
The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis?

 ,
,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction

2. Summary of Study Findings: tRAS and Its Tole in Human Tissues
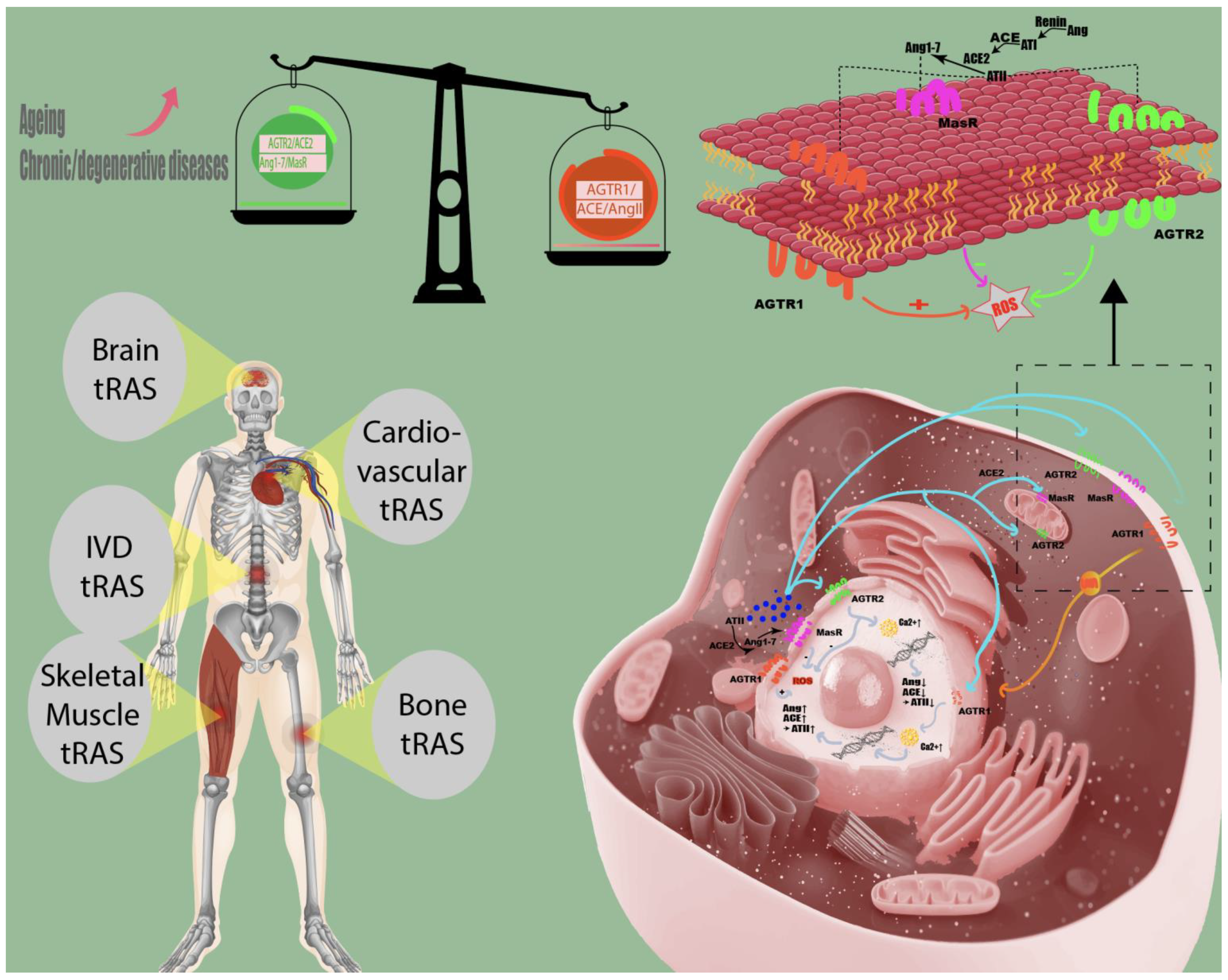
2.1. The Role of tRAS in Aging and Age-Related Diseases
2.2. The Role of tRAS in Autoimmune and Inflammatory Diseases

{kind=link}
{kind=link}
| Drug | Chemical Structures | Therapeutic Pathway | Inflammatory Disease | Outcome | Reference |
|---|---|---|---|---|---|
| Ramipril |  | ACE-inhibitor | Rheumatoid arthritis | Improved endothelial function | [87] |
| Losartan/Ramipril |   | ARB/ACE-inhibitor | Rheumatoid arthritis | Lower erythrocyte sedimentation rate | [88] |
| Enalapril |  | ACE-inhibitor | Rheumatoid arthritis | Reduction of arterial stiffness | [89] |
| Ramipril |  | ACE-inhibitor | Atherosclerosis | Beneficial effects on atherosclerosis progression | [90] |
| Ramipril |  | ACE-inhibitor | Atherosclerosis | Reduces high-sensitivity C-reactive protein concentration | [91] |
| Fosinopril |  | ACE-inhibitor | Atherosclerosis | Stopped the progression of atherosclerosis compared to hydrochlorothiazide | [92] |
| Perindopril |  | ACE-inhibitor | Atherosclerosis | Reductions of TNF-alpha and D-dimer | [93] |
| Enalapril/Losartan |   | ACE-inhibitor/ARB | Atherosclerosis | Enalapril and losartan decreased the plasma adhesion molecules clCAM-1, cVCAM-1 | [78] |
| Captopril/Valsartan |   | ACE-inhibitor/ARB | Atherosclerosis | Captopril and Valsartan were similarly effective in reducing atherosclerotic events | [94] |
| Irbesartan |  | ARB | Atherosclerosis | Reduction of VCAM-1, solubilized TNF-alpha receptor II and superoxide levels | [95] |
| Losartan/Candesartan/Irbesartan |    | ARB | Atherosclerosis | Reduction of tissue factor and plasminogen activator inhibitor type-1 | [96] |
| Olmesartan |  | ARB | Atherosclerosis | Increased circulating endothelial progenitor cells and serum levels of eNOS and NO | [97] |
| Olmesartan |  | ARB | Atherosclerosis | lower rate of coronary atheroma progression | [98] |
| Olmesartan |  | ARB | Atherosclerosis | Higher event-free survival | [99] |
| Olmesartan |  | ARB | Atherosclerosis | Decreased intima-media thickness, and reduced volume of larger atherosclerotic plaques | [100] |
| Valsartan |  | ARB | Atherosclerosis | Regression in carotid atherosclerosis | [101] |
| Eprosartan |  | ARB | Atherosclerosis | Reduction in neutrophil superoxide anion generating capacity, soluble monocyte chemotactic protein-1, and soluble vascular cell adhesion molecule | [102] |
| Losartan |  | ARB | Atherosclerosis | Protecting the progression of atherosclerosis of the carotid artery | [103] |
| Valsartan |  | ARB | Atherosclerosis | Reduced ROS generation by polymorphonuclear and mononuclear cells, NF-kappa β binding activity, expression of total cellular p65, and c-reactive protein. Increase in inhibitor kappa β | [79] |
| Valsartan |  | ARB | Atherosclerosis | Decreased high-sensitivity CRP, VCAM-1, and increased antioxidant status and glutathione peroxidase | [104] |
| Losartan |  | ARB | NASH | Reduction of blood markers of hepatic fibrosis, plasma TGF-beta1, and serum ferritin concentration. Improvement of serum aminotransferase levels, hepatic necroinflammation, reduction of hepatic fibrosis, and disappearance of iron depositions | [80] |
| Losartan |  | ARB | NASH | Decreased steatosis degree, subcutaneous adipose tissue, and visceral adipose tissue diameter | [105] |
| Olmesartan |  | ARB | NASH | Decreased TGF-beta1 but not hepatic fibrosis markers | [106] |
| Losartan |  | ARB | IgA Nephritis | Reduced proteinuria and preserved renal functions | [107] |
| Losartan/enalapril |   | ARB/ACE-inhibitor | Glomerulonephritis | Reduced proteinuria and tubular injury extent | [108] |
| Irbesartan |  | ARB | Glomerulonephritis | Reduced proteinuria and the urine protein/creatinine ratio, concentrations of adiponectin, and high sensitivity c-reactive protein | [109] |
2.3. The Role of tRAS in Cardiovascular Diseases
2.4. tRAS and (Neuropathic) Pain: Vulvodynia
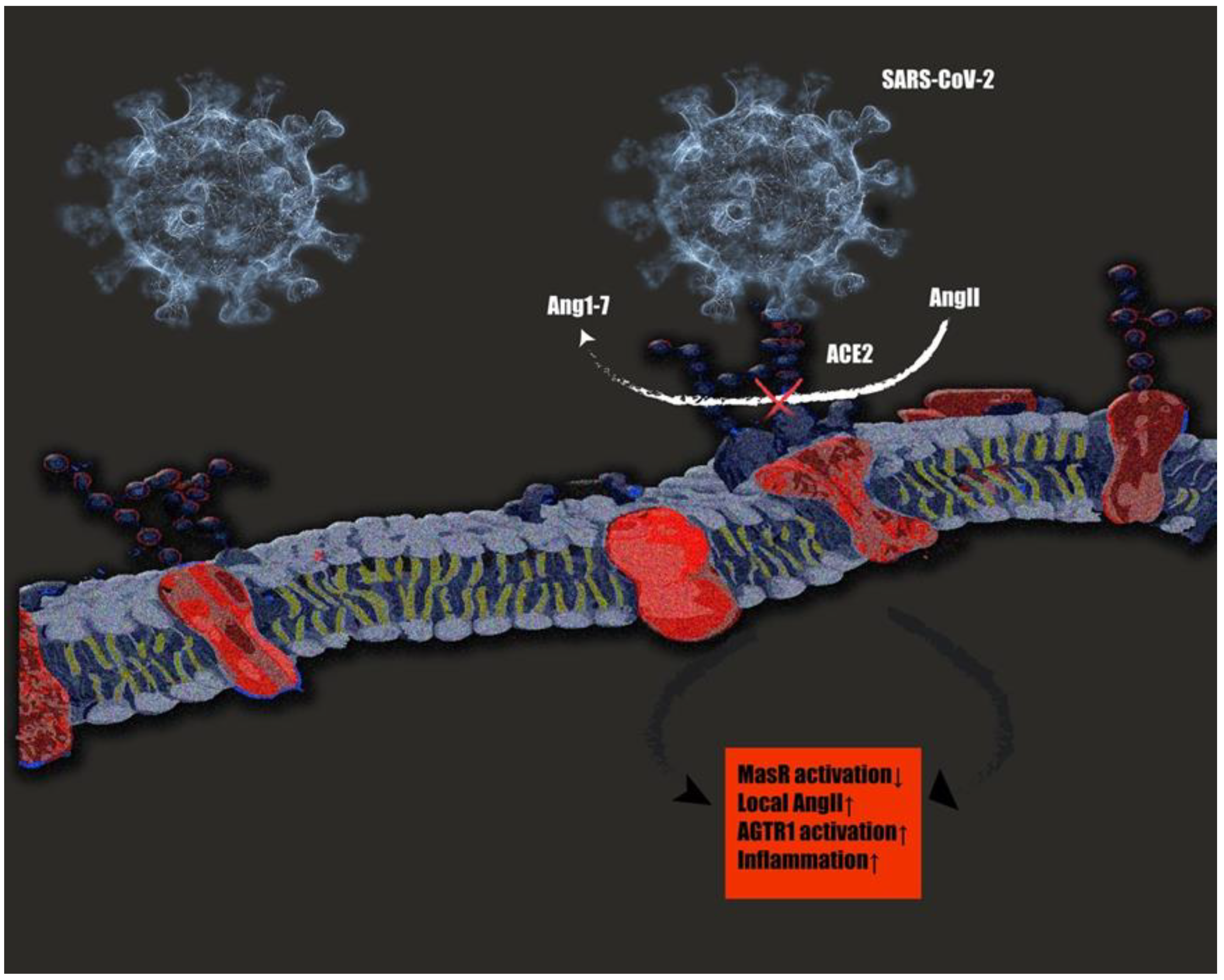
2.5. The Role of tRAS in the COVID-19 Pandemic

3. Therapeutic Implications and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tigerstedt, R.; Bergman, P.Q. Niere und kreislauf. Skand. Archiv Physiol. 1898, 8, 223–271. [Google Scholar] [CrossRef]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Yoon, H.E.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Jang, I.-A.; Ban, T.H.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Choi, B.S. Age-associated changes in the vascular renin-angiotensin system in mice. Oxid. Med. Cell Longev. 2016, 2016, 6731093. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D.; Luft, F.C.; Bader, M.; Ganten, D.; Andrade-Navarro, M.A. Emergence and evolution of the renin-angiotensin-aldosterone system. J. Mol. Med. 2012, 90, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehme, A.; Zouein, F.A.; Deris Zayeri, Z.; Zibara, K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, T.; Steckelings, U.M.; dos Santos, R.S. The Protective Arm of the Renin Angiotensin: Functional Aspects and Therapeutic Implications; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 978-0-12-801485-1. [Google Scholar]
- Villar-Cheda, B.; Costa-Besada, M.A.; Valenzuela, R.; Perez-Costas, E.; Melendez-Ferro, M.; Labandeira-Garcia, J.L. The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 2017, 8, e3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan Danser, A.H. Local renin–angiotensin systems: The unanswered questions. Int. J. Biochem. Cell Biol. 2003, 35, 759–768. [Google Scholar] [CrossRef]
- Filipeanu, C.M.; Henning, R.H.; Nelemans, S.A.; de Zeeuw, D. Review: Intracellular angiotensin II: From myth to reality? J. Renin. Angiotensin. Aldosterone Syst. 2001, 2, 219–226. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Ahmad, S.; Varagic, J.; Cheng, C.P.; Groban, L.; Wang, H.; Collawn, J.F.; Dell′Italia, L.J. Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H404–H414. [Google Scholar] [CrossRef] [Green Version]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Cao, Z.; Kelly, D.J.; Cox, A.; Casley, D.; Forbes, J.M.; Martinello, P.; Dean, R.; Gilbert, R.E.; Cooper, M.E. Angiotensin Type 2 receptor is expressed in the adult rat kidney and promotes cellular proliferation and apoptosis. Kidney Int. 2000, 58, 2437–2451. [Google Scholar] [CrossRef] [Green Version]
- Lenkei, Z.; Palkovits, M.; Corvol, P.; Llorens-Cortes, C. Distribution of angiotensin ii type-2 receptor (AT2) MRNA expression in the adult rat brain. J. Comp. Neurol. 1996, 373, 322–339. [Google Scholar] [CrossRef]
- Yu, L.; Zheng, M.; Wang, W.; Rozanski, G.J.; Zucker, I.H.; Gao, L. Developmental changes in AT1 and AT2 receptor-protein expression in rats. J. Renin. Angiotensin. Aldosterone Syst. 2010, 11, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Terenzi, R.; Manetti, M.; Rosa, I.; Romano, E.; Galluccio, F.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Angiotensin II type 2 receptor (AT2R) as a novel modulator of inflammation in rheumatoid arthritis synovium. Sci. Rep. 2017, 7, 13293. [Google Scholar] [CrossRef]
- Xu, X.; He, H.; Hu, S.; Han, J.; Huang, L.; Xu, J.; Xie, J.; Liu, A.; Yang, Y.; Qiu, H. Ang II-AT2R increases mesenchymal stem cell migration by signaling through the FAK and RhoA/Cdc42 pathways in vitro. Stem. Cell Res. Ther. 2017, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izu, Y.; Mizoguchi, F.; Kawamata, A.; Hayata, T.; Nakamoto, T.; Nakashima, K.; Inagami, T.; Ezura, Y.; Noda, M. Angiotensin II type 2 receptor blockade increases bone mass. J. Biol. Chem. 2009, 284, 4857–4864. [Google Scholar] [CrossRef] [PubMed]
- Galindo, M.; Santiago, B.; Palao, G.; Gutierrez-Cañas, I.; Ramirez, J.C.; Pablos, J.L. Coexpression of AT1 and AT2 receptors by human fibroblasts is associated with resistance to angiotensin II. Peptides 2005, 26, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.S.; Vinh, A.; McCarthy, C.A.; Gaspari, T.A.; Widdop, R.E. AT2 Receptors: Functional relevance in cardiovascular disease. Pharmacol. Ther. 2008, 120, 292–316. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wystrach, L.; Bernstein, A.; Grad, S.; Alini, M.; Richards, R.; Kubosch, D.; Südkamp, N.; Izadpanah, K.; Kubosch, E.; et al. The tissue-renin-angiotensin-system of the human intervertebral disc. eCM 2020, 40, 115–132. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Mogi, M. Effect of renin–angiotensin system on senescence. Geriatr. Gerontol. Int. 2020, 20, 520–525. [Google Scholar] [CrossRef]
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr. Hypertens Rep. 2018, 20, 100. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Stone, R.E.; Liu, S.; Levy, A.M.; Kashani, N.; Louie, S.G.; Rodgers, K.E.; Kelland, E.E.; Lund, B.T. Activation of the protective arm of the renin angiotensin system in demyelinating disease. J. Neuroimmune. Pharmacol. 2020, 15, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Delatorre, N.; Hurst, C.; Rodgers, K.E. Targeting the protective arm of the renin-angiotensin system to reduce systemic lupus erythematosus related pathologies in MRL-Lpr mice. Front. Immunol. 2020, 11, 1572. [Google Scholar] [CrossRef]
- Namsolleck, P.; Moll, G.N. Does activation of the protective renin-angiotensin system have therapeutic potential in COVID-19? Mol. Med. 2020, 26, 80. [Google Scholar] [CrossRef]
- Mascolo, A.; Scavone, C.; Rafaniello, C.; Ferrajolo, C.; Racagni, G.; Berrino, L.; Paolisso, G.; Rossi, F.; Capuano, A. Renin-angiotensin system and Coronavirus disease 2019: A narrative review. Front. Cardiovasc. Med. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. A Forkhead in the road to longevity: The molecular basis of lifespan becomes clearer. J. Hypertens. 2005, 23, 1285–1309. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M. The frail renin-angiotensin system. Clin. Geriatr. Med. 2011, 27, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.M.; Oyama, T.T.; Kelly, F.J.; Kennefick, T.M.; Anderson, S. Activity and responsiveness of the renin-angiotensin system in the aging rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1787–R1794. [Google Scholar] [CrossRef]
- Anderson, S. Ageing and the renin-angiotensin system. Nephrol. Dialysis Transplantat. 1997, 12, 1093–1094. [Google Scholar] [CrossRef]
- Baylis, C. Renal responses to acute angiotensin II inhibition and administered angiotensin II in the aging, conscious, chronically catheterized rat. Am. J. Kidney Dis. 1993, 22, 842–850. [Google Scholar] [CrossRef]
- Wang, M.; Takagi, G.; Asai, K.; Resuello, R.G.; Natividad, F.F.; Vatner, D.E.; Vatner, S.F.; Lakatta, E.G. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 2003, 41, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymes, C.; Silvestre, J.-S.; Llorens-Cortes, C.; Marotte, F.; Chevalier, B.; Levy, B.I.; Swynghedauw, B.; Samuel, J.-L. Cardiac senescence is associated with enhanced expression of angiotensin II receptor subtypes. Endocrinology 1998, 139, 2579–2587. [Google Scholar] [CrossRef]
- Diz, D.I.; Varagic, J.; Groban, L. Aging and the brain renin–angiotensin system: Relevance to Age-related decline in cardiac function. Future Cardiol. 2008, 4, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Min, L.-J.; Mogi, M.; Iwai, M.; Horiuchi, M. Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing. Res. Rev. 2009, 8, 113–121. [Google Scholar] [CrossRef]
- Delafontaine, P.; Yoshida, T. The renin-angiotensin system and the biology of skeletal muscle: Mechanisms of muscle wasting in chronic disease states. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 245–258. [Google Scholar]
- Yamamoto, K.; Takeshita, H.; Rakugi, H. ACE2, Angiotensin 1-7 and skeletal muscle: Review in the era of COVID-19. Clin. Sci. 2020, 134, 3047–3062. [Google Scholar] [CrossRef] [PubMed]
- Semprun-Prieto, L.C.; Sukhanov, S.; Yoshida, T.; Rezk, B.M.; Gonzalez-Villalobos, R.A.; Vaughn, C.; Michael Tabony, A.; Delafontaine, P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem. Biophys. Res. Commun. 2011, 409, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Galvez, S.; Tiwari, S.; Rezk, B.M.; Semprun-Prieto, L.; Higashi, Y.; Sukhanov, S.; Yablonka-Reuveni, Z.; Delafontaine, P. Angiotensin II inhibits satellite cell proliferation and prevents skeletal muscle regeneration. J. Biol. Chem. 2013, 288, 23823–23832. [Google Scholar] [CrossRef] [Green Version]
- Tabony, A.M.; Yoshida, T.; Galvez, S.; Higashi, Y.; Sukhanov, S.; Chandrasekar, B.; Mitch, W.E.; Delafontaine, P. Angiotensin II upregulates protein phosphatase 2Cα and inhibits AMP-activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension 2011, 58, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, H.; Yamamoto, K.; Nozato, S.; Takeda, M.; Fukada, S.-I.; Inagaki, T.; Tsuchimochi, H.; Shirai, M.; Nozato, Y.; Fujimoto, T.; et al. Angiotensin-converting enzyme 2 deficiency accelerates and angiotensin 1-7 restores age-related muscle weakness in mice. J. Cachexia Sarcopenia Muscle 2018, 9, 975–986. [Google Scholar] [CrossRef]
- Gomes-Santos, I.L.; Fernandes, T.; Couto, G.K.; Ferreira-Filho, J.C.A.; Salemi, V.M.C.; Fernandes, F.B.; Casarini, D.E.; Brum, P.C.; Rossoni, L.V.; de Oliveira, E.M.; et al. Effects of exercise training on circulating and skeletal muscle renin-angiotensin system in chronic heart failure rats. PLoS ONE 2014, 9, e98012. [Google Scholar] [CrossRef] [Green Version]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Yabumoto, C.; Akazawa, H.; Yamamoto, R.; Yano, M.; Kudo-Sakamoto, Y.; Sumida, T.; Kamo, T.; Yagi, H.; Shimizu, Y.; Saga-Kamo, A.; et al. Angiotensin II receptor blockade promotes repair of skeletal muscle through down-regulation of aging-promoting C1q expression. Sci. Rep. 2015, 5, 14453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, E.L.; de Picoli Souza, K.; da Silva, E.D.; Batista, E.C.; Martins, P.J.F.; D’Almeida, V.; Pesquero, J.B. Long term treatment with ACE inhibitor enalapril decreases body weight gain and increases life span in rats. Biochem. Pharmacol. 2009, 78, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Linz, W.; Heitsch, H.; Schölkens, B.A.; Wiemer, G. Long-term angiotensin II type 1 receptor blockade with fonsartan doubles lifespan of hypertensive rats. Hypertension 2000, 35, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Linz, W.; Jessen, T.; Becker, R.H.A.; Schölkens, B.A.; Wiemer, G. Long-term ACE inhibition doubles lifespan of hypertensive rats. Circulation 1997, 96, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Spindler, S.R.; Mote, P.L.; Flegal, J.M. Combined statin and angiotensin-converting enzyme (ACE) inhibitor treatment increases the lifespan of long-lived F1 male mice. Age 2016, 38, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Basso, N.; Cini, R.; Pietrelli, A.; Ferder, L.; Terragno, N.A.; Inserra, F. Protective effect of long-term angiotensin II inhibition. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1351–H1358. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, R.; Ichiki, T.; Hashimoto, T.; Inanaga, K.; Imayama, I.; Sadoshima, J.; Sunagawa, K. SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. ATVB 2008, 28, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Xu, T.-T.; Lu, J.; Li, L.; Xu, J.; Hao, D.-L.; Chen, H.-Z.; Liu, D.-P. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. 2014, 92, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, K.-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.E.; Choi, B.S. The renin-angiotensin system and aging in the kidney. Korean J. Intern. Med. 2014, 29, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Ruiz, C.; Rodriguez-Perez, A.I.; Beiroa, D.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. Reciprocal regulation between sirtuin-1 and angiotensin-II in the substantia nigra: Implications for aging and neurodegeneration. Oncotarget 2015, 6, 26675–26689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Pallares, J.; Rey, P.; Parga, J.A.; Muñoz, A.; Guerra, M.J.; Labandeira-Garcia, J.L. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol. Dis. 2008, 31, 58–73. [Google Scholar] [CrossRef]
- Rey, P.; Lopez-Real, A.; Sanchez-Iglesias, S.; Muñoz, A.; Soto-Otero, R.; Labandeira-Garcia, J.L. Angiotensin type-1-receptor antagonists reduce 6-hydroxydopamine toxicity for dopaminergic neurons. Neurobiol. Aging. 2007, 28, 555–567. [Google Scholar] [CrossRef]
- Grammatopoulos, T.N.; Jones, S.M.; Ahmadi, F.A.; Hoover, B.R.; Snell, L.D.; Skoch, J.; Jhaveri, V.V.; Poczobutt, A.M.; Weyhenmeyer, J.A.; Zawada, W.M. Angiotensin type 1 receptor antagonist losartan, reduces MPTP-induced degeneration of dopaminergic neurons in substantia nigra. Mol. Neurodegener. 2007, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Zawada, W.M.; Banninger, G.P.; Thornton, J.; Marriott, B.; Cantu, D.; Rachubinski, A.L.; Das, M.; Griffin, W.S.T.; Jones, S.M. Generation of reactive oxygen species in 1-Methyl-4-Phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J. Neuroinflamm. 2011, 8, 129. [Google Scholar] [CrossRef] [Green Version]
- Villar-Cheda, B.; Valenzuela, R.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol. Aging. 2012, 33, 204.e1–204.e11. [Google Scholar] [CrossRef]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Valenzuela, R.; Granado, N.; Moratalla, R.; Labandeira-Garcia, J.L. Aging-related dysregulation of dopamine and angiotensin receptor interaction. Neurobiol. Aging. 2014, 35, 1726–1738. [Google Scholar] [CrossRef] [Green Version]
- Labandeira-García, J.L.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Valenzuela, R.; Borrajo, A.; Rodríguez-Perez, A.I. Brain renin-angiotensin system and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Kawas, L.H.; Harding, J.W. A Role for the brain RAS in Alzheimer’s and Parkinson’s diseases. Front. Endocrinol. 2013, 4, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labandeira-Garcia, J.L.; Rodriguez-Pallares, J.; Dominguez-Meijide, A.; Valenzuela, R.; Villar-Cheda, B.; Rodríguez-Perez, A.I. Dopamine-angiotensin interactions in the basal ganglia and their relevance for Parkinson’s disease. Mov. Disord. 2013, 28, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Patel, V.B.; Ramprasath, T.; Alrob, O.A.; DesAulniers, J.; Scholey, J.W.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1–7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am. J. Physiol. Renal Physiol. 2014, 306, F812–F821. [Google Scholar] [CrossRef]
- Benigni, A.; Orisio, S.; Noris, M.; Iatropoulos, P.; Castaldi, D.; Kamide, K.; Rakugi, H.; Arai, Y.; Todeschini, M.; Ogliari, G.; et al. Variations of the angiotensin II type 1 receptor gene are associated with extreme human longevity. Age 2013, 35, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nataraj, C.; Oliverio, M.I.; Mannon, R.B.; Mannon, P.J.; Audoly, L.P.; Amuchastegui, C.S.; Ruiz, P.; Smithies, O.; Coffman, T.M. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J. Clin. Invest. 1999, 104, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Ruiz-Ortega, M.; Gomez-Guerrero, C.; Tomino, Y.; Egido, J. Angiotensin II, the immune system and renal diseases: Another road for RAS? Nephrol. Dial. Transplant. 2003, 18, 1423–1426. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Egido, J. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 2003, 35, 881–900. [Google Scholar] [CrossRef]
- Hahn, A.W.; Jonas, U.; Bühler, F.R.; Resink, T.J. Activation of human peripheral monocytes by angiotensin II. FEBS Lett. 1994, 347, 178–180. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Wei, W. Angiotensin II in inflammation, immunity and rheumatoid arthritis: Angiotensin II and rheumatoid arthritis. Clin. Exp. Immunol. 2015, 179, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Liu, J.; Liu, X.; Li, M. Angiotensin II induces c-reactive protein expression through ERK1/2 and JNK signaling in human aortic endothelial cells. Atherosclerosis 2010, 212, 206–212. [Google Scholar] [CrossRef]
- Schupp, M.; Janke, J.; Clasen, R.; Unger, T.; Kintscher, U. Angiotensin type 1 receptor blockers induce peroxisome proliferator–activated receptor-γ activity. Circulation 2004, 109, 2054–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-M.; Yang, S.-P.; Ho, L.-J.; Tsao, T.-P.; Chang, D.-M.; Lai, J.-H. Irbesartan inhibits human T-Lymphocyte activation through downregulation of activator protein-1: Suppression of T-cell activation by irbesartan. Br. J. Pharmacol. 2004, 142, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor ΚB. Hypertension 2010, 55, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Graninger, M.; Reiter, R.; Drucker, C.; Minar, E.; Jilma, B. Angiotensin receptor blockade decreases markers of vascular inflammation. J. Cardiovasc. Pharmacol. 2004, 44, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Kumar, V.; Aljada, A.; Ghanim, H.; Syed, T.; Hofmayer, D.; Mohanty, P.; Tripathy, D.; Garg, R. Angiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear Factor-ΚB, in mononuclear cells of normal subjects: Evidence of an antiinflammatory action. J. Clin. Endocrinol. Metab. 2003, 88, 4496–4501. [Google Scholar] [CrossRef] [Green Version]
- Yokohama, S.; Yoneda, M.; Haneda, M.; Okamoto, S.; Okada, M.; Aso, K.; Hasegawa, T.; Tokusashi, Y.; Miyokawa, N.; Nakamura, K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004, 40, 1222–1225. [Google Scholar] [CrossRef]
- Okada, H.; Watanabe, Y.; Inoue, T.; Kobayashi, T.; Kikuta, T.; Kanno, Y.; Ban, S.; Suzuki, H. Angiotensin II type 1 receptor blockade attenuates renal fibrogenesis in an immune-mediated nephritic kidney through counter-activation of angiotensin II Type 2 receptor. Biochem. Biophys. Res. Commun. 2004, 314, 403–408. [Google Scholar] [CrossRef]
- Hammer, A.; Yang, G.; Friedrich, J.; Kovacs, A.; Lee, D.-H.; Grave, K.; Jörg, S.; Alenina, N.; Grosch, J.; Winkler, J.; et al. Role of the receptor mas in macrophage-mediated inflammation in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 14109–14114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuohashish, H.M.; Ahmed, M.M.; Sabry, D.; Khattab, M.M.; Al-Rejaie, S.S. Angiotensin (1-7) ameliorates the structural and biochemical alterations of ovariectomy-induced osteoporosis in rats via activation of ACE-2/Mas receptor axis. Sci. Rep. 2017, 7, 2293. [Google Scholar] [CrossRef] [Green Version]
- Saravi, B.; Lang, G.; Ülkümen, S.; Burchard, T.; Weihrauch, V.; Patzelt, S.; Boeker, M.; Li, Z.; Woelber, J.P. The tissue renin-angiotensin system (TRAS) and the impact of its inhibition on inflammation and bone loss in the periodontal tissue. Eur. Cell Mater. 2020, 40, 203–226. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; Santos, A.C.P.M.; Galvao, I.; Souto, G.R.; Mesquita, R.A.; Sa, M.A.; Ferreira, A.J. The angiotensin converting enzyme 2/angiotensin-(1-7)/mas receptor axis as a key player in alveolar bone remodeling. Bone 2019, 128, 115041. [Google Scholar] [CrossRef]
- Saravi, B.; Li, Z.; Pfannkuche, J.; Wystrach, L.; Albers, C.E.; Grad, S.; Alini, M.; Richards, R.G.; Lang, C.; Südkamp, N.; et al. Angiotensin II Type 1 receptor antagonist losartan inhibits TNF-α induced inflammation and degeneration processes in human nucleus pulposus cells. Preprints 2020. [Google Scholar] [CrossRef]
- Flammer, A.J.; Sudano, I.; Hermann, F.; Gay, S.; Forster, A.; Neidhart, M.; Künzler, P.; Enseleit, F.; Périat, D.; Hermann, M.; et al. Angiotensin-converting enzyme inhibition improves vascular function in rheumatoid arthritis. Circulation 2008, 117, 2262–2269. [Google Scholar] [CrossRef]
- Perry, M.E.; Chee, M.M.; Ferrell, W.R.; Lockhart, J.C.; Sturrock, R.D. Angiotensin receptor blockers reduce erythrocyte sedimentation rate levels in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 1646–1647. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vazquez, F.; Bäck, M.; Chavarria-Avila, E.; Gomez-Bañuelos, E.; Ramos-Becerra, C.G.; Pizano-Martínez, Ó.; Salazar-Páramo, M.; Grover-Páez, F.; Nava-Zavala, A.H.; Cardona-Muñoz, E.G.; et al. Enalapril influence on arterial stiffness in rheumatoid arthritis women: A randomized clinical trial. Front. Med. 2019, 6, 341. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.M.; Yusuf, S.; Dzavik, V.; Doris, C.I.; Yi, Q.; Smith, S.; Moore-Cox, A.; Bosch, J.; Riley, W.A.; Teo, K.K. Effects of ramipril and vitamin E on atherosclerosis: The study to evaluate carotid ultrasound changes in patients treated with ramipril and vitamin E (SECURE). Circulation 2001, 103, 919–925. [Google Scholar] [CrossRef]
- Mitrovic, V.; Klein, H.H.; Krekel, N.; Kreuzer, J.; Fichtlscherer, S.; Schirmer, A.; Paar, W.D.; Hamm, C.W. Influence of the angiotensin converting enzyme inhibitor ramipril on high-sensitivity C-reactive protein (Hs-CRP) in patients with documented atherosclerosis. Z. Kardiol. 2005, 94, 336–342. [Google Scholar] [CrossRef]
- Zanchetti, A.; Crepaldi, G.; Bond, M.G.; Gallus, G.; Veglia, F.; Mancia, G.; Ventura, A.; Baggio, G.; Sampieri, L.; Rubba, P.; et al. Different effects of antihypertensive regimens based on fosinopril or hydrochlorothiazide with or without lipid lowering by pravastatin on progression of asymptomatic carotid atherosclerosis: Principal results of PHYLLIS—A randomized double-blind trial. Stroke 2004, 35, 2807–2812. [Google Scholar] [CrossRef] [Green Version]
- Ceconi, C.; Fox, K.M.; Remme, W.J.; Simoons, M.L.; Deckers, J.W.; Bertrand, M.; Parrinello, G.; Kluft, C.; Blann, A.; Cokkinos, D.; et al. ACE inhibition with perindopril and biomarkers of atherosclerosis and thrombosis: Results from the PERTINENT study. Atherosclerosis 2009, 204, 273–275. [Google Scholar] [CrossRef]
- McMurray, J.; Solomon, S.; Pieper, K.; Reed, S.; Rouleau, J.; Velazquez, E.; White, H.; Howlett, J.; Swedberg, K.; Maggioni, A.; et al. The effect of valsartan, captopril, or both on atherosclerotic events after acute myocardial infarction: An analysis of the valsartan in acute myocardial infarction trial (VALIANT). J. Am. Coll Cardiol. 2006, 47, 726–733. [Google Scholar] [CrossRef]
- Navalkar, S.; Parthasarathy, S.; Santanam, N.; Khan, B.V. Irbesartan, an angiotensin type 1 receptor inhibitor, regulates markers of inflammation in patients with premature atherosclerosis. J. Am. Coll. Cardiol. 2001, 37, 440–444. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.K.; Chung, W.-J.; Ahn, J.Y.; Han, S.H.; Kang, W.C.; Seo, Y.-H.; Ahn, T.H.; Choi, I.S.; Shin, E.K. Angiotensin II type 1 receptor blockers reduce tissue factor activity and plasminogen activator inhibitor type-1 antigen in hypertensive patients: A randomized, double-blind, placebo-controlled study. Atherosclerosis 2004, 177, 155–160. [Google Scholar] [CrossRef]
- Gong, X.; Shao, L.; Fu, Y.-M.; Zou, Y. Effects of olmesartan on endothelial progenitor cell mobilization and function in carotid atherosclerosis. Med. Sci. Monit. 2015, 21, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, A.; Yamamoto, K.; Miyoshi, T.; Hatanaka, K.; Hirohata, S.; Yamawaki, H.; Komatsubara, I.; Murakami, M.; Hirose, E.; Sato, S.; et al. Impact of olmesartan on progression of coronary atherosclerosis. J. Am. Coll. Cardiol. 2010, 55, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Hirohata, A.; Yamamoto, K.; Miyoshi, T.; Hatanaka, K.; Hirohata, S.; Yamawaki, H.; Komatsubara, I.; Hirose, E.; Kobayashi, Y.; Ohkawa, K.; et al. Four-year clinical outcomes of the OLIVUS-Ex (impact of olmesartan on progression of coronary atherosclerosis: Evaluation by intravascular ultrasound) extension trial. Atherosclerosis 2012, 220, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, K.O.; Agabiti-Rosei, E.; Zielinski, T.; Schremmer, D.; Scholze, J.; Laeis, P.; Schwandt, P.; Ludwig, M. MORE study investigators carotid intima-media thickness and plaque volume changes following 2-year angiotensin II-receptor blockade. The multicentre olmesartan atherosclerosis regression evaluation (MORE) study. Ther. Adv. Cardiovasc. Dis. 2007, 1, 97–106. [Google Scholar] [CrossRef]
- Ramadan, R.; Dhawan, S.S.; Binongo, J.N.G.; Alkhoder, A.; Jones, D.P.; Oshinski, J.N.; Quyyumi, A.A. Effect of angiotensin II Type i receptor blockade with valsartan on carotid artery atherosclerosis: A double blind randomized clinical trial comparing valsartan and placebo (EFFERVESCENT). Am. Heart J. 2016, 174, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.T.; Lauten, W.B.; Khan, Q.A.; Navalkar, S.; Parthasarathy, S.; Khan, B.V. Effects of eprosartan versus hydrochlorothiazide on markers of vascular oxidation and inflammation and blood pressure (renin-angiotensin system antagonists, oxidation, and inflammation). Am. J. Cardiol. 2002, 89, 686–690. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ozaki, H.; Takayasu, K.; Akehi, N.; Fukui, S.; Sakai, A.; Kodama, M.; Shimonagata, T.; Kobayashi, K.; Ota, M.; et al. The effect of losartan and amlodipine on left ventricular diastolic function and atherosclerosis in japanese patients with mild-to-moderate hypertension (J-ELAN) study. Hypertens. Res. 2011, 34, 325–330. [Google Scholar] [CrossRef]
- Janić, M.; Lunder, M.; Prezelj, M.; Šabovič, M. A Combination of low-dose fluvastatin and valsartan decreases inflammation and oxidative stress in apparently healthy middle-aged males. J. Cardiopulm. Rehabil. Prev. 2014, 34, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Fogari, R.; Maffioli, P.; Mugellini, A.; Zoppi, A.; Lazzari, P.; Derosa, G. Effects of losartan and amlodipine alone or combined with simvastatin in hypertensive patients with nonalcoholic hepatic steatosis. Eur. J. Gastroenterol. Hepatol. 2012, 24, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, H.; Nakazawa, T.; Shibuya, A.; Minamino, T.; Takada, J.; Tanaka, Y.; Okuwaki, Y.; Watanabe, M.; Koizumi, W. Effects of 1-Year administration of olmesartan on portal pressure and TGF-Beta1 in selected patients with cirrhosis: A randomized controlled trial. J. Gastroenterol. 2011, 46, 1316–1323. [Google Scholar] [CrossRef]
- Woo, K.-T.; Chan, C.-M.; Choong, H.-L.; Tan, H.-K.; Foo, M.; Lee, E.J.C.; Tan, C.-C.; Lee, G.S.L.; Tan, S.-H.; Vathsala, A.; et al. High dose losartan and ACE gene polymorphism in IgA nephritis. HUGO J. 2008, 2, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Tylicki, L.; Renke, M.; Rutkowski, P.; Rutkowski, B.; Lysiak-Szydłowska, W. Randomized, controlled study of the effects of losartan versus enalapril in small doses on proteinuria and tubular injury in primary glomerulonephritis. Med. Sci. Monit. 2005, 11, PI31–PI37. [Google Scholar]
- Tsuruoka, S.; Kai, H.; Usui, J.; Morito, N.; Saito, C.; Yoh, K.; Yamagata, K. Effects of irbesartan on inflammatory cytokine concentrations in patients with chronic glomerulonephritis. Intern. Med. 2013, 52, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the heart failure society of America. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failurethe task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Docherty, K.F.; Vaduganathan, M.; Solomon, S.D.; McMurray, J.J.V. Sacubitril/valsartan: Neprilysin inhibition 5 years after PARADIGM-HF. JACC Heart Fail. 2020, 8, 800–810. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dostal, D.E.; Rothblum, K.N.; Chernin, M.I.; Cooper, G.R.; Baker, K.M. Intracardiac detection of angiotensinogen and renin: A localized renin-angiotensin system in neonatal rat heart. Am. J. Physiol. Cell Physiol. 1992, 263, C838–C850. [Google Scholar] [CrossRef]
- Endo-Mochizuki, Y.; Mochizuki, N.; Sawa, H.; Takada, A.; Okamoto, H.; Kawaguchi, H.; Nagashima, K.; Kitabatake, A. Expression of renin and angiotensin-converting enzyme in human hearts. Heart Vessels 1995, 10, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Sawa, H.; Tokuchi, F.; Mochizuki, N.; Endo, Y.; Furuta, Y.; Shinohara, T.; Takada, A.; Kawaguchi, H.; Yasuda, H.; Nagashima, K. Expression of the angiotensinogen gene and localization of its protein in the human heart. Circulation 1992, 86, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Raizada, V.; Skipper, B.; Luo, W.; Griffith, J. Intracardiac and intrarenal renin-angiotensin systems: Mechanisms of cardiovascular and renal effects. J. Investig. Med. 2007, 55, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Nakamura, S.; Ito, T.; Nakayama, M.; Harada, E.; Mizuno, Y.; Sakamoto, T.; Yamamuro, M.; Saito, Y.; Nakao, K.; et al. Expression of aldosterone synthase gene in failing human heart: Quantitative analysis using modified real-time polymerase chain reaction. J. Clin. Endocrinol. Metab. 2002, 87, 3936–3940. [Google Scholar] [CrossRef]
- Lilly, L.S.; Pratt, R.E.; Alexander, R.W.; Larson, D.M.; Ellison, K.E.; Gimbrone, M.A.; Dzau, V.J. Renin expression by vascular endothelial cells in culture. Circ. Res. 1985, 57, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- De Mello, W.C. Local renin angiotensin aldosterone systems and cardiovascular diseases. Med. Clin. North Am. 2017, 101, 117–127. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Patel, M.P.; Chappell, M.; Butany, J.; Backx, P.H.; Tsushima, R.G.; Scholey, J.W.; Khokha, R.; Penninger, J.M. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc. Res. 2007, 75, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassiri, Z.; Zhong, J.; Guo, D.; Basu, R.; Wang, X.; Liu, P.P.; Scholey, J.W.; Penninger, J.M.; Oudit, G.Y. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ. Heart Fail. 2009, 2, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascolo, A.; Urbanek, K.; De Angelis, A.; Sessa, M.; Scavone, C.; Berrino, L.; Rosano, G.M.C.; Capuano, A.; Rossi, F. Angiotensin II and angiotensin 1–7: Which is their role in atrial fibrillation? Heart Fail Rev. 2020, 25, 367–380. [Google Scholar] [CrossRef]
- Alghamri, M.S.; Weir, N.M.; Anstadt, M.P.; Elased, K.M.; Gurley, S.B.; Morris, M. Enhanced angiotensin II-induced cardiac and aortic remodeling in ACE2 knockout mice. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Serneri, G.N.N.; Boddi, M.; Cecioni, I.; Vanni, S.; Coppo, M.; Papa, M.L.; Bandinelli, B.; Bertolozzi, I.; Polidori, G.; Toscano, T.; et al. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function. Circ. Res. 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.B.; Bodiga, S.; Fan, D.; Das, S.K.; Wang, Z.; Wang, W.; Basu, R.; Zhong, J.; Kassiri, Z.; Oudit, G.Y. Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1–7 in experimental heart failure in angiotensin-converting enzyme 2-null mice. Hypertension 2012, 59, 1195–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728, 18 p following 728. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, L.; van Esch, J.H.M.; Roks, A.J.M.; van den Meiracker, A.H.; Danser, A.H. Jan hypertension. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Lo, J.; Patel, V.B.; Wang, Z.; Levasseur, J.; Kaufman, S.; Penninger, J.M.; Oudit, G.Y. Angiotensin-converting enzyme 2 antagonizes angiotensin II-induced pressor response and NADPH oxidase activation in wistar-kyoto rats and spontaneously hypertensive rats. Exp. Physiol. 2013, 98, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Moreira de Macêdo, S.; Guimarães, T.A.; Feltenberger, J.D.; Sousa Santos, S.H. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 2014, 62, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Patel, V.B.; Abo Alrob, O.; Basu, R.; Altamimi, T.; Desaulniers, J.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1–7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart Fail. 2014, 7, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krähenbühl, S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef]
- Becker, L.K.; Totou, N.; Moura, S.; Kangussu, L.; Millán, R.D.S.; Campagnole-Santos, M.J.; Coelho, D.; Motta-Santos, D.; Santos, R.A.S. Eccentric overload muscle damage is attenuated by a novel angiotensin- (1–7) treatment. Int. J. Sports Med. 2018, 39, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Cassis, L.A.; Police, S.B.; Yiannikouris, F.; Thatcher, S.E. Local adipose tissue renin-angiotensin system. Curr. Hypertens. Rep. 2008, 10, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, K.; Wu, Y.; Okamoto, Y.; Pratt, R.E.; Dzau, V.J. Local Renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension 2006, 48, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Yoshimoto, T.; Hirono, Y.; Tateno, T.; Sugiyama, T.; Hirata, Y. Angiotensin II induces monocyte chemoattractant protein-1 expression via a nuclear factor-kappab-dependent pathway in rat preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E771–E778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurata, A.; Nishizawa, H.; Kihara, S.; Maeda, N.; Sonoda, M.; Okada, T.; Ohashi, K.; Hibuse, T.; Fujita, K.; Yasui, A.; et al. Blockade of angiotensin II type-1 receptor reduces oxidative stress in adipose tissue and ameliorates adipocytokine dysregulation. Kidney Int. 2006, 70, 1717–1724. [Google Scholar] [CrossRef] [Green Version]
- Harlow, B.L.; Stewart, E.G. A Population-based assessment of chronic unexplained vulvar pain: Have we underestimated the prevalence of vulvodynia? J. Am. Med. Womens Assoc. 2003, 58, 82–88. [Google Scholar]
- Meana, M.; Binik, Y.M.; Khalife, S.; Cohen, D.R. Biopsychosocial profile of women with dyspareunia. Obstet. Gynecol. 1997, 90, 583–589. [Google Scholar] [CrossRef]
- Reed, B.D.; Haefner, H.K.; Punch, M.R.; Roth, R.S.; Gorenflo, D.W.; Gillespie, B.W. Psychosocial and sexual functioning in women with vulvodynia and chronic pelvic pain. A comparative evaluation. J. Reprod. Med. 2000, 45, 624–632. [Google Scholar]
- Bazin, S.; Bouchard, C.; Brisson, J.; Morin, C.; Meisels, A.; Fortier, M. Vulvar vestibulitis syndrome: An exploratory case-control study. Obstet. Gynecol. 1994, 83, 47–50. [Google Scholar] [PubMed]
- Witkin, S.S.; Gerber, S.; Ledger, W.J. Differential characterization of women with vulvar vestibulitis syndrome. Am. J. Obstet. Gynecol. 2002, 187, 589–594. [Google Scholar] [CrossRef]
- Liao, Z.; Chakrabarty, A.; Mu, Y.; Bhattacherjee, A.; Goestch, M.; Leclair, C.M.; Smith, P.G. A Local inflammatory renin-angiotensin system drives sensory axon sprouting in provoked vestibulodynia. J. Pain 2017, 18, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Gianotten, W.L.; Drogendijk, A.C.; Weijmar Schultz, W.C.; Blindeman, L.A.; van der Meijden, W.I. Histopathologic features of vulvar vestibulitis. Int. J. Gynecol. Pathol. 1998, 17, 7–11. [Google Scholar] [CrossRef]
- Bohm-Starke, N.; Hilliges, M.; Falconer, C.; Rylander, E. Increased intraepithelial innervation in women with vulvar vestibulitis syndrome. Gynecol. Obstet. Invest. 1998, 46, 256–260. [Google Scholar] [CrossRef]
- Tympanidis, P.; Terenghi, G.; Dowd, P. Increased innervation of the vulval vestibule in patients with vulvodynia. Br. J. Dermatol. 2003, 148, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Côté, F.; Do, T.H.; Laflamme, L.; Gallo, J.M.; Gallo-Payet, N. Activation of the AT(2) receptor of angiotensin ii induces neurite outgrowth and cell migration in microexplant cultures of the cerebellum. J. Biol. Chem. 1999, 274, 31686–31692. [Google Scholar] [CrossRef] [Green Version]
- Gallinat, S.; Yu, M.; Dorst, A.; Unger, T.; Herdegen, T. Sciatic nerve transection evokes lasting up-regulation of angiotensin AT2 and AT1 receptor MRNA in adult rat dorsal root ganglia and sciatic nerves. Brain Res. Mol. Brain Res. 1998, 57, 111–122. [Google Scholar] [CrossRef]
- Reinecke, K.; Lucius, R.; Reinecke, A.; Rickert, U.; Herdegen, T.; Unger, T. Angiotensin II accelerates functional recovery in the rat sciatic nerve in vivo: Role of the AT2 receptor and the transcription factor NF-KappaB. FASEB J. 2003, 17, 2094–2096. [Google Scholar] [CrossRef]
- Anand, U.; Yiangou, Y.; Sinisi, M.; Fox, M.; MacQuillan, A.; Quick, T.; Korchev, Y.E.; Bountra, C.; McCarthy, T.; Anand, P. Mechanisms underlying clinical efficacy of angiotensin II Type 2 Receptor (AT2R) antagonist EMA401 in neuropathic pain: Clinical tissue and in vitro studies. Mol. Pain 2015, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Anand, U.; Facer, P.; Yiangou, Y.; Sinisi, M.; Fox, M.; McCarthy, T.; Bountra, C.; Korchev, Y.E.; Anand, P. Angiotensin II type 2 receptor (AT2 R) localization and antagonist-mediated inhibition of capsaicin responses and neurite outgrowth in human and rat sensory neurons. Eur. J. Pain 2013, 17, 1012–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarty, A.; Blacklock, A.; Svojanovsky, S.; Smith, P.G. Estrogen elicits dorsal root ganglion axon sprouting via a renin-angiotensin system. Endocrinology 2008, 149, 3452–3460. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Liao, Z.; Mu, Y.; Smith, P.G. Inflammatory renin-angiotensin system disruption attenuates sensory hyperinnervation and mechanical hypersensitivity in a rat model of provoked vestibulodynia. J. Pain 2018, 19, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Muthuraman, A.; Kaur, P. Renin-angiotensin-aldosterone system: A current drug target for the management of neuropathic pain. Curr. Drug Targets 2016, 17, 178–195. [Google Scholar] [CrossRef]
- Bali, A.; Singh, N.; Jaggi, A.S. Renin-angiotensin system in pain: Existing in a double life? J. Renin. Angiotensin. Aldosterone Syst. 2014, 15, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessaguet, F.; Magy, L.; Desmoulière, A.; Demiot, C. The Therapeutic potential of renin angiotensin aldosterone system (RAAS) in chronic pain: From preclinical studies to clinical trials. Expert Rev. Neurother. 2016, 16, 331–339. [Google Scholar] [CrossRef]
- Smith, M.T.; Wyse, B.D.; Edwards, S.R. Small molecule angiotensin II type 2 receptor (AT₂R) antagonists as novel analgesics for neuropathic pain: Comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain Med. 2013, 14, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: A comprehensive review. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sama, I.E.; Ravera, A.; Santema, B.T.; van Goor, H.; Ter Maaten, J.M.; Cleland, J.G.F.; Rienstra, M.; Friedrich, A.W.; Samani, N.J.; Ng, L.L.; et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur. Heart J. 2020, 41, 1810–1817. [Google Scholar] [CrossRef]
- Pang, X.; Cui, Y.; Zhu, Y. Recombinant human ACE2: Potential therapeutics of SARS-CoV-2 infection and its complication. Acta Pharmacol. Sin. 2020, 41, 1255–1257. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS Coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Bogaerts, P.; Rooman, M. Modeling the molecular impact of SARS-CoV-2 infection on the renin-angiotensin system. Viruses 2020, 12, 1367. [Google Scholar] [CrossRef] [PubMed]
- Van Lier, D.; Kox, M.; Santos, K.; van der Hoeven, H.; Pillay, J.; Pickkers, P. Increased blood angiotensin converting enzyme 2 activity in critically Ill COVID-19 patients. ERJ Open Res. 2021, 00848–02020. [Google Scholar] [CrossRef]
- Nagy, B.; Fejes, Z.; Szentkereszty, Z.; Sütő, R.; Várkonyi, I.; Ajzner, É.; Kappelmayer, J.; Papp, Z.; Tóth, A.; Fagyas, M. A dramatic rise in serum ACE2 activity in a critically Ill COVID-19 patient. Int. J. Infect. Dis. 2021, 103, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A Mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9, e59177. [Google Scholar] [CrossRef] [PubMed]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. COVID-19: ACE2centric infective disease? Hypertension 2020, 76, 294–299. [Google Scholar] [CrossRef]
- Kai, H.; Kai, M. Interactions of Coronaviruses with ACE2, Angiotensin II, and RAS inhibitors—Lessons from available evidence and insights into COVID-19. Hypertens. Res. 2020, 43, 648–654. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-NCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.B.J.; Khalil, N. Angiotensin II type 1 receptor blocker inhibits pulmonary injury. Clin. Invest. Med. 2005, 28, 118–126. [Google Scholar]
- Kintscher, U.; Slagman, A.; Domenig, O.; Röhle, R.; Konietschke, F.; Poglitsch, M.; Möckel, M. Plasma angiotensin peptide profiling and ACE (Angiotensin-Converting Enzyme)-2 activity in COVID-19 patients treated with pharmacological blockers of the renin-angiotensin system. Hypertension 2020, 76, e34–e36. [Google Scholar] [CrossRef]
- Rieder, M.; Wirth, L.; Pollmeier, L.; Jeserich, M.; Goller, I.; Baldus, N.; Schmid, B.; Busch, H.-J.; Hofmann, M.; Kern, W.; et al. Serum ACE-2, angiotensin II, and aldosterone levels are unchanged in patients with COVID-19. Am. J. Hypertens. 2020. [Google Scholar] [CrossRef]
- Vicenzi, M.; Di Cosola, R.; Ruscica, M.; Ratti, A.; Rota, I.; Rota, F.; Bollati, V.; Aliberti, S.; Blasi, F. The liaison between respiratory failure and high blood pressure: Evidence from COVID-19 patients. Eur. Respir. J. 2020, 56, 2001157. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. Risks of ACE inhibitor and ARB usage in COVID-19: Evaluating the evidence. Clin. Pharmacol. Ther. 2020, 108, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.D.; Macedo, A.V.S.; de Barros E Silva, P.G.M.; Moll-Bernardes, R.J.; Feldman, A.; D’Andréa Saba Arruda, G.; de Souza, A.S.; de Albuquerque, D.C.; Mazza, L.; Santos, M.F.; et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)--the brace Corona trial. Am. Heart J. 2020, 226, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, G.T.M.; Laghmani, E.H.; Fidder, M.; Sengers, R.M.A.; de Visser, Y.P.; de Vries, L.; Rink, R.; Roks, A.J.M.; Folkerts, G.; Walther, F.J. Agonists of MAS oncogene and angiotensin II type 2 receptors attenuate cardiopulmonary disease in rats with neonatal hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L341–L351. [Google Scholar] [CrossRef]
- Foulquier, S.; Steckelings, U.M.; Unger, T. Impact of the AT(2) receptor agonist C21 on blood pressure and beyond. Curr. Hypertens. Rep. 2012, 14, 403–409. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A Potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Uhal, B.D.; Li, X.; Xue, A.; Gao, X.; Abdul-Hafez, A. Regulation of alveolar epithelial cell survival by the ACE-2/Angiotensin 1–7/Mas Axis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L269–L274. [Google Scholar] [CrossRef] [Green Version]
- Dalbeth, N. The non-thiol angiotensin-converting enzyme inhibitor quinapril suppresses inflammatory arthritis. Rheumatology 2005, 44, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, G.C.B.; de Menezes, M.S.S.; de Araújo, A.A.; de Araújo Júnior, R.F.; de Medeiros, C.A.C.X. Olmesartan prevented intra-articular inflammation induced by zymosan in rats. Biol. Pharm. Bull. 2016, 39, 1793–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagawa, K.; Nagatani, K.; Komagata, Y.; Yamamoto, K. Angiotensin receptor blockers suppress antigen-specific T cell responses and ameliorate collagen-induced arthritis in mice. Arthritis Rheum. 2005, 52, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Refaat, R.; Salama, M.; Abdel Meguid, E.; El Sarha, A.; Gowayed, M. Evaluation of the effect of losartan and methotrexate combined therapy in adjuvant-induced arthritis in rats. Eur. J. Pharmacol. 2013, 698, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Namisaki, T.; Kitade, M.; Kaji, K.; Shirai, Y.; Yoshii, J.; Yanase, K.; Yamazaki, M.; et al. Losartan, an angiotensin-II type 1 receptor blocker, attenuates the liver fibrosis development of non-alcoholic steatohepatitis in the rat. BMC Res. Notes 2009, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Kurita, S.; Takamura, T.; Ota, T.; Matsuzawa-Nagata, N.; Kita, Y.; Uno, M.; Nabemoto, S.; Ishikura, K.; Misu, H.; Ando, H.; et al. Olmesartan ameliorates a dietary rat model of non-alcoholic steatohepatitis through its pleiotropic effects. Eur. J. Pharmacol. 2008, 588, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Kudo, H.; Yata, Y.; Takahara, T.; Kawai, K.; Nakayama, Y.; Kanayama, M.; Oya, T.; Morita, S.; Sasahara, M.; Mann, D.A.; et al. Telmisartan attenuates progression of steatohepatitis in mice: Role of hepatic macrophage infiltration and effects on adipose tissue. Liver. Int. 2009, 29, 988–996. [Google Scholar] [CrossRef]
- Kuwashiro, S.; Terai, S.; Oishi, T.; Fujisawa, K.; Matsumoto, T.; Nishina, H.; Sakaida, I. Telmisartan improves nonalcoholic steatohepatitis in medaka (Oryzias Latipes) by reducing macrophage infiltration and fat accumulation. Cell Tissue Res. 2011, 344, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Abajo, F.J. Renin–angiotensin system inhibitors and COVID-19: Overwhelming evidence against an association. Lancet Digit. Health 2020. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saravi, B.; Li, Z.; Lang, C.N.; Schmid, B.; Lang, F.K.; Grad, S.; Alini, M.; Richards, R.G.; Schmal, H.; Südkamp, N.; et al. The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis? Cells 2021, 10, 650. https://doi.org/10.3390/cells10030650
Saravi B, Li Z, Lang CN, Schmid B, Lang FK, Grad S, Alini M, Richards RG, Schmal H, Südkamp N, et al. The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis? Cells. 2021; 10(3):650. https://doi.org/10.3390/cells10030650
Chicago/Turabian StyleSaravi, Babak, Zhen Li, Corinna N. Lang, Bonaventura Schmid, Frauke K. Lang, Sibylle Grad, Mauro Alini, Robert Geoffrey Richards, Hagen Schmal, Norbert Südkamp, and et al. 2021. "The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis?" Cells 10, no. 3: 650. https://doi.org/10.3390/cells10030650