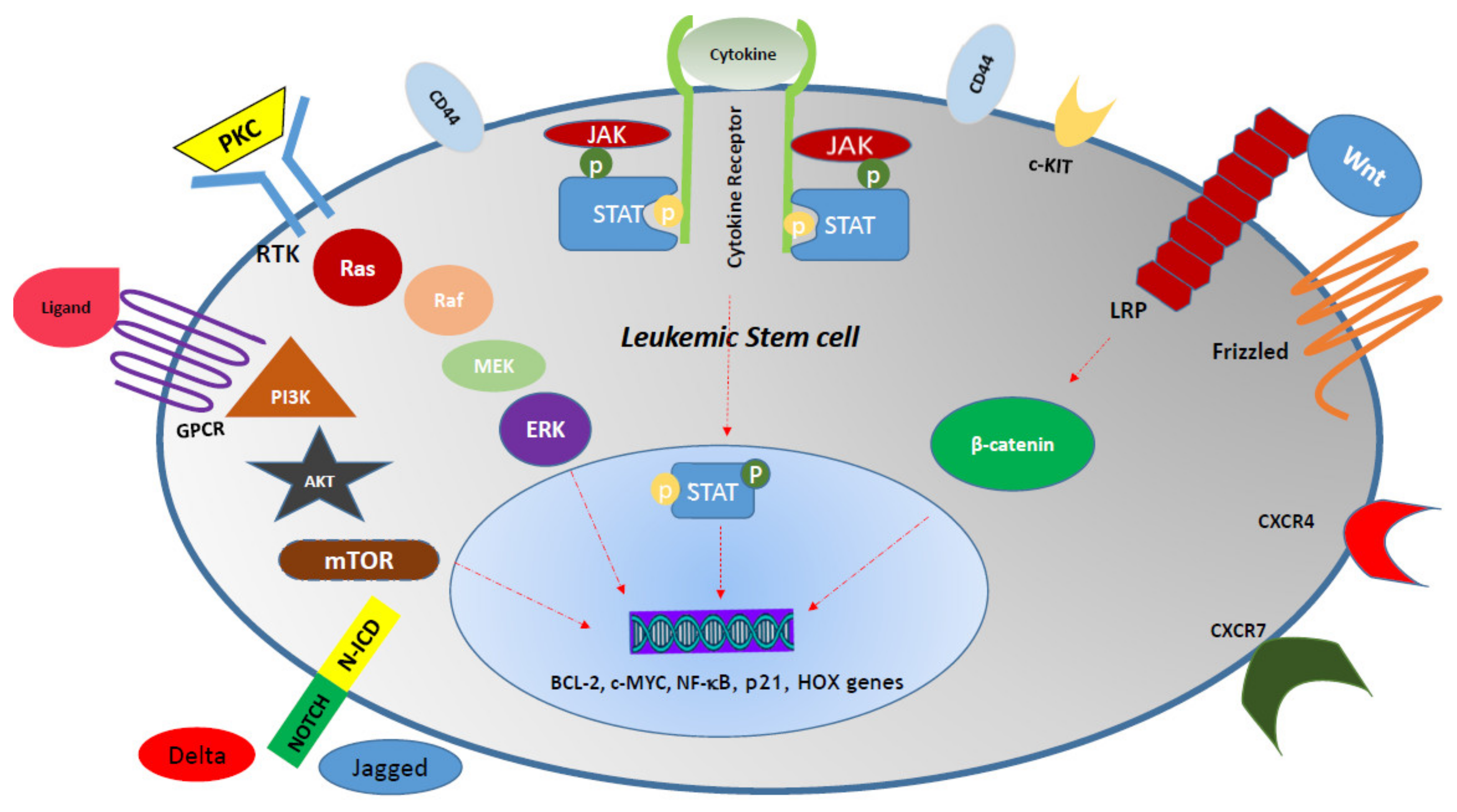
7. Metabolic Pathways, AML LSC Survival, and Resistance to Therapy
8. Concluding Thoughts
Author Contributions
Funding
Conflicts of Interest
References
- Kaushansky, K.; Zhan, H. The regulation of normal and neoplastic hematopoiesis is dependent on microenvironmental cells. Adv. Biol. Regul. 2018, 69, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Korn, C.; Méndez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.E.; Wong, J.J.L.; Rasko, J.E. Micro RNA s in myeloid malignancies. Br. J. Haematol. 2013, 162, 162–176. [Google Scholar] [CrossRef]
- Potts, K.S.; Bowman, T.V. Modeling myeloid malignancies using zebrafish. Front. Oncol. 2017, 7, 297. [Google Scholar] [CrossRef]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Papayannidis, C.; Sartor, C.; Marconi, G.; Fontana, M.C.; Nanni, J.; Cristiano, G.; Parisi, S.; Paolini, S.; Curti, A. Acute Myeloid Leukemia Mutations: Therapeutic Implications. Int. J. Mol. Sci. 2019, 20, 2721. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Kadia, T.; Ravandi, F. Identifying effective drug combinations for patients with acute myeloid leukemia. Expert Rev. Anticancer Ther. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Cancer Stat Facts: Leukemia-Acute Myeloid Leukemia (AML). 2017. Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 17 September 2021).
- Yi, M.; Li, A.; Zhou, L.; Chu, Q.; Song, Y.; Wu, K. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: Estimates based on the global burden of disease study 2017. J. Hematol. Oncol. 2020, 13, 1–16. [Google Scholar] [CrossRef]
- Puumala, S.E.; Ross, J.A.; Aplenc, R.; Spector, L.G. Epidemiology of childhood acute myeloid leukemia. Pediatr. Blood Cancer 2013, 60, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Pan, J.; Wang, S.; Hong, S.; Hong, S.; He, S. The Epidemiological Trend of Acute Myeloid Leukemia in Childhood: A Population-Based Analysis. J. Cancer 2019, 10, 4824. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Noone, A.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D. Surveillance, Epidemiology, and End Results (SEER) Program Cancer Statistics Review, 1975-2015; National Cancer Institute: Bethesda, MD, USA, 2018.
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wölfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine dose in the consolidation treatment of AML: A systematic review and meta-analysis. Blood 2017, 130, 946–948. [Google Scholar] [CrossRef] [Green Version]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Gabra, M.M.; Salmena, L. microRNAs and acute myeloid leukemia chemoresistance: A mechanistic overview. Front. Oncol. 2017, 7, 255. [Google Scholar] [CrossRef]
- van Gils, N.; Denkers, F.; Smit, L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 659253. [Google Scholar] [CrossRef] [PubMed]
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, D.; Weinberg, O.K. How I investigate acute myeloid leukemia. Int. J. Lab. Hematol. 2020, 42, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2016, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Matarraz, S.; Almeida, J.; Flores-Montero, J.; Lécrevisse, Q.; Guerri, V.; López, A.; Bárrena, S.; Van Der Velden, V.H.; Te Marvelde, J.G.; Van Dongen, J.J. Introduction to the diagnosis and classification of monocytic-lineage leukemias by flow cytometry. Cytometry B Clin. Cytom. 2017, 92, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Demirer, S.; Özdemir, H.; Şencan, M.; Marakoḡlu, I. Gingival hyperplasia as an early diagnostic oral manifestation in acute monocytic leukemia: A case report. Eur. J. Dent. 2007, 1, 111. [Google Scholar] [CrossRef] [Green Version]
- Reikvam, H.; Hatfield, K.J.; Kittang, A.O.; Hovland, R.; Bruserud, Ø. Acute myeloid leukemia with the t (8; 21) translocation: Clinical consequences and biological implications. J. Biomed. Biotechnol. 2011, 2011, 104631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Galera, P.K.; Jiang, C.; Braylan, R. Immunophenotyping of Acute Myeloid Leukemia. Methods Mol. Biol. 2019, 2032, 281–296. [Google Scholar] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidori, A.; Salvestrini, V.; Ciciarello, M.; Loscocco, F.; Visani, G.; Parisi, S.; Lecciso, M.; Ocadlikova, D.; Rossi, L.; Gabucci, E. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev. Hematol. 2014, 7, 807–818. [Google Scholar] [CrossRef]
- Bose, P.; Vachhani, P.; Cortes, J.E. Treatment of relapsed/refractory acute myeloid leukemia. Curr. Treat. Options Oncol. 2017, 18, 17. [Google Scholar] [CrossRef]
- Kampen, K.R.; Ter Elst, A.; de Bont, E.S. Vascular endothelial growth factor signaling in acute myeloid leukemia. Cell. Mol. Life Sci. 2013, 70, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts and Figures 2005; American Cancer Society: Atlanta, GA, USA, 2005. [Google Scholar]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 1937. [Google Scholar] [CrossRef] [Green Version]
- Briot, T.; Roger, E.; Thépot, S.; Lagarce, F. Advances in treatment formulations for acute myeloid leukemia. Drug Discov. Today 2018, 23, 1936–1949. [Google Scholar] [CrossRef] [Green Version]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Acheampong, D.O.; Adokoh, C.K.; Asante, D.-B.; Asiamah, E.A.; Barnie, P.A.; Bonsu, D.O.; Kyei, F. Immunotherapy for acute myeloid leukemia (AML): A potent alternative therapy. Biomed. Pharmacother. 2018, 97, 225–232. [Google Scholar] [CrossRef]
- Forghieri, F.; Comoli, P.; Marasca, R.; Potenza, L.; Luppi, M. Minimal/Measurable Residual Disease Monitoring in NPM1-Mutated Acute Myeloid Leukemia: A Clinical Viewpoint and Perspectives. Int. J. Mol. Sci. 2018, 19, 3492. [Google Scholar] [CrossRef] [Green Version]
- Kassim, A.A.; Savani, B.N. Hematopoietic stem cell transplantation for acute myeloid leukemia: A review. Hematol. Oncol. Stem Cell Ther. 2017, 10, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Craddock, C.; Raghavan, M. Which patients with acute myeloid leukemia in CR1 can be spared an allogeneic transplant? Curr. Opin. Hematol. 2019, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L. Leukemia: Management of relapse after allogeneic bone marrow transplantation. J. Clin. Oncol. 1994, 12, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Yee, G.; McGuire, T. Allogeneic bone marrow transplantation in the treatment of hematologic diseases. Clin. Pharm. 1985, 4, 149–160. [Google Scholar]
- Cerrano, M.; Itzykson, R. New treatment options for acute myeloid leukemia in 2019. Curr. Oncol. Rep. 2019, 21, 16. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Sivaloganathan, H.; Pepper, A.; Chevassut, T. Acute Myeloid Leukaemia in Its Niche: The Bone Marrow Microenvironment in Acute Myeloid Leukaemia. Curr. Oncol. Rep. 2020, 22, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Geng, Z.; Deng, T.; Wang, D.; Jiang, L. Tumor Necrosis Factor Receptor Type 1-Associated Death Domain Protein Is a Potential Prognostic Biomarker in Acute Myeloid Leukemia. Am. J. Med. Sci. 2019, 357, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Tiong, I.S.; Wei, A.H. New drugs creating new challenges in acute myeloid leukemia. Genes Chromosomes Cancer 2019, 58, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Elshoury, A.; Przespolewski, A.; Baron, J.; Wang, E.S. Advancing treatment of acute myeloid leukemia: The future of FLT3 inhibitors. Expert Rev. Anticancer Ther. 2019, 19, 273–286. [Google Scholar] [CrossRef]
- Kopmar, N.E.; Estey, E.H. New Drug Approvals in Acute Myeloid Leukemia: An Unprecedented Paradigm Shift. Clin. Adv. Hematol. Oncol. 2019, 17, 569–575. [Google Scholar]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Lovly, C.M.; Shaw, A.T. Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [Green Version]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Tabe, Y.; Konopleva, M. Role of microenvironment in resistance to therapy in AML. Curr. Hematol. Malig. Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Chopra, M.; Bohlander, S.K. The cell of origin and the leukemia stem cell in acute myeloid leukemia. Genes Chromosomes Cancer 2019, 58, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA. 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, S.; Chen, J.L. Understanding of leukemic stem cells and their clinical implications. Mol. Cancer 2017, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef]
- Camacho, V.; McClearn, V.; Patel, S.; Welner, R.S. Regulation of normal and leukemic stem cells through cytokine signaling and the microenvironment. Int. J. Hematol. 2017, 105, 566–577. [Google Scholar] [CrossRef]
- Behrmann, L.; Wellbrock, J.; Fiedler, W. The bone marrow stromal niche: A therapeutic target of hematological myeloid malignancies. Expert Opin. Ther. Targets 2020, 24, 451–462. [Google Scholar] [CrossRef]
- Lane, S.W.; Wang, Y.J.; Celso, C.L.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.; DiGiusto, D. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glait-Santar, C.; Desmond, R.; Feng, X.; Bat, T.; Chen, J.; Heuston, E.; Mizukawa, B.; Mulloy, J.C.; Bodine, D.M.; Larochelle, A. Functional niche competition between normal hematopoietic stem and progenitor cells and myeloid leukemia cells. Stem Cells 2015, 33, 3635–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-A.; Shim, J.-S.; Lee, G.-Y.; Yim, H.W.; Kim, T.-M.; Kim, M.; Leem, S.-H.; Lee, J.-W.; Min, C.-K.; Oh, I.-H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.S.; Takei, H. Transcription factor-based therapies for acute myeloid leukemia. Rinsho Ketsueki 2018, 59, 922–931. [Google Scholar]
- Karathedath, S.; Rajamani, B.M.; Musheer Aalam, S.M.; Abraham, A.; Varatharajan, S.; Krishnamurthy, P.; Mathews, V.; Velayudhan, S.R.; Balasubramanian, P. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS ONE 2017, 12, e0177227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, H.; Kobayashi, S.S. Targeting transcription factors in acute myeloid leukemia. Int. J. Hematol. 2019, 109, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Seipel, K.; Marques, M.T.; Bozzini, M.-A.; Meinken, C.; Mueller, B.U.; Pabst, T. Inactivation of the p53–KLF4–CEBPA Axis in Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Wahlin, A. Accumulating evidence for a role of p53 in multiple drug resistant Acute Myeloid Leukemia. Leuk. Lymphoma 2008, 49, 383–384. [Google Scholar] [CrossRef]
- Pan, X.-N.; Chen, J.-J.; Wang, L.-X.; Xiao, R.-Z.; Liu, L.-L.; Fang, Z.-G.; Liu, Q.; Long, Z.-J.; Lin, D.-J. Inhibition of c-Myc overcomes cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. PLoS ONE 2014, 9, e105381. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Schneeweiss, M.; Eisenwort, G.; Berger, D.; Herrmann, H.; Blatt, K.; Greiner, G.; Byrgazov, K.; Hoermann, G.; Konopleva, M. Combined targeting of STAT3 and STAT5: A novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica 2017, 102, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesbahi, Y.; Zekri, A.; Ghaffari, S.H.; Tabatabaie, P.S.; Ahmadian, S.; Ghavamzadeh, A. Blockade of JAK2/STAT3 intensifies the anti-tumor activity of arsenic trioxide in acute myeloid leukemia cells: Novel synergistic mechanism via the mediation of reactive oxygen species. Eur. J. Pharmacol. 2018, 834, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.A.; Cummings, C.L.; Korb, B.; Boaglio, S.; Oehler, V.G. Deregulated KLF4 expression in myeloid leukemias alters cell proliferation and differentiation through microRNA and gene targets. Mol. Cell. Biol. 2016, 36, 559–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safa, M.; Mousavizadeh, K.; Noori, S.; Pourfathollah, A.; Zand, H. cAMP protects acute promyelocytic leukemia cells from arsenic trioxide-induced caspase-3 activation and apoptosis. Eur. J. Pharmacol. 2014, 736, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Shankar, D.B.; Cheng, J.C.; Sakamoto, K.M. Role of cyclic AMP response element binding protein in human leukemias. Cancer 2005, 104, 1819–1824. [Google Scholar] [CrossRef]
- Mitton, B.; Chae, H.-D.; Hsu, K.; Dutta, R.; Aldana-Masangkay, G.; Ferrari, R.; Davis, K.; Tiu, B.C.; Kaul, A.; Lacayo, N. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia 2016, 30, 2302–2311. [Google Scholar] [CrossRef]
- Mueller, B.U.; Pabst, T.; Osato, M.; Asou, N.; Johansen, L.M.; Minden, M.D.; Behre, G.; Hiddemann, W.; Ito, Y.; Tenen, D.G. Heterozygous PU. 1 mutations are associated with acute myeloid leukemia. Blood 2002, 100, 998–1007. [Google Scholar] [CrossRef] [Green Version]
- Goyama, S.; Schibler, J.; Cunningham, L.; Zhang, Y.; Rao, Y.; Nishimoto, N.; Nakagawa, M.; Olsson, A.; Wunderlich, M.; Link, K.A.; et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Investig. 2013, 123, 3876–3888. [Google Scholar] [CrossRef] [Green Version]
- Darwish, N.H.; Sudha, T.; Godugu, K.; Bharali, D.J.; Elbaz, O.; El-ghaffar, H.A.A.; Azmy, E.; Anber, N.; Mousa, S.A. Novel targeted nano-parthenolide molecule against NF-kB in Acute Myeloid Leukemia. Molecules 2019, 24, 2103. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ching, Y.Q.; Chng, W.J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef]
- Asada, N.; Takeishi, S.; Frenette, P.S. Complexity of bone marrow hematopoietic stem cell niche. Int. J. Hematol. 2017, 106, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573. [Google Scholar] [CrossRef]
- Ghiaur, G.; Wroblewski, M.; Loges, S. Acute myelogenous leukemia and its microenvironment: A molecular conversation. Semin. Hematol. 2015, 52, 200–206. [Google Scholar]
- Shafat, M.S.; Gnaneswaran, B.; Bowles, K.M.; Rushworth, S.A. The bone marrow microenvironment–Home of the leukemic blasts. Blood Rev. 2017, 31, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Bakker, S.T.; Passegué, E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 2013, 41, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Ostanin, A.; Petrovskii, Y.L.; Shevela, E.Y.; Chernykh, E. Multiplex analysis of cytokines, chemokines, growth factors, MMP-9 and TIMP-1 produced by human bone marrow, adipose tissue, and placental mesenchymal stromal cells. Bull. Exp. Biol. Med. 2011, 151, 133–141. [Google Scholar] [CrossRef]
- Kondo, M.; Wagers, A.J.; Manz, M.G.; Prohaska, S.S.; Scherer, D.C.; Beilhack, G.F.; Shizuru, J.A.; Weissman, I.L. Biology of hematopoietic stem cells and progenitors: Implications for clinical application. Annu. Rev. Immunol. 2003, 21, 759–806. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Riether, C.; Schürch, C.; Ochsenbein, A. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrmann, L.; Wellbrock, J.; Fiedler, W. Acute myeloid leukemia and the bone marrow niche—Take a closer look. Front. Oncol. 2018, 8, 444. [Google Scholar] [CrossRef] [Green Version]
- Yu, V.; Scadden, D. Hematopoietic stem cell and its bone marrow niche. Curr. Top. Dev. Biol. 2016, 118, 21–44. [Google Scholar]
- Le, P.M.; Andreeff, M.; Battula, V.L. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018, 103, 1945–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangialardi, G.; Cordaro, A.; Madeddu, P. The bone marrow pericyte: An orchestrator of vascular niche. Regen. Med. 2016, 11, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Tabe, Y.; Konopleva, M. Leukemia stem cells microenvironment. In Stem Cell Microenvironments and Beyond, Springer: 2017; pp 19-32.
- Yilmaz, Ö.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Bromberg, J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 2012, 30, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The role of CXC chemokine receptors 1–4 on immune cells in the tumor microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef]
- Cho, B.-S.; Kim, H.-J.; Konopleva, M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: From bench to bedside. Korean J. Intern. Med. 2017, 32, 248. [Google Scholar] [CrossRef]
- Rashidi, A.; Uy, G.L. Targeting the microenvironment in acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2015, 10, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Gruszka, A.M.; Valli, D.; Restelli, C.; Alcalay, M. Adhesion deregulation in acute myeloid leukaemia. Cells 2019, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, M.; Bendas, G. Contribution of very late antigen-4 (VLA-4) integrin to cancer progression and metastasis. Cancer Metastasis Rev. 2015, 34, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Dong-Feng, Z.; Ting, L.; Yong, Z.; Cheng, C.; Xi, Z.; Pei-Yan, K. The TPO/c-MPL pathway in the bone marrow may protect leukemia cells from chemotherapy in AML patients. Pathol. Oncol. Res. 2014, 20, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Wang, L.L.; Ma, D.C. Effects of vascular endothelial growth factors and their receptors on megakaryocytes and platelets and related diseases. Br. J. Haematol. 2018, 180, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Baaten, B.J.; Li, C.-R.; Deiro, M.F.; Lin, M.M.; Linton, P.J.; Bradley, L.M. CD44 regulates survival and memory development in Th1 cells. Immunity 2010, 32, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro-and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Rep. 2016, 6, 897–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boraschi, D.; Tagliabue, A. The interleukin-1 receptor family. Sem. Immunol. 2013, 25, 394–407. [Google Scholar]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.S.; Ghoreschi, K. The interleukin-1 family. In Regulation of Cytokine Gene Expression in Immunity and Diseases; Springer: Berlin, Germany, 2016; pp. 21–29. [Google Scholar]
- Estrov, Z.; Kurzrock, R.; Talpaz, M. Role of Interleukin-1 Inhibitory Molecules in Therapy of Acute and Chronic Myelogenous Leukemia. Leuk. Lymphoma 1993, 10, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; del Mar Arriero, M.; Villatoro, A. Interleukin-1β as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Maleknia, M.; Valizadeh, A.; Pezeshki, S.; Saki, N. Immunomodulation in leukemia: Cellular aspects of anti-leukemic properties. Clin. Transl. Oncol. 2020, 22, 1–10. [Google Scholar] [CrossRef]
- Medler, J.; Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Z.; Zhou, J. Tumor necrosis factor α in the onset and progression of leukemia. Exp. Hematol. 2017, 45, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Londino, J.D.; Gulick, D.L.; Lear, T.B.; Suber, T.L.; Weathington, N.M.; Masa, L.S.; Chen, B.B.; Mallampalli, R.K. Post-translational modification of the interferon-gamma receptor alters its stability and signaling. Biochem. J. 2017, 474, 3543–3557. [Google Scholar] [CrossRef] [Green Version]
- Kursunel, M.A.; Esendagli, G. The untold story of IFN-γ in cancer biology. Cytokine Growth Factor Rev. 2016, 31, 73–81. [Google Scholar] [CrossRef]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors—distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef]
- Nursal, A.F.; Pehlivan, M.; Sahin, H.H.; Pehlivan, S. The Associations of IL-6, IFN-c, TNF-a, IL-10, and TGF-b1 Functional Variants with Acute Myeloid Leukemia in Turkish Patients. Genet. Test. Mol. Biomark. 2016, 20, 544–551. [Google Scholar] [CrossRef]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Ma, Z.; Cheng, Y.; Hu, W.; Deng, C.; Jiang, S.; Li, T.; Chen, F.; Yang, Y. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol. Cancer 2018, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.-J.; Chang, W.-S.; Hsu, H.-F.; Ji, H.-X.; Hsiao, C.-L.; Tsai, C.-W.; Yeh, S.-P.; Chen, C.-M.; Bau, D.-T. Significant association of interleukin-10 polymorphisms with childhood leukemia susceptibility in Taiwan. In Vivo 2016, 30, 265–269. [Google Scholar]
- Carson, W.E.; Lindemann, M.J.; Baiocchi, R.; Linett, M.; Tan, J.C.; Chou, C.-C.; Narula, S.; Caligiuri, M. The functional characterization of interleukin-10 receptor expression on human natural killer cells. Blood 1995, 85, 3577–3585. [Google Scholar] [CrossRef] [Green Version]
- Bruserud, Ø.; Tore, B.G.; Brustugun, O.T.; Bassøe, C.; Nesthus, I.; Espen, P.A.; Bühring, H.; Pawelec, G. Effects of interleukin 10 on blast cells derived from patients with acute myelogenous leukemia. Leukemia 1995, 9, 1910–1920. [Google Scholar]
- Wu, Y.; Su, M.; Zhang, S.; Cheng, Y.; Liao, X.Y.; Lin, B.Y.; Chen, Y.Z. Abnormal expression of TGF-beta type II receptor isoforms contributes to acute myeloid leukemia. Oncotarget 2017, 8, 10037–10049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, W.P. Interleukin-1 Receptor Antagonist. Adv. Immunol. 1993, 54, 167–227. [Google Scholar]
- Arend, W.P.; Guthridge, C.J. Biological role of interleukin 1 receptor antagonist isoforms. Ann. Rheum. Dis. 2000, 59, i60–i64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Li, C.; Zhang, X.; Xiong, H.; Deng, H. The Role of IL-35 in Regulating Tumor Immunity. Adv. Mod. Oncol. Res. 2018, 4, 8–16. [Google Scholar]
- Ok, C.Y.; Young, K.H. Checkpoint inhibitors in hematological malignancies. J. Hematol. Oncol. 2017, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandsemb, E.N.; Kim, T.K.; Zeidan, A.M. Will deeper characterization of the landscape of immune checkpoint molecules in acute myeloid leukemia bone marrow lead to improved therapeutic targeting? Cancer 2019, 125, 1410. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.-i.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell 2015, 17, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannopoulos, K. Targeting immune signaling checkpoints in acute myeloid leukemia. J. Clin. Med. 2019, 8, 236. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Dias, S.; Choy, M.; Alitalo, K.; Rafii, S. Vascular endothelial growth factor (VEGF)–C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood 2002, 99, 2179–2184. [Google Scholar] [CrossRef]
- Yang, X.; Sexauer, A.; Levis, M. Bone marrow stroma-mediated resistance to FLT 3 inhibitors in FLT 3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br. J. Haematol. 2014, 164, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Mak, P.Y.; Wang, X.; Tao, W.; Ruvolo, V.; Mak, D.; Mu, H.; Burks, J.K.; Andreeff, M. An ARC-Regulated IL1β/Cox-2/PGE2/β-Catenin/ARC Circuit Controls Leukemia–Microenvironment Interactions and Confers Drug Resistance in AML. Cancer Res. 2019, 79, 1165–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, C.; Jia, G.; Jingjing, Z.; Yapeng, H.; Zhi, L.; Guanghui, X. miR-486 is involved in the pathogenesis of acute myeloid leukemia by regulating JAK-STAT signaling. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 394, 177–187. [Google Scholar] [CrossRef]
- Venugopal, S.; Bar-Natan, M.; Mascarenhas, J.O. JAKs to STATs: A tantalizing therapeutic target in acute myeloid leukemia. Blood Rev. 2020, 40, 100634. [Google Scholar] [CrossRef] [PubMed]
- Takam Kamga, P.; Collo, G.D.; Resci, F.; Bazzoni, R.; Mercuri, A.; Quaglia, F.M.; Tanasi, I.; Delfino, P.; Visco, C.; Bonifacio, M. Notch Signaling Molecules as Prognostic Biomarkers for Acute Myeloid Leukemia. Cancers 2019, 11, 1958. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhao, Y.; Xu, M.; Dai, Q.; Meng, W.; Yang, J.; Qin, R. Activation of Notch signal pathway is associated with a poorer prognosis in acute myeloid leukemia. Med. Oncol. 2011, 28, 483–489. [Google Scholar] [CrossRef]
- Terao, T.; Minami, Y. Targeting hedgehog (Hh) pathway for the acute myeloid leukemia treatment. Cells 2019, 8, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Knight, T.; Irving, J.A. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front. Oncol. 2014, 4, 160. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [Green Version]
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt signalling in acute myeloid leukaemia. Cells 2019, 8, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashihara, E.; Takada, T.; Maekawa, T. Targeting the canonical Wnt/β-catenin pathway in hematological malignancies. Cancer Sci. 2015, 106, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Miraki-Moud, F.; Anjos-Afonso, F.; Hodby, K.A.; Griessinger, E.; Rosignoli, G.; Lillington, D.; Jia, L.; Davies, J.K.; Cavenagh, J.; Smith, M.; et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 13576–13581. [Google Scholar] [CrossRef] [Green Version]
- Waclawiczek, A.; Hamilton, A.; Rouault-Pierre, K.; Abarrategi, A.; Albornoz, M.G.; Miraki-Moud, F.; Bah, N.; Gribben, J.; Fitzgibbon, J.; Taussig, D.; et al. Mesenchymal niche remodeling impairs hematopoiesis via stanniocalcin 1 in acute myeloid leukemia. J. Clin. Investig. 2020, 130, 3038–3050. [Google Scholar] [CrossRef]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-β pathways impinge on Hedgehog signaling complexity: An open window on cancer. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.Y.; Jordan, C.T. Leukemia stem cells and microenvironment: Biology and therapeutic targeting. J. Clin. Oncol. 2011, 29, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Bakker, E.; Qattan, M.; Mutti, L.; Demonacos, C.; Krstic-Demonacos, M. The role of microenvironment and immunity in drug response in leukemia. Biochim. Biophys. Acta 2016, 1863, 414–426. [Google Scholar] [CrossRef]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Grant, S.; Saleiro, D.; Crispino, J.D.; Hijiya, N.; Giles, F.; Platanias, L.; Eklund, E.A. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol. Genet. Metab. 2015, 114, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrigoni, E.; Del Re, M.; Galimberti, S.; Restante, G.; Rofi, E.; Crucitta, S.; Baratè, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Concise review: Chronic myeloid leukemia: Stem cell niche and response to pharmacologic treatment. Stem Cells Transl. Med. 2018, 7, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, D.; Garcia-Fernandez, M.; Sanchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martin-Perez, D.; Lopez, J.A.; Costa, A.S.H.; Tronci, L.; Nikitopoulou, E.; et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab. 2020, 32, 829–843.e9. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Sanchez-Aguilera, A.; Martin-Perez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntion, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Kouzi, F.; Zibara, K.; Bourgeais, J.; Picou, F.; Gallay, N.; Brossaud, J.; Dakik, H.; Roux, B.; Hamard, S.; Le Nail, L.-R. Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene 2020, 39, 1198–1212. [Google Scholar] [CrossRef]
- Nehrbas, J.; Butler, J.T.; Chen, D.-W.; Kurre, P. Extracellular vesicles and chemotherapy resistance in the AML microenvironment. Front. Oncol. 2020, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudgapalli, N.; Nallasamy, P.; Chava, H.; Chava, S.; Pathania, A.S.; Gunda, V.; Gorantla, S.; Pandey, M.K.; Gupta, S.C.; Challagundla, K.B. The role of exosomes and MYC in therapy resistance of acute myeloid leukemia: Challenges and opportunities. Mol. Aspects Med. 2019, 70, 21–32. [Google Scholar] [CrossRef]
- Pando, A.; Reagan, J.L.; Quesenberry, P.; Fast, L.D. Extracellular vesicles in leukemia. Leuk. Res. 2018, 64, 52–60. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Whiteside, T.L. Exosomes in acute myeloid leukemia inhibit hematopoiesis. Curr. Opin. Hematol. 2018, 25, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Minhajuddin, M.; Krug, A.; Pei, S.; Chou, C.H.; Culp-Hill, R.; Ponder, J.; De Bloois, E.; Schniedewind, B.; Amaya, M.L.; et al. The Hepatic Microenvironment Uniquely Protects Leukemia Cells through Induction of Growth and Survival Pathways Mediated by LIPG. Cancer Discov. 2021, 11, 500–519. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740 e4. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Corydon, T.J.; Bross, P.; Holst, H.U.; Neve, S.; Kristiansen, K.; Gregersen, N.; Bolund, L. A human homologue of Escherichia coli ClpP caseinolytic protease: Recombinant expression, intracellular processing and subcellular localization. Biochem. J. 1998, 331, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Reddy, M.M.; Fernandes, M.S.; Salgia, R.; Levine, R.L.; Griffin, J.D.; Sattler, M. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 2011, 25, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, R.; Arora, D.; Bauer, R.; Stopp, S.; Muller, J.P.; Heinrich, T.; Bohmer, S.A.; Dagnell, M.; Schnetzke, U.; Scholl, S.; et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/ PTPRJ. Blood 2012, 119, 4499–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, C.C.; Fiol, C.R.; Baker, M.J.; Nadal-Melsio, E.; Yebra-Fernandez, E.; Bicalho, L.; Chowdhury, A.; Albert, M.; Reid, A.G.; Claudiani, S.; et al. Identification of genetic targets in acute myeloid leukaemia for designing targeted therapy. Br. J. Haematol. 2021, 192, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Adane, B.; Ye, H.; Khan, N.; Pei, S.; Minhajuddin, M.; Stevens, B.M.; Jones, C.L.; D’Alessandro, A.; Reisz, J.A.; Zaberezhnyy, V.; et al. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep. 2019, 27, 238–254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Number | Genomic Classification of AML | Rate |
|---|---|---|
| 1 | NPM1-mutated AML | 27% |
| 2 | AML with mutated chromatin and/or RNA-splicing genes which include (RUNX1, MLL, SRSF2, ASXL1, STAG2) | 18% |
| 3 | AML with TP53 mutations and/or chromosomal aneuploidy | 13% |
| 4 | AML with inv (16) (p13.1q22) or t(16;16) (p13.1; q22); CBFB–MYH11 | 5% |
| 5 | AML with biallelic CEBPA mutations | 4% |
| 6 | AML with t (15;17) (q22; q12); PML–RARA | 4% |
| 7 | AML with t (8;21) (q22; q22); RUNX1–RUNX1T1 | 4% |
| 8 | AML with MLL fusion genes; t(x;11) (x; q23) | 3% |
| 9 | AML with inv (3) (q21q26.2) or t (3;3) (q21; q26.2); GATA2, MECOM (EVI1) | 1% |
| 10 | AML with IDH2R172 mutations and no other class-defining lesions | 1% |
| 11 | AML with t (6;9) (p23; q34); DEK–NUP214 | 1% |
| Function | Name | Target | Mechanism | FDA Approved | Refs |
|---|---|---|---|---|---|
| IDH1 inhibitor | Ivosidenib | IDH1 | Myeloblast differentiation induction through isocitrate dehydrogenase 1 (IDH1) inhibition and 2-hydroxyglutarate (2-HG) blockage | Yes | [46] |
| IDH2 inhibitor | Enasidenib | IDH2 | Myeloblast differentiation induction through isocitrate dehydrogenase 2 (IDH2) inhibition and 2-HG blockage | Yes | [46] |
| FLT3 inhibitor | Gilteritinib | FLT3-TKD |
| Yes | [47] |
| Quizartinib | FLT3-ITD |
| No | [47,48] | |
| Antibody drug conjugate (ABDC) | Gemtuzumab ozogamicin (GO) | CD33 | Anti-CD33 monoclonal antibody conjugated with cytotoxin | Yes | [46] |
| Selective E-selectin antagonist | Uproleselan (GMI-1271) | E-selectin | Chemotherapy sensitizer | No | [46] |
| TF | Effects | Therapeutics | Refs |
|---|---|---|---|
| NF-E2 related factor-2 (NRF2) | 1. Reactive oxygen species (ROS) neutralization 2. Chemotherapy resistant 3. Antioxidant response element (ARE) up-regulation | Brusatol | [70,71] |
| CCAAT/enhancer binding protein alpha (C/EBPα) | 1. Tumor suppressor 2. Activated by TP53-KLF4 3. Down-regulated in AML due to TP53 down-regulation 4. Drug resistance 5. CSF3R, MPO, and ELANE up-regulation | ICCB280 NSC23766 OICR-9429 C/EBPA-siRNA | [71,72] |
| TP53 | 1. Tumor suppressor 2. Down-regulated in AML 3. Severe drug resistance 4. BAX and CDKN1A up-regulation | PRIMA-1 PRIMA-1MET SAR405838 AM-8553 AMG232 MK-8242 DS-3032b CGM097 | [71,73] |
| c-MYC | Up-regulated in AML 1. Leukemic cells proliferation enhancement 2. Chemotherapy resistance 3. BCL-2, CDKN1A and CCND1 up-regulation | IIA6B17 NY2267 MYRA-A 10074-G5 Mycro3 JQ-1 | [71,74] |
| STAT3 | Up-regulated in AML 1. Chemotherapy resistance 2. Pro-survival 3. Proliferation enhancement 4. Anti-apoptotic 5. BCL-2, BCL-XL, Mc1-1, cyclin D1, and c-MYC up-regulation | Galiellalactone | [71,75,76] |
| Krüppel-like factor 4 (KLF4) | 1. Tumor suppressor 2. Cell cycle arrest by CDKN1A suppression 3. Down-regulated in AML (NPM1-mutant) 4. Down-regulation is correlated with chemoresistance 5. P21, P27 up-regulation 6. Suppressed by metal-regulatory transcription factor 1 (MTF-1) | APTO-253 | [69,71,72,77] |
| cAMP response element-binding protein (CREB) | Up-regulated in AML 1. Pro-survival 2. Anti-apoptotic 3. Chemotherapy resistance 4. Up-regulates BCL-2 5. Up regulates transcription of numerous gens such as c-fos, junB, and egr-1 | STF-017794 STF-038533 STF-046536 STF-046728 STF-055910 | [69,71,78,79,80] |
| PU.1 | Up-regulated in AML 1. Up-regulates CSF1R, IL7R, CD11b, M-CSFR, GM-CSFR, G-CSFR 2. Hematopoiesis defect in AML | DB2313 DB2115 DB1976 | [71,81] |
| Runt-related transcription factor 1 (RUNX1) | Up-regulated in AML 1. Up-regulates C/EBPα, PU.1, and cell cycle progression 2. Down-regulates TP53 | Chb-M Chb-50 | [71,82] |
| NF-κB | Up-regulated in AML Poor prognostic factor 1. Up-regulates BCL-2 and BCL-XL 2. Pro-survival 3. Feed-back positive effect with TNF-α in AML | Bortezomib (FDA) | [83,84,85] |
| Receptor | Cell(s) | Ligand | Ligand Source | Normal Function | Expression in AML | Refs |
|---|---|---|---|---|---|---|
| CXCR4 | 1. Most immune cells 2. AML leukemic cells | SDF-1 (CXCL12) | 1. MSC 2. Leukemic cells | 1.Chemotaxis 2. Migration 3. Pro-survival | 1. Chemotherapy resistance 2. Pro-survivalthrough PI3K/AKT and MEK/ERK activation | [44,95,102,103,104] |
| VCAM-1 (CD106, fibronectin) | Stromal cells | Very late antigen 4 (VLA-4) | 1. HSC and hematopoietic progenitors 2. Monocytes (MO) 3. Leukemic cells 4. Myeloid cells 5. Immature dendritic cells 6. Neutrophils 7. Eosinophils 8. Immature mast cells 9. Endothelial cells | 1. Adhesion 2. Pro-survival 3. Proliferation | 1. Pro-survival 2. Proliferative 3. NF-κB activation 4. Chemotherapy resistance 5. MRD and relapse | [62,95,105,106] |
| RANK | NK cell | RANKL or Tumor necrosis factor-receptor (TNF-R) | 1. Stromal cells 2. Osteoblast 3. Activated lymphocyte 4. Leukemic cells | Bone remodeling | NK cell inhibitory | [44] |
| c-MPL (CD 110) | 1. HSC 2. Megakaryocyte (MK) 3. Chronic myeloid leukemia (CML) 4. AML leukemic cells | TPO | 1. Liver 2. Kidney | 1. HSC quiescence 2. Thrombopoiesis | Chemotherapy resistance | [87,107] |
| Vascular endothelial growth factor receptor(VEGFR) | 1. MO 2. MQ 3. Vascularendothelial cells (VEC) 4. Lymphoid endothelial cells (LEC) 5. HSC | 1. VEGF 2. PIGF | 1. Stromal cell 2. MK 3. HSC 4. Leukemic cells | 1. GM-CSF stimulation 2. Angiogenesis 3. Metabolichomeostasis 4. Proliferation 5. Migration 6. Tubulogenesis | 1. Anti-apoptotic 2. Chemotherapy resistance | [32,95,108] |
| E-Selectin | 1. Endothelial cells 2. Stromal cell | CD44 | 1. HSC and Hematopoietic progenitors 2. T cells 3. Leukemic stem cells 4. Stromal cells | 1. HSC pro-survival 2. Proliferation of HSCs | 1. E-selectin: chemotherapy resistance 2. CD44: Pro-survival | [95,104,105,109] |
| IL-1R1 | 1. Most hematopoietic and non-hematopoietic cells 2. AML leukemic cells | IL-1β | 1. Myeloid lineage 2. Leukemic cells 3. EC 4. MSC 5. MQ | 1. Pro-inflammatory 2. Hematopoiesis regulation | 1. Pro-survival 2. Pro-proliferative 3. Sometimes feedback positive 4. Association with endogenous IL-1β related to apoptosis resistance | [110,111,112,113,114,115,116] |
| TNFαRI (p55 or p60) | A broad spectrum of different cell types like AML cells | TNF-α | 1. CD8/ CD4 T cell 2. NKT cells 3. Neutrophils 4. Macrophage 1 (MQ1) 5. LSCs 6. MSCs | Pro-inflammatory | 1. Pro-survival 2. Chemotherapy resistance 3. NF-κB activation | [44,110,113,117,118,119,120] |
| IFNGR1,2 | 1. Widelydistributed on various cell types 2. LSCs | IFN-ϒ | Most immune cells | Pro-inflammatory | 1. Anti-leukemic 2. Anti-proliferative 3. Antigen presentation through MHC I/II augment 4. Nitric oxide (NO) and reactive oxygen species (ROS) mediators, NADPH, and inducible nitric oxide synthase (INOS) production | [110,118,121,122,123] |
| IL-10R | 1. AML leukemic cells 2. T cells 3. B cells 4. NK cells 5. Epithelial cells 6. Endothelial cells 7. Plasmacytoid DCs 8. Peripheral blood mononuclear cells (PBMCs) | IL-10 | 1. T helper 2 (TH 2) 2. BM-MSCs 3. Macrophage 2 (MQ2) 4. T-reg 5. B cells 6. MO 7. Thymocytes | Anti-inflammatory TH1 suppressor | 1. Growth arrest-specific gene 6 (Gas6) up-regulation 2. Pro-survival 3. Chemotherapy resistance | [118,123,124,125,126,127,128,129] |
| TGF-βR | 1. T cell 2. Hematopoietic progenitor cells 3. AML leukemic cells | TGF-β | 1. T-reg 2. MQ2 3. MSC 4. Endothelial cells 5. Platelets 6. PBMCs | 1. Anti-inflammatory 2. Proliferation 3. Migration 4. Pro-survival 5. Growth and differentiation inhibition of hematopoietic progenitor cells | 1. Anti-proliferative 2. IL-1β, IL-6, GM-CSF, and granulocyte colony-stimulating factor (G-CSF) production 3. Reduction in AML | [110,118,126,130] |
| IL1R1 | 1. Most hematopoietic and non-hematopoietic cells 2. AML leukemic cells | IL-1Ra | 1. MQ 2 2. MO 4. Neu 6. Fibroblasts 7. Chondrocytes | 1. Anti-inflammatory 2. IL-1 antagonist | Leukemic cell colonization inhibitor | [110,112,131,132] |
| IL-35R | 1. Effector T cells 2. CD4+ T-reg 3. AML leukemic cells | IL-35 | 1. T-reg 2. DCs 3. B-reg 4. sometimes in endothelial cells, monocytes and smooth muscle cells | 1. Anti-inflammatory 2. Inhibits T cell proliferation 3. Transformation of T cells to iTreg | 1. Anti-apoptotic 2. Proliferation 3. Weak prognosis 4. AML progression | [110,118,133] |
| PD1 (CD279) | Lymphocytes | Programmed death-ligand 1 (PDL1) (CD274) (B7-H1) | 1. T-reg 2. Follicular T cells (FTC) 3.MQ 4. Dendritic cell (DC) 5. placental syncytiotrophoblasts 6. MO 7. AML leukemic cells | T cell activation and proliferation inhibitor | 1. Pro-survival 2. Weak prognosis | [118,134] |
| Lymphocyte activation gene-3 (LAG3) | T cell | MHC II | APCs | T cells inhibitory | 1. Correlation with programmed death-1 (PD1) 2. Increased activity of leukemic cells | [118,135] |
| Galectin-9 (Gal-9) | 1. AML LSC 2. Lymphocytes 3. Spleen 4. Thymus | T-cell immunoglobin mucin-3 (TIM-3) | 1. AML leukemic cells 2. MO 3. DC 4. Some of T cells 5. NK cells 6. Myeloid pre-leukemic progenitors Not in normal HSCs | 1. TH1 inhibitory 2. DC maturation 3. TNF-α secretion from monocytes 4. Innate immune system activation | Strong self-renewal signaling through TIM-3/Gal-9 autocrine loop, NF-κβ and β-catenin signaling Up-regulated in pre-leukemic disorders | [136] |
| Cytotoxic T-lymphocyte antigen-4 (CTLA-4) or (CD152) | 1. T cells 2. AML leukemic cells | Β7-1 Β7-2 | Antigen-presenting cells (APCs) | T-cell inhibitory and tolerance induction | 1. AML relapse and MRD 2. Immune evasion Blockage leads to sensitivity to cytotoxic T lymphocytes (CTL) | [134,137] |
| Signaling Pathway | Leukemic Effect | Mechanism | Therapeutics | Activator Ligand (L) Receptor (R) | Mediators (M) Target (T) | Refs |
|---|---|---|---|---|---|---|
| JAK/STAT | Chemo-therapy resistance | 1. Proliferation 2. Pro-survival | 1. Ruxolitinib (FDA) 2. Ruxolitinib 3. Pacritinib 4. Lestaurtinib 5. Fedratinib 6. Momelotinib | L: TPO/MPL/G-CSF R: Cytokine receptor superfamily | M: JAK2, STAT3, STAT5, TYK2 T: p21, Mcl-1, PIM1, BCL-2, BCL-XL | [142,143] |
| Notch1 | 1. Poor prognosis 2. Chemotherapy resistance | 1. Rb phosphorylation 2. C-MYC and BCL-2 up-regulation 3. Pro-survival 4. Proliferation 5. Connection to Delta-1 leads to NF-κB pathway activation | GSIs (GSI-IX and GSI-XII) | L: Deltalike1,4 Jagged1 R: NOTCH1 | M: Notch intracellular domain of Notch (N-ICN) T: 1. CSL activity Hes family: HES1, HES5 Hes-related repressor proteins (Herps) family: HERP2 2. DELTEX1 | [144,145] |
| Hedgehog (Hh) | 1. Poor prognosis 2. Chemotherapy resistance | Activated in AML through GLI1 and SMO up-regulation | 1. LDE225 (Sonidegib) 2. PF-04449913 (Glasdegib) 3. Vismodegib (GDC-0449) 4. BMS-833923 (XL139) 5. GANT-61 | L: Hh proteins R: PTCH1 and SMO | M: GLI1 T: BCL-2, SNAIL, RAS, TGF-β, c-MYC | [146] |
| Ras/Raf/MEK/ERK | 1. Chemotherapy resistance 2. Leukemic cell survival | 1. Anti-apoptotic 2. Pro-survival through Raf-1 downstream molecule phosphorylation | 1. L-779,450 2. ZM 336372 3. Bay 43-9006 4. Geldanamycin 5. Coumermycin 5. Dasatinib 6. PD98059 7. U0126 8. PD184352 9. ARRY142886 | L: 1. Ras proteins (Ha-Ras, N-Ras, Ki-Ras 4A, Ki-Ras 4B) 2. Protein kinase C (PKC) R: Receptor tyrosine kinases (RTK) | M: Raf-1, A-Raf and B-Raf T: 1. Transcription factors, including Ets-1, c-Jun and c-MYC CREB NF-κB 2. Bad, Bim, Mcl-1, caspase 9, BCL-2 | [147,148] |
| Phosphatidy-linositol 3-kinase (PI3K)/Akt/mTOR | 1. Poor prognosis 2. Chemotherapy resistance | 1. Glycolysis up-regulation 2. Proliferation 3. Pro-survival | 1. Ridaforolimus 2. Sirolimus (Rapamycin) 3. Everolimus 4. Temsirolimus | L: Wide variety of extracellular stimuli R: G-protein-coupled receptors (GPCRs) RTK, various integrins, B and T cell receptors | M: Akt, mTOR T: p70S6K, S6RP, 4EBP1 | [53,149] |
| Wnt | 1. Poor prognosis 2. Chemotherapy resistance | 1. LSC self- renewal 2. AML progression | 1. Celecoxib 2. CWP232291 3. LY2090314 4. PRI-724 5. Sulindac | L: Wnt1 Wnt3a, PCP R: Frizzled (FZD) and lipoprotein receptor-related protein (LRP) | M: β-catenin, Ca2+ T: cyclin D1, c-MYC, Hox genes, MLL/ENL | [150,151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolandi, S.M.; Pakjoo, M.; Beigi, P.; Kiani, M.; Allahgholipour, A.; Goudarzi, N.; Khorashad, J.S.; Eiring, A.M. A Role for the Bone Marrow Microenvironment in Drug Resistance of Acute Myeloid Leukemia. Cells 2021, 10, 2833. https://doi.org/10.3390/cells10112833
Bolandi SM, Pakjoo M, Beigi P, Kiani M, Allahgholipour A, Goudarzi N, Khorashad JS, Eiring AM. A Role for the Bone Marrow Microenvironment in Drug Resistance of Acute Myeloid Leukemia. Cells. 2021; 10(11):2833. https://doi.org/10.3390/cells10112833
Chicago/Turabian StyleBolandi, Seyed Mohammadreza, Mahdi Pakjoo, Peyman Beigi, Mohammad Kiani, Ali Allahgholipour, Negar Goudarzi, Jamshid S. Khorashad, and Anna M. Eiring. 2021. "A Role for the Bone Marrow Microenvironment in Drug Resistance of Acute Myeloid Leukemia" Cells 10, no. 11: 2833. https://doi.org/10.3390/cells10112833