
Etoposide Triggers Cellular Senescence by Inducing Multiple Centrosomes and Primary Cilia in Adrenocortical Tumor Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. Flow Cytometry
2.3. EdU Incorporation Assay
2.4. Immunofluorescence Microscopy
2.5. Western Blotting Assay
2.6. Antibodies
2.7. RNA Interference (RNAi)
2.8. Mitotic Index
2.9. Statistical Analysis
3. Results
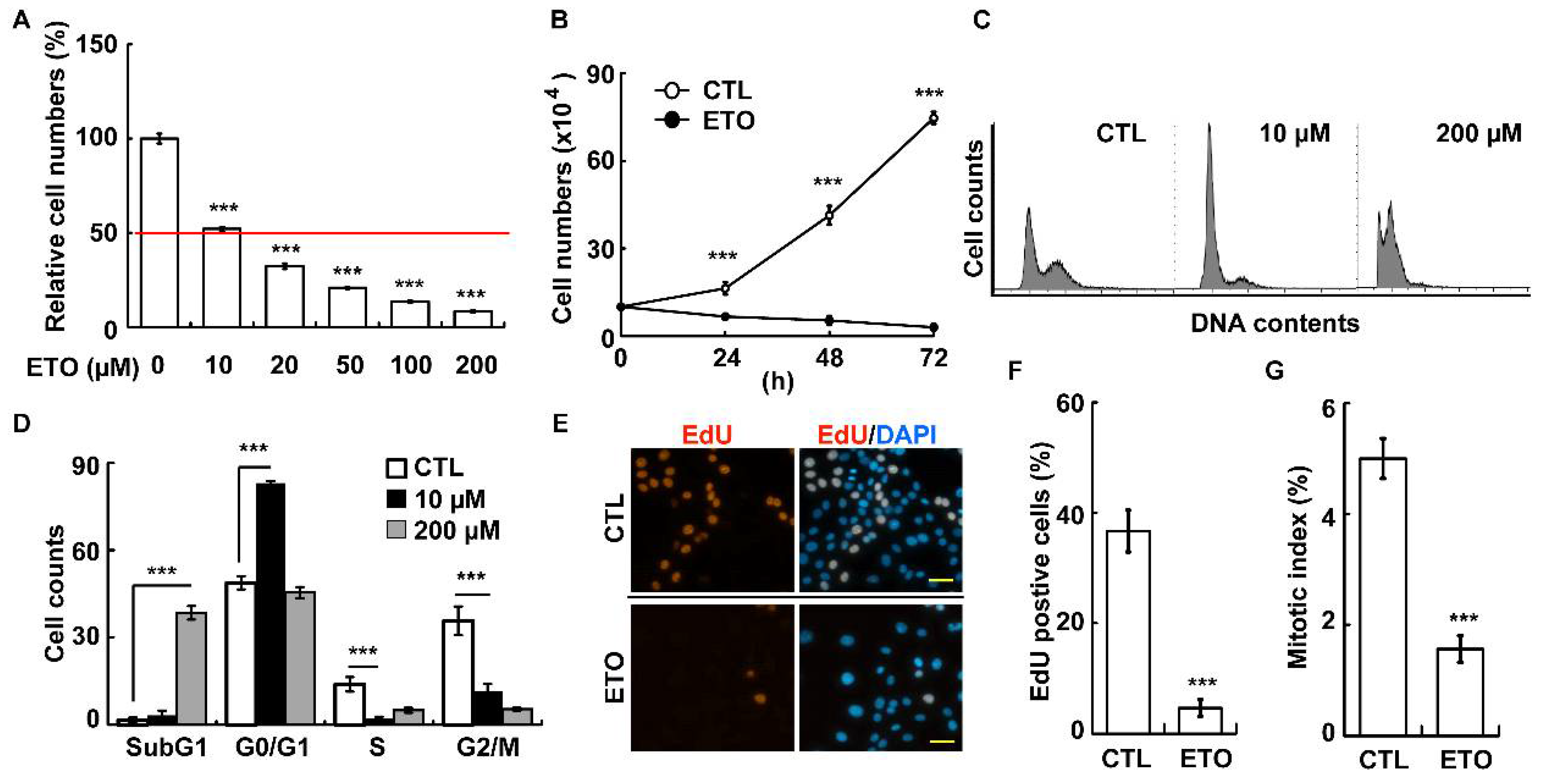
3.1. Etoposide Inhibits Adrenocortical Tumor Cell Growth
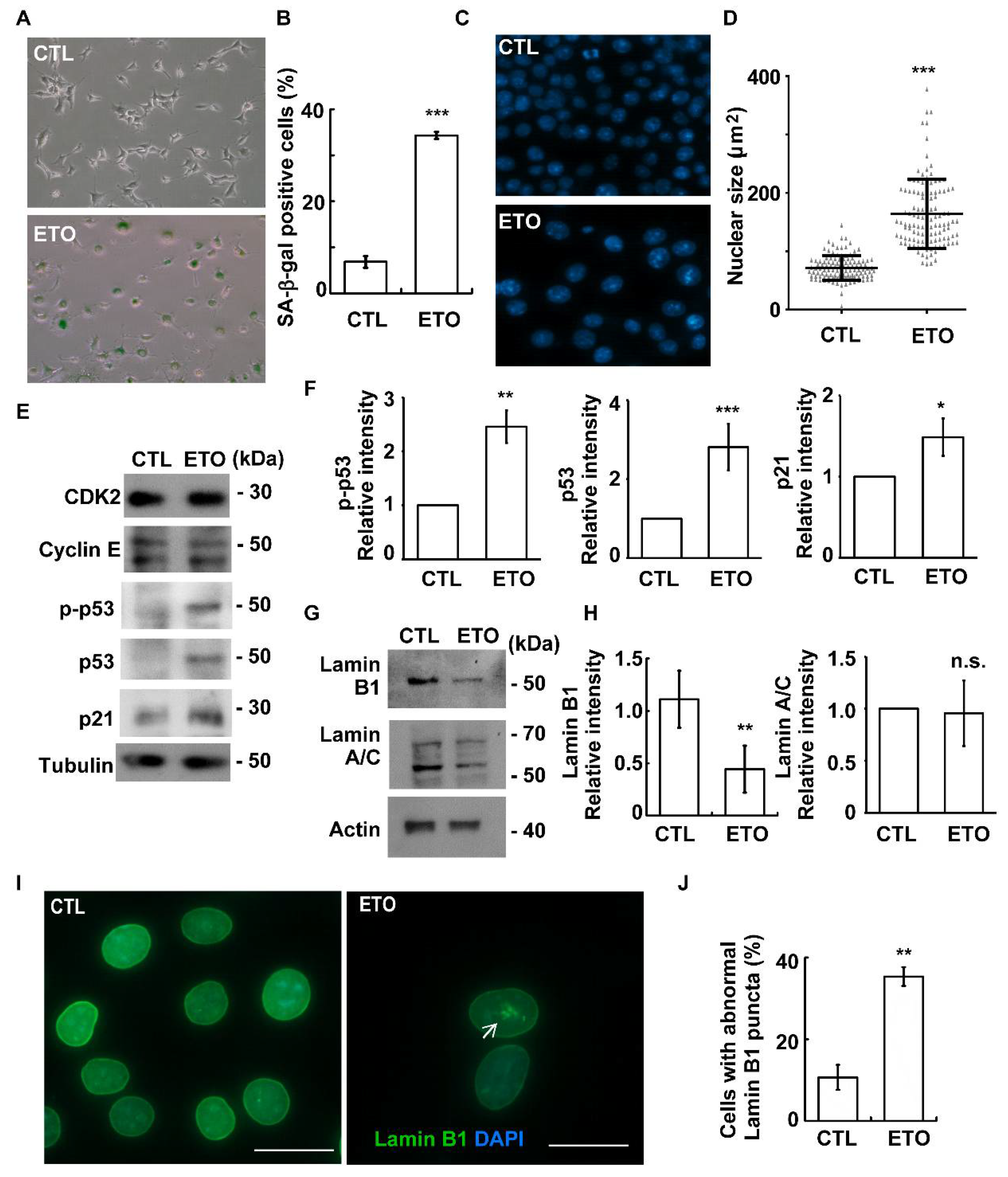
3.2. Etoposide Induces Cellular Senescence in Adrenocortical Tumor Cells
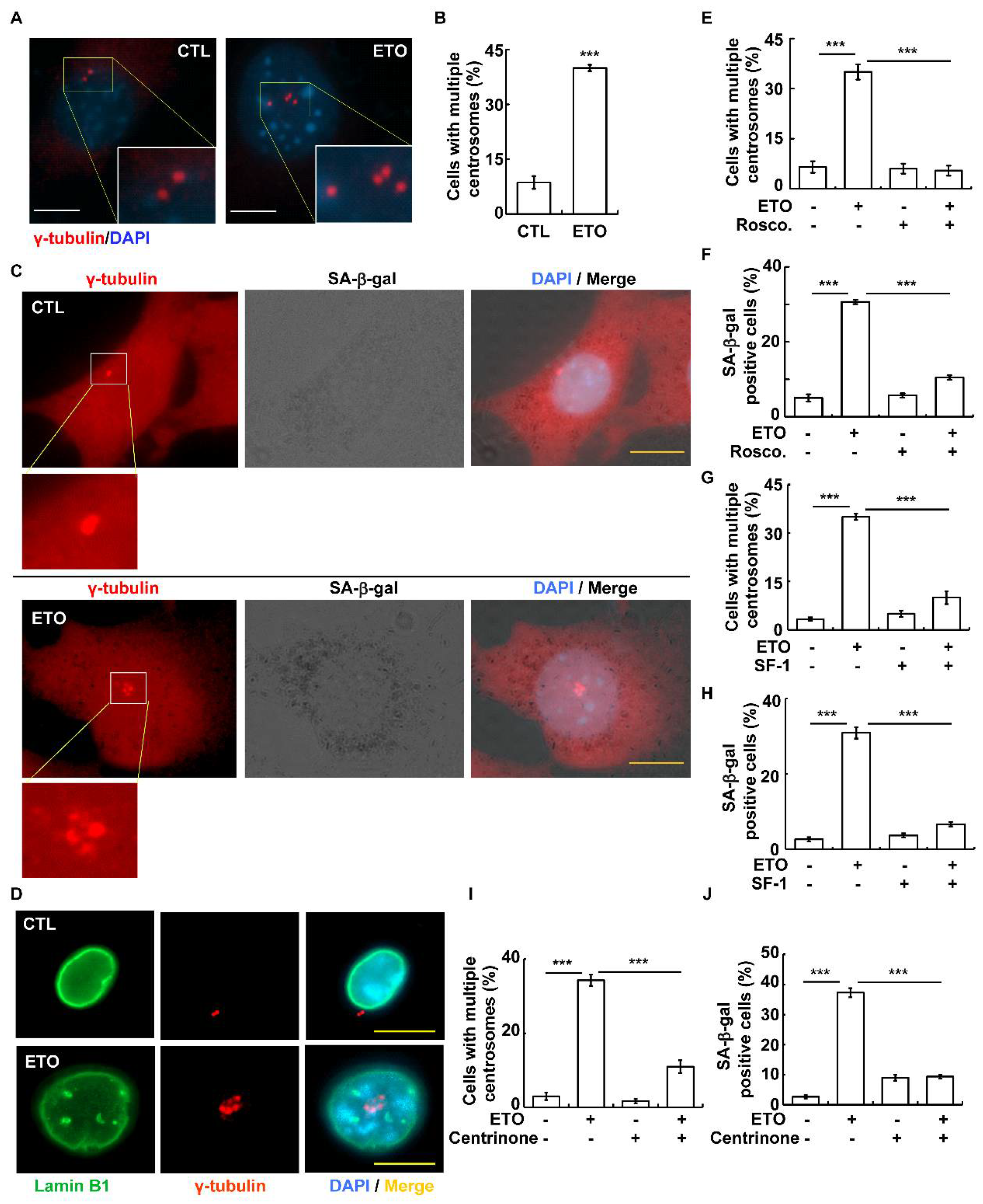
3.3. Etoposide Promotes Cellular Senescence by Inducing Centrosome Amplification
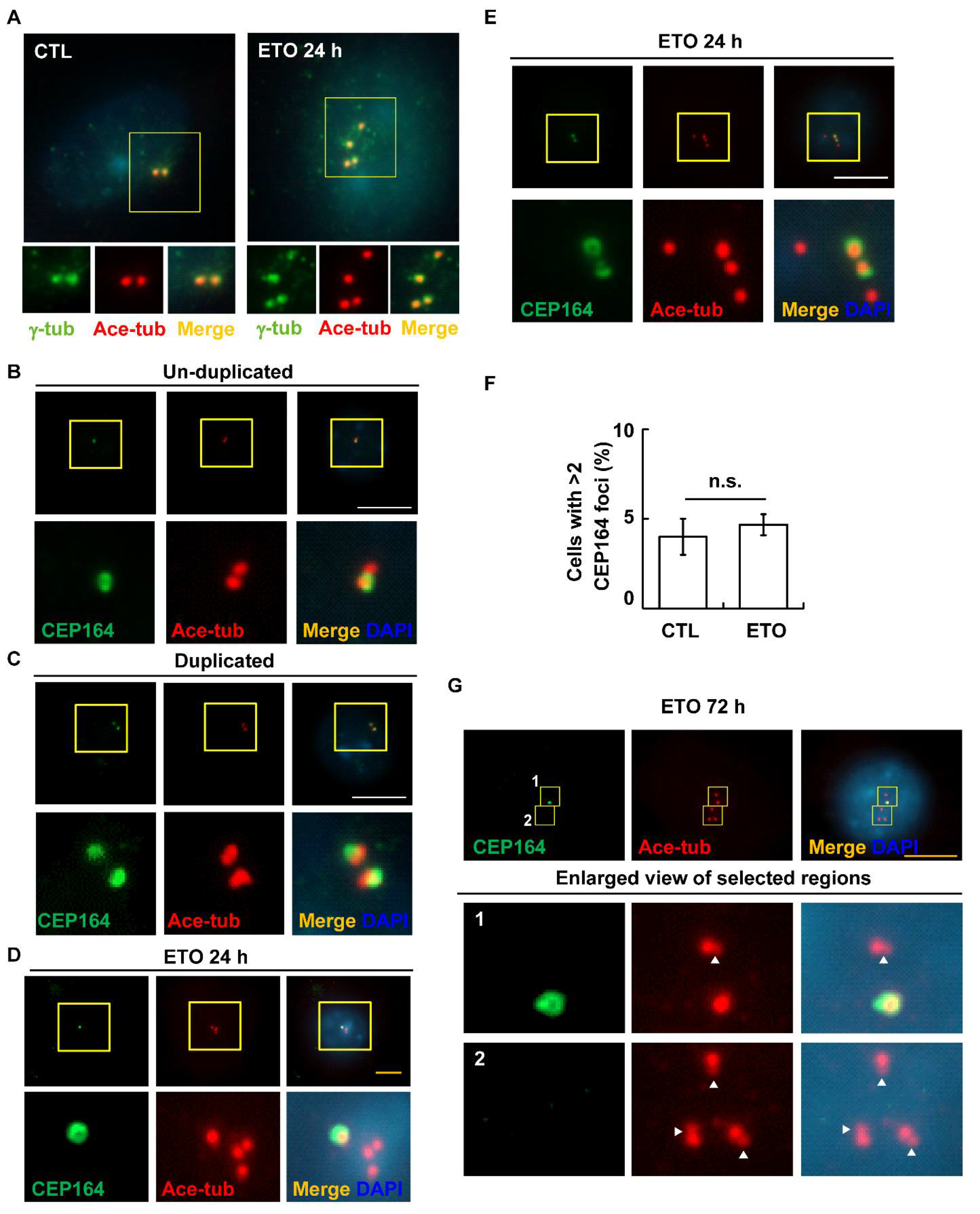
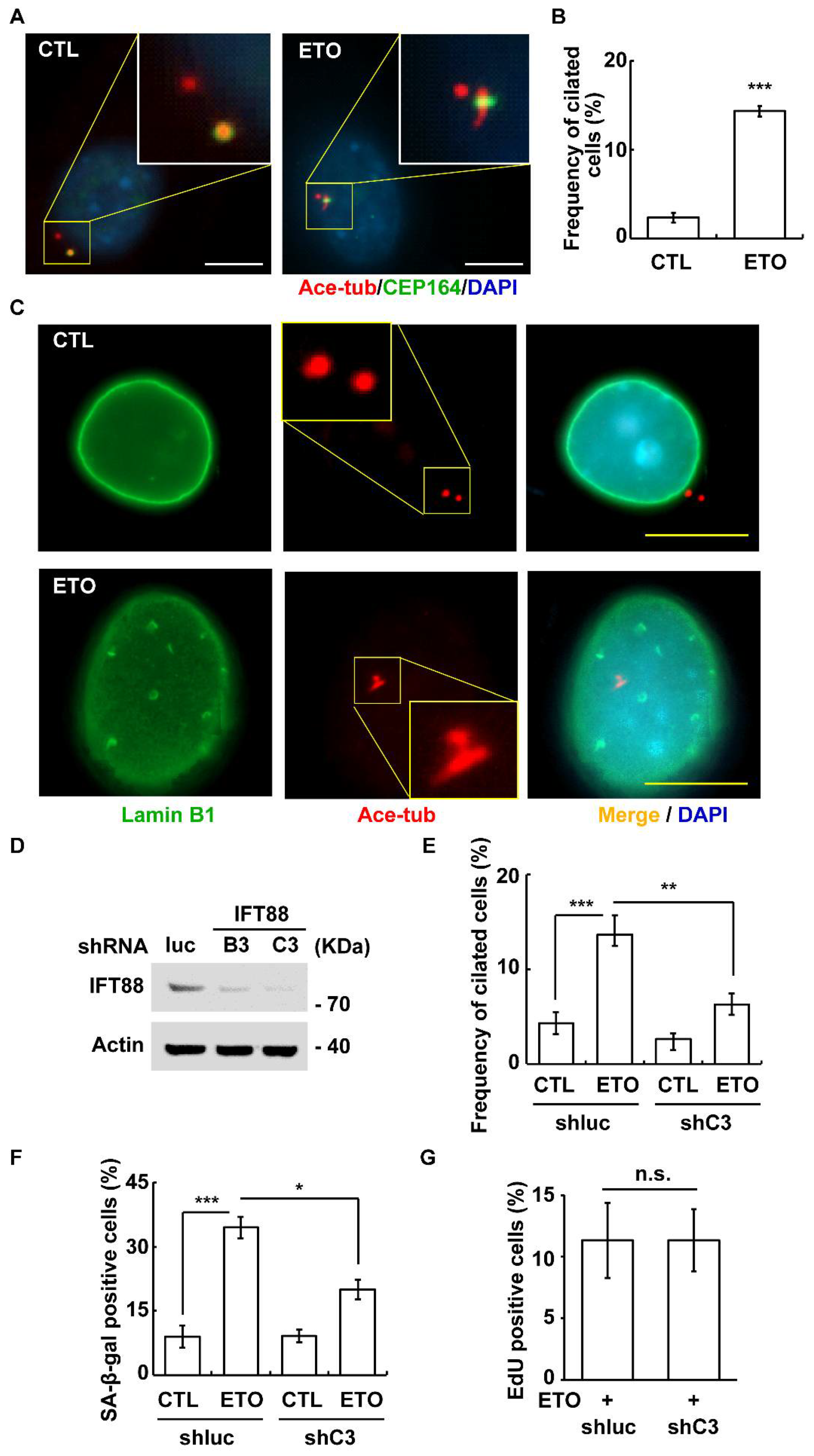
3.4. ETO Induces Primary Cilia Formation
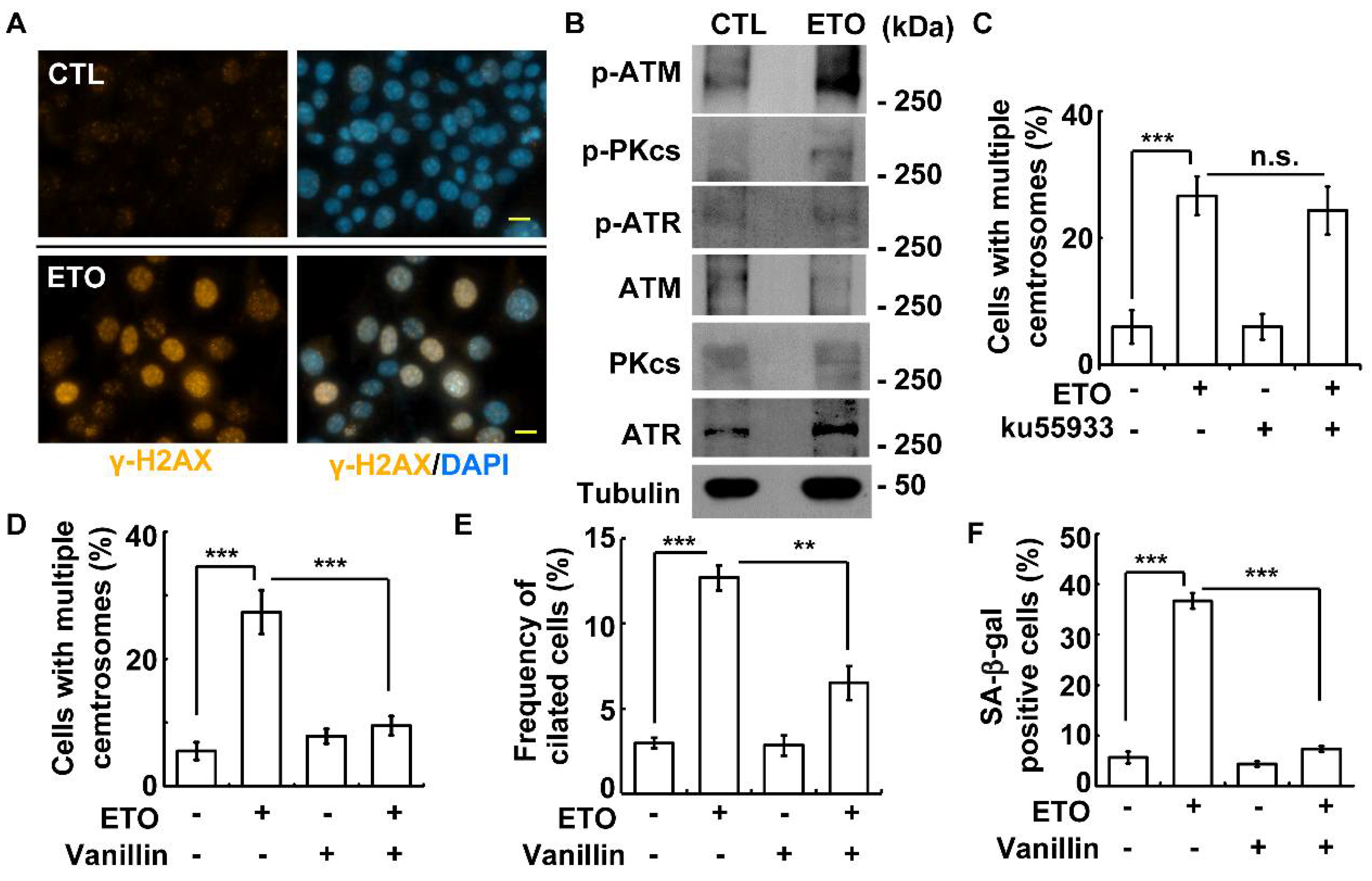
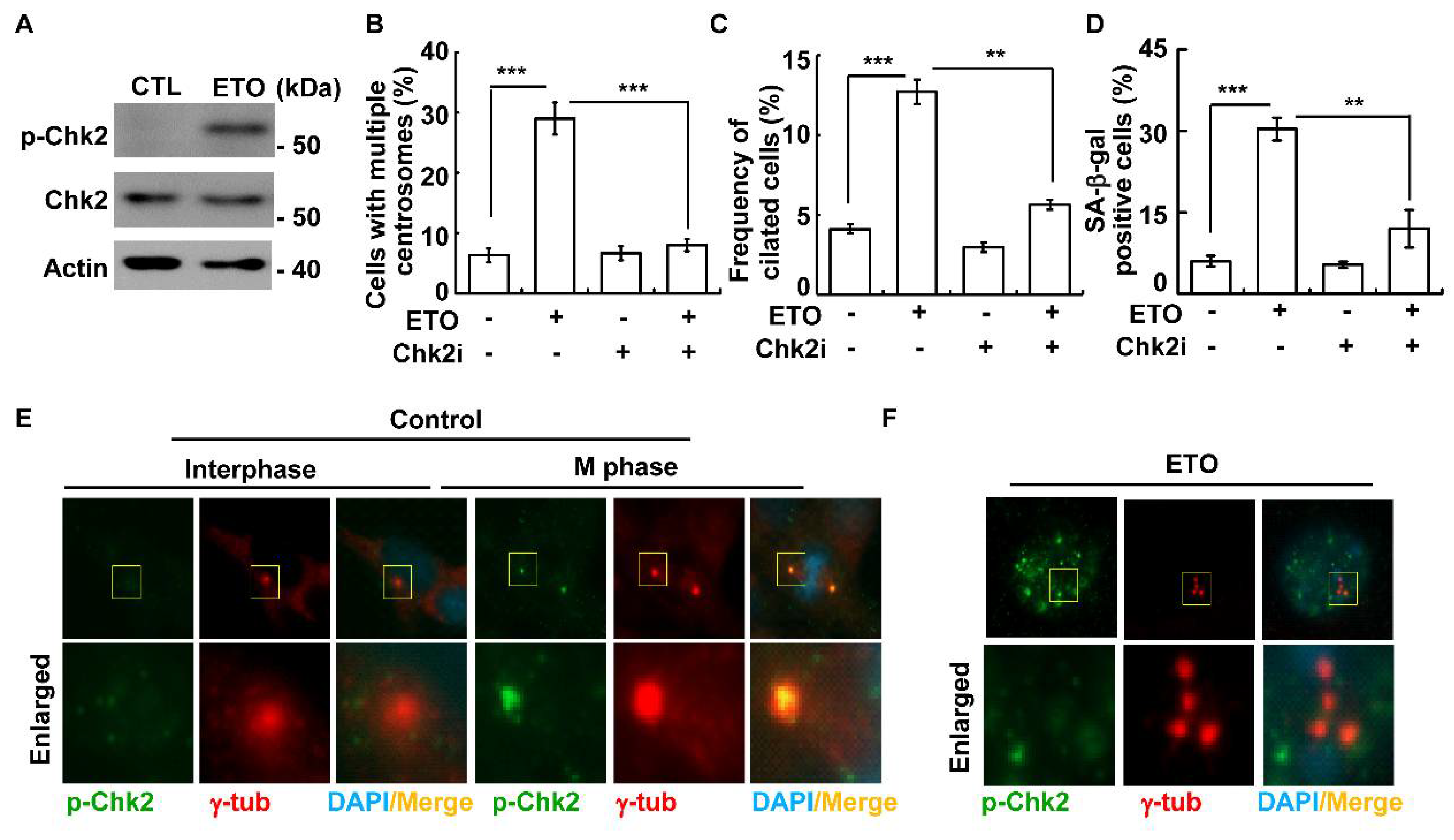
3.5. DNA-PK-Chk2 Axis Triggers Multiple Centrosomes and Primary Cilia
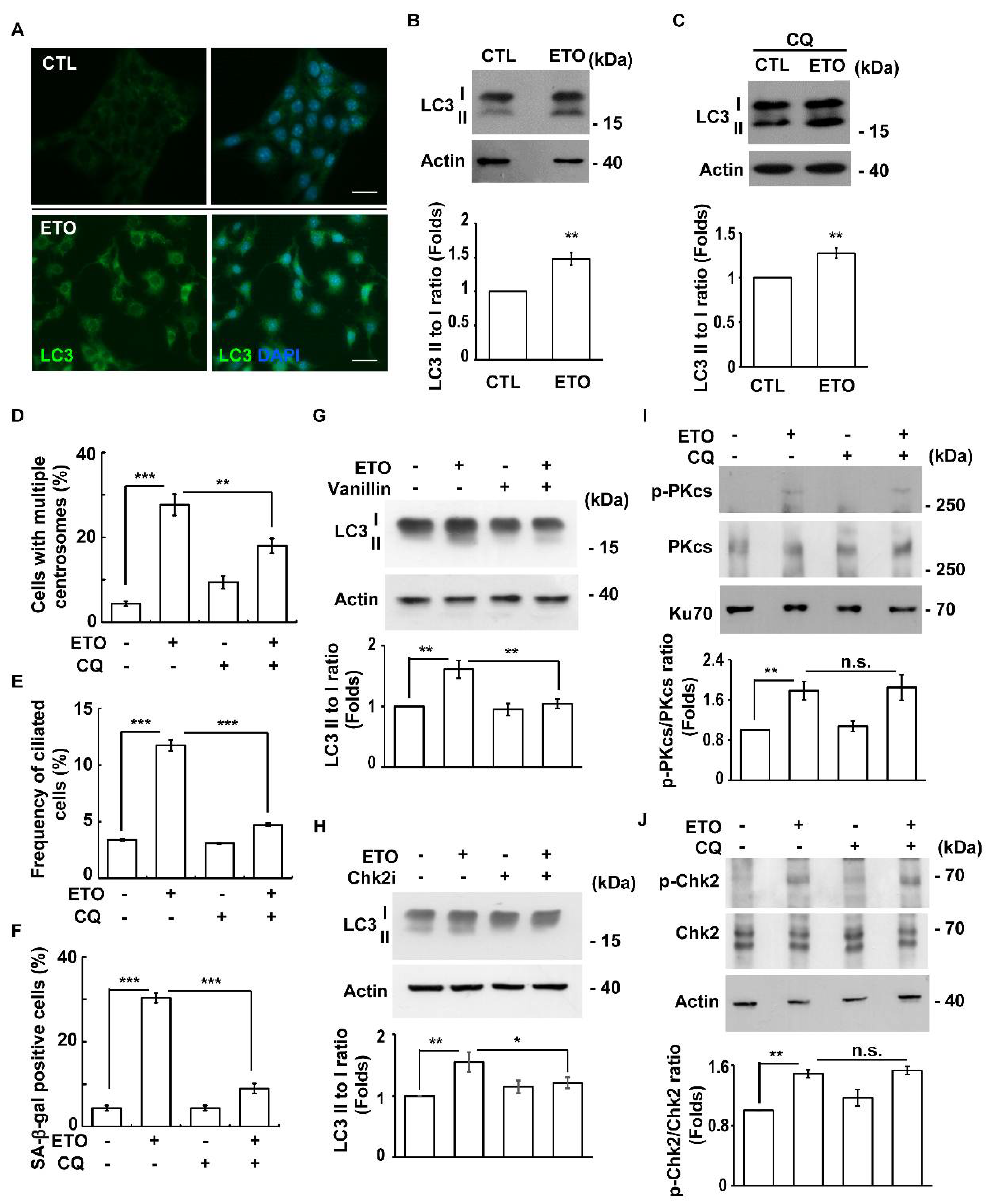
3.6. ETO-Activated Autophagy Induces Multiple Centrosomes and Primary Cilia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erickson, L.A.; Rivera, M.; Zhang, J. Adrenocortical carcinoma: Review and update. Adv. Anat. Pathol. 2014, 21, 151–159. [Google Scholar] [CrossRef]
- Baudin, E.; Endocrine Tumor Board of Gustave Roussy. Adrenocortical carcinoma. Endocrinol. Metab. Clin. N. Am. 2015, 44, 411–434. [Google Scholar] [CrossRef]
- Figueiredo, B.C.; Cavalli, L.R.; Pianovski, M.A.; Lalli, E.; Sandrini, R.; Ribeiro, R.C.; Zambetti, G.; DeLacerda, L.; Rodrigues, G.A.; Haddad, B.R. Amplification of the steroidogenic factor 1 gene in childhood adrenocortical tumors. J. Clin. Endocrinol. Metab. 2005, 90, 615–619. [Google Scholar] [CrossRef]
- Boulle, N.; Logie, A.; Gicquel, C.; Perin, L.; Le Bouc, Y. Increased levels of insulin-like growth factor II (IGF-II) and IGF-binding protein-2 are associated with malignancy in sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 1998, 83, 1713–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthon, A.; Sahut-Barnola, I.; Lambert-Langlais, S.; de Joussineau, C.; Damon-Soubeyrand, C.; Louiset, E.; Taketo, M.M.; Tissier, F.; Bertherat, J.; Lefrancois-Martinez, A.M.; et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet. 2010, 19, 1561–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Terzolo, M.; Allolio, B.; Baudin, E.; Haak, H.; Berruti, A.; Welin, S.; Schade-Brittinger, C.; Lacroix, A.; Jarzab, B.; et al. Combination chemotherapy in advanced adrenocortical carcinoma. N. Engl. J. Med. 2012, 366, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Paragliola, R.M.; Corsello, A.; Locantore, P.; Papi, G.; Pontecorvi, A.; Corsello, S.M. Medical Approaches in Adrenocortical Carcinoma. Biomedicines 2020, 8, 551. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [Green Version]
- Lara, L.I.; Fenner, S.; Ratcliffe, S.; Isidro-Llobet, A.; Hann, M.; Bax, B.; Osheroff, N. Coupling the core of the anticancer drug etoposide to an oligonucleotide induces topoisomerase II-mediated cleavage at specific DNA sequences. Nucleic Acids Res. 2018, 46, 2218–2233. [Google Scholar] [CrossRef]
- Wang, C.Y.; Huang, E.Y.; Huang, S.C.; Chung, B.C. DNA-PK/Chk2 induces centrosome amplification during prolonged replication stress. Oncogene 2015, 34, 1263–1269. [Google Scholar] [CrossRef]
- Squatrito, M.; Holland, E.C. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011, 71, 5945–5949. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Hagan, I.M.; Glover, D.M. The centrosome and its duplication cycle. Cold Spring Harb. Perspect. Biol. 2015, 7, a015800. [Google Scholar] [CrossRef] [Green Version]
- Schatten, H.; Sun, Q.Y. Functions and dysfunctions of the mammalian centrosome in health, disorders, disease, and aging. Histochem. Cell Biol. 2018, 150, 303–325. [Google Scholar] [CrossRef]
- Cheng, H.W.; Hsiao, C.T.; Chen, Y.Q.; Huang, C.M.; Chan, S.I.; Chiou, A.; Kuo, J.C. Centrosome guides spatial activation of Rac to control cell polarization and directed cell migration. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Stearns, T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011, 13, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Pagan, J.K.; Marzio, A.; Jones, M.J.; Saraf, A.; Jallepalli, P.V.; Florens, L.; Washburn, M.P.; Pagano, M. Degradation of Cep68 and PCNT cleavage mediate Cep215 removal from the PCM to allow centriole separation, disengagement and licensing. Nat. Cell Biol. 2015, 17, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, S.; Chen, Q.; Duan, X.; Li, G.; Huang, Q.; Zhu, X.; Yan, X. Cep57 and Cep57l1 function redundantly to recruit the Cep63-Cep152 complex for centriole biogenesis. J. Cell Sci. 2020, 133, jcs241836. [Google Scholar] [CrossRef]
- Mullee, L.I.; Morrison, C.G. Centrosomes in the DNA damage response—The hub outside the centre. Chromosome Res. 2016, 24, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.Y.; Wang, C.Y.; Chen, W.Y.; Kao, Y.H.; Tsai, H.M.; Tachibana, T.; Chang, W.C.; Chung, B.C. Steroidogenic Factor 1 (NR5A1) resides in centrosomes and maintains genomic stability by controlling centrosome homeostasis. Cell Death Differ. 2011, 18, 1836–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Joukov, V.; De Nicolo, A. The Centrosome and the Primary Cilium: The Yin and Yang of a Hybrid Organelle. Cells 2019, 8, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wloga, D.; Joachimiak, E.; Louka, P.; Gaertig, J. Posttranslational Modifications of Tubulin and Cilia. Cold Spring Harb. Perspect. Biol. 2017, 9, a028159. [Google Scholar] [CrossRef] [PubMed]
- Craft, J.M.; Harris, J.A.; Hyman, S.; Kner, P.; Lechtreck, K.F. Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J. Cell Biol. 2015, 208, 223–237. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.Y.; Huang, B.M.; Tang, T.K.; Chao, Y.Y.; Xiao, X.Y.; Lee, P.R.; Yang, L.Y.; Wang, C.Y. Genotoxic stress-activated DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis to maintain the DNA damage response. Cell Death Differ. 2021, 28, 1865–1879. [Google Scholar] [CrossRef]
- Jeffries, E.P.; Di Filippo, M.; Galbiati, F. Failure to reabsorb the primary cilium induces cellular senescence. FASEB J. 2019, 33, 4866–4882. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Diaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.Y.; Lin, T.C.; Kuo, P.L.; Chen, Z.R.; Cheng, H.L.; Chao, Y.Y.; Syu, J.S.; Lu, F.I.; Wang, C.Y. Septin 7 is a centrosomal protein that ensures S phase entry and microtubule nucleation by maintaining the abundance of p150(glued). J. Cell Physiol. 2020, 236, 2706–2724. [Google Scholar] [CrossRef]
- Chen, T.Y.; Syu, J.S.; Lin, T.C.; Cheng, H.L.; Lu, F.L.; Wang, C.Y. Chloroquine alleviates etoposide-induced centrosome amplification by inhibiting CDK2 in adrenocortical tumor cells. Oncogenesis 2015, 4, e180. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Liu, G.; Luckhardt, T.; Antony, V.; Zhou, Y.; Carter, A.B.; Thannickal, V.J.; Liu, R.M. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell 2017, 16, 1114–1124. [Google Scholar] [CrossRef]
- Dulic, V.; Kaufmann, W.K.; Wilson, S.J.; Tlsty, T.D.; Lees, E.; Harper, J.W.; Elledge, S.J.; Reed, S.I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994, 76, 1013–1023. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Hayashi, K.; Nishida, E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 1999, 9, 429–432. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Kao, Y.H.; Lai, P.Y.; Chen, W.Y.; Chung, B.C. Steroidogenic factor 1 (NR5A1) maintains centrosome homeostasis in steroidogenic cells by restricting centrosomal DNA-dependent protein kinase activation. Mol. Cell Biol. 2013, 33, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, J.A.; Kao, L.R.; Hammer, R.E.; Seemann, J.; Fuchs, J.L.; Megraw, T.L. CDK5RAP2 regulates centriole engagement and cohesion in mice. Dev. Cell 2010, 18, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Lai, P.Y.; Chen, T.Y.; Chung, B.C. NR5A1 prevents centriole splitting by inhibiting centrosomal DNA-PK activation and beta-catenin accumulation. Cell Commun. Signal. 2014, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douthwright, S.; Sluder, G. Link between DNA damage and centriole disengagement/reduplication in untransformed human cells. J. Cell Physiol. 2014, 229, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, L.; Prosser, S.L.; Cuffe, S.; Morrison, C.G. Ciliary abnormalities in senescent human fibroblasts impair proliferative capacity. Cell Cycle 2014, 13, 2773–2779. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Mizumoto, K.; Nakamura, M.; Tanaka, M. Radiation-induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp. Cell Res. 2000, 255, 321–326. [Google Scholar] [CrossRef]
- Li, Y.; Yang, D.Q. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol. Cancer Ther. 2010, 9, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Durant, S.; Karran, P. Vanillins—A novel family of DNA-PK inhibitors. Nucleic Acids Res. 2003, 31, 5501–5512. [Google Scholar] [CrossRef] [PubMed]
- Chouinard, G.; Clement, I.; Lafontaine, J.; Rodier, F.; Schmitt, E. Cell cycle-dependent localization of CHK2 at centrosomes during mitosis. Cell Div. 2013, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Honda, S.; Konishi, A.; Arakawa, S.; Murohashi, M.; Yamaguchi, H.; Torii, S.; Tanabe, M.; Tanaka, S.; Warabi, E.; et al. Autophagy controls centrosome number by degrading Cep63. Nat. Commun. 2016, 7, 13508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehme, K.A.; Kulikov, R.; Blattner, C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc. Natl. Acad. Sci. USA 2008, 105, 7785–7790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourke, E.; Dodson, H.; Merdes, A.; Cuffe, L.; Zachos, G.; Walker, M.; Gillespie, D.; Morrison, C.G. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 2007, 8, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, H.; Bourke, E.; Jeffers, L.J.; Vagnarelli, P.; Sonoda, E.; Takeda, S.; Earnshaw, W.C.; Merdes, A.; Morrison, C. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 2004, 23, 3864–3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef]
- Wang, L.; Xu, C.; Johansen, T.; Berger, S.L.; Dou, Z. SIRT1—A new mammalian substrate of nuclear autophagy. Autophagy 2020, 17, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Z.; Cai, Z.; Shi, S.; Jiang, H.; Shang, Y.R.; Ma, N.; Wang, J.J.; Guan, D.X.; Chen, T.W.; Rong, Y.F.; et al. Cilia loss sensitizes cells to transformation by activating the mevalonate pathway. J. Exp. Med. 2018, 215, 177–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenks, A.D.; Vyse, S.; Wong, J.P.; Kostaras, E.; Keller, D.; Burgoyne, T.; Shoemark, A.; Tsalikis, A.; de la Roche, M.; Michaelis, M.; et al. Primary Cilia Mediate Diverse Kinase Inhibitor Resistance Mechanisms in Cancer. Cell Rep. 2018, 23, 3042–3055. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Company | Catalog Number |
|---|---|---|
| phospho-p53 (Ser15) | Cell signaling | #9284 |
| p53 | Santa Cruz | sc-6243 |
| p21 | Abcam | ab7960-1 |
| actin (AC-15) | GeneTex | GTX26276 |
| Lamin B1 | Abcam | ab16048 |
| Lamin A/C | Genetex | GTX101127 |
| γ-tubulin | Sigma | T6557 |
| tubulin-acetylated | Sigma | T6793 |
| γ-H2A.X (Ser139) | Abcam | ab2893 |
| ATM | GeneTex | GTX70103 |
| phospho-ATM (Ser1981) | Abcam | ab81292 |
| DNA-PKcs (H163) | Santa Cruz | sc-9051 |
| phospho-DNA-PKcs (Thr2609) | Santa Cruz | sc-101664 |
| ATR | Cell signaling | #2790 |
| phospho-ATR | Cell signaling | #2853 |
| Chk2 | Cell signaling | #3440 |
| phospho-Chk2 (Thr68) | Cell signaling | #2661 |
| LC3 A/B | Cell signaling | #12741 |
| CEP164 | NOVUS | NBP1-81445 |
| Phosphor-Chk1 (Ser317) (D12H3) | Cell signaling | #12302 |
| Chk1 (2G1D5) | Cell signaling | #2360 |
| Akt (C67E7) | Cell signaling | #4691 |
| p-Akt (S473) | Cell signaling | #4060 |
| α-tubulin | GeneTex | GTX112141 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, Y.-N.; Chang, H.-C.; Chao, Y.-Y.; Cheng, H.-L.; Lien, W.-C.; Wang, C.-Y. Etoposide Triggers Cellular Senescence by Inducing Multiple Centrosomes and Primary Cilia in Adrenocortical Tumor Cells. Cells 2021, 10, 1466. https://doi.org/10.3390/cells10061466
Teng Y-N, Chang H-C, Chao Y-Y, Cheng H-L, Lien W-C, Wang C-Y. Etoposide Triggers Cellular Senescence by Inducing Multiple Centrosomes and Primary Cilia in Adrenocortical Tumor Cells. Cells. 2021; 10(6):1466. https://doi.org/10.3390/cells10061466
Chicago/Turabian StyleTeng, Yen-Ni, Huei-Cih Chang, Yu-Ying Chao, Hui-Ling Cheng, Wei-Chih Lien, and Chia-Yih Wang. 2021. "Etoposide Triggers Cellular Senescence by Inducing Multiple Centrosomes and Primary Cilia in Adrenocortical Tumor Cells" Cells 10, no. 6: 1466. https://doi.org/10.3390/cells10061466