
Genetically Raised Circulating Bilirubin Levels and Risk of Ten Cancers: A Mendelian Randomization Study

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
Statistical Analyses
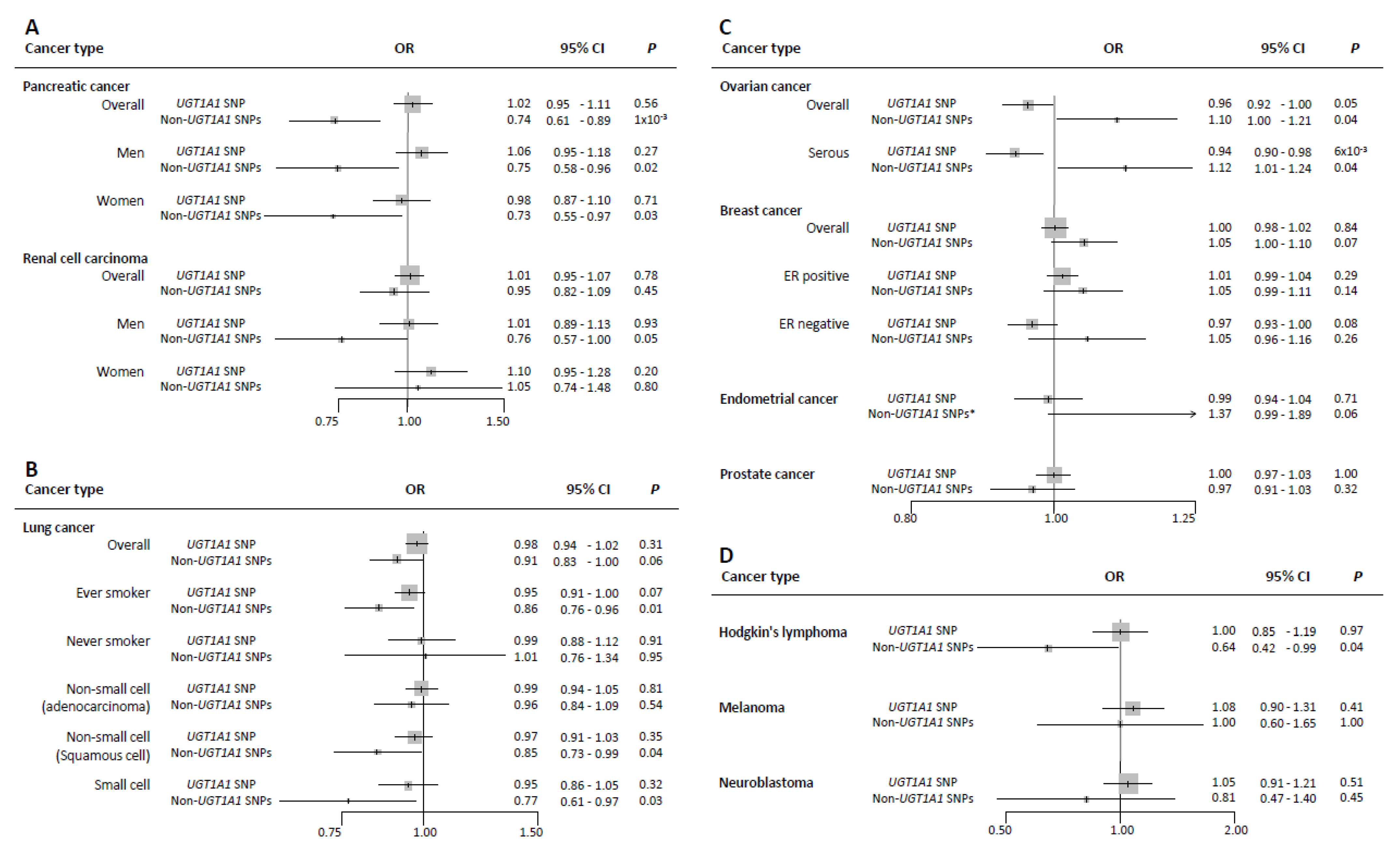
3. Results
Sensitivity Analyses
4. Discussion
4.1. Lung Cancer
4.2. Hodgkin’s Lymphoma
4.3. Breast Cancer and Other Hormone-Related Cancers
4.4. Pancreatic Cancer
4.5. Other Cancers (Renal Cell Cancer, Prostate Cancer, Melanoma, and Neuroblastoma)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
Abbreviations
| CI | confidence interval |
| CRC | colorectal cancer |
| ER | estrogen receptor |
| GS | Gilbert’s syndrome |
| GWAS | genome-wide association studies |
| IVW | inverse-variance weighted |
| MR | Mendelian randomization |
| 1-SD | one-standard deviation |
| OR | odds ratio |
| SNP | single-nucleotide polymorphism |
| TA | thymine–adenine |
| UGT1A1 | uridine-diphosphoglucuronate glucuronosyltransferase1A1 |
| UKB | UK Biobank |
Appendix A
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Bock, K.W. Human AHR functions in vascular tissue: Pro- and anti-inflammatory responses of AHR agonists in atherosclerosis. Biochem. Pharmacol. 2019, 159, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, H.; Kang, S.; Lee, J.; Park, J.; Jon, S. Bilirubin Nanoparticles as a Nanomedicine for Anti-inflammation Therapy. Angew. Chem. Int. Ed. 2016, 55, 7460–7463. [Google Scholar] [CrossRef] [PubMed]
- Zelenka, J.; Dvořák, A.; Alán, L.; Zadinová, M.; Haluzík, M.; Vítek, L. Hyperbilirubinemia Protects against Aging-Associated Inflammation and Metabolic Deterioration. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deming, S.L.; Zheng, W.; Xu, W.-H.; Cai, Q.; Ruan, Z.; Xiang, Y.-B.; Shu, X.-O. UGT1A1 Genetic Polymorphisms, Endogenous Estrogen Exposure, Soy Food Intake, and Endometrial Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2008, 17, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.D.; Kavousi, M.; Smith, A.V.; Chen, M.-H.; Dehghan, A.; Aspelund, T.; Lin, J.-P.; Van Duijn, C.M.; Harris, T.B.; Cupples, L.A.; et al. Genome-wide association meta-analysis for total serum bilirubin levels. Hum. Mol. Genet. 2009, 18, 2700–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stender, S.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Grande, P.; Tybjaerg-Hansen, A. Genetically elevated bilirubin and risk of ischaemic heart disease: Three Mendelian randomization studies and a meta-analysis. J. Intern. Med. 2012, 273, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Duguay, Y.; McGrath, M.; Lépine, J.; Gagné, J.-F.; Hankinson, S.E.; Colditz, G.A.; Hunter, D.J.; Plante, M.; Têtu, B.; Bélanger, A.; et al. The Functional UGT1A1 Promoter Polymorphism Decreases Endometrial Cancer Risk. Cancer Res. 2004, 64, 1202–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, S.K.; Rossing, M.A.; Malone, K.E.; Schwartz, S.M.; Weiss, N.S.; Chen, C. Ovarian Cancer Risk and Polymorphisms Involved in Estrogen Catabolism. Cancer Epidemiol. Biomark. Prev. 2007, 16, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, Y.; Kanai, M.; Narahara, M.; Tamon, A.; Brown, J.B.; Taneishi, K.; Nakatsui, M.; Okamoto, K.; Uneno, Y.; Yamaguchi, D.; et al. Association between UGT1A1*28*28 genotype and lung cancer in the Japanese population. Int. J. Clin. Oncol. 2016, 22, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Horsfall, L.J.; Burgess, S.; Hall, I.; Nazareth, I. Genetically raised serum bilirubin levels and lung cancer: A cohort study and Mendelian randomisation using UK Biobank. Thorax 2020, 75, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Tsezou, A.; Tzetis, M.; Giannatou, E.; Gennatas, C.; Pampanos, A.; Kanavakis, E.; Kitsiou-Tzeli, S. Genetic Polymorphisms in theUGT1A1Gene and Breast Cancer Risk in Greek Women. Genet. Test. 2007, 11, 303–306. [Google Scholar] [CrossRef]
- Karatzas, A.; Giannatou, E.; Tzortzis, V.; Gravas, S.; Aravantinos, E.; Moutzouris, G.; Melekos, M.; Tsezou, A. Genetic polymorphisms in the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene and prostate cancer risk in Caucasian men. Cancer Epidemiol. 2010, 34, 345–349. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.; Butterworth, A.S.; Thompson, J.R. Beyond Mendelian randomization: How to interpret evidence of shared genetic predictors. J. Clin. Epidemiol. 2016, 69, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnott-Armstrong, N.; Gen, F.; Tanigawa, Y.; Amar, D.; Mars, N.; Benner, C.; Aguirre, M.; Venkataraman, G.R.; Wainberg, M.; Ollila, H.M.; et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 2021, 53, 185–194. [Google Scholar] [CrossRef]
- Collins, R. What makes UK Biobank special? Lancet (Lond. Engl.) 2012, 379, 1173–1174. [Google Scholar] [CrossRef]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute. Available online: https://ldlinkncinihgov/?tab=ldpair (accessed on 10 February 2020).
- Burgess, S.; Thompson, S.G. CRP CHD Genetics Collaboration Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Petersen, G.M.; Amundadottir, L.; Fuchs, C.S.; Kraft, P.; Stolzenberg-Solomon, R.Z.; Jacobs, K.B.; Arslan, A.A.; Bueno-De-Mesquita, H.B.; Gallinger, S.; Gross, M.D.; et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat. Genet. 2010, 42, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Childs, E.J.; Mocci, E.; Campa, D.; Bracci, P.M.; Gallinger, S.; Goggins, M.; Li, D.; Neale, R.E.; Olson, S.H.; Scelo, G.; et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat. Genet. 2015, 47, 911–916. [Google Scholar] [CrossRef]
- Amundadottir, L.; Kraft, P.; Stolzenberg-Solomon, R.Z.; Fuchs, C.S.; Petersen, G.M.; Arslan, A.A.; Bueno-De-Mesquita, H.B.; Gross, M.D.; Helzlsouer, K.J.; Jacobs, E.J.; et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat. Genet. 2009, 41, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Scelo, G.; Purdue, M.P.; Brown, K.M.; Johansson, M.; Wang, Z.; Eckel-Passow, J.E.; Ye, Y.; Hofmann, J.N.; Choi, J.; Foll, M.; et al. Genome-wide association study identifies multiple risk loci for renal cell carcinoma. Nat. Commun. 2017, 8, 15724. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.D.; SpiroMeta Consortium; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef]
- Phelan, C.M.; AOCS Study Group; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [Green Version]
- O’Mara, T.A.; Glubb, D.M.; Amant, F.; Annibali, D.; Ashton, K.; Attia, J.; Auer, P.L.; Beckmann, M.W.; Black, A.; Bolla, M.K.; et al. Identification of nine new susceptibility loci for endometrial cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Collaborators, N.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Urayama, K.Y.; Jarrett, R.F.; Hjalgrim, H.; Diepstra, A.; Kamatani, Y.; Chabrier, A.; Gaborieau, V.; Boland, A.; Nieters, A.; Becker, N.; et al. Genome-Wide Association Study of Classical Hodgkin Lymphoma and Epstein–Barr Virus Status–Defined Subgroups. J. Natl. Cancer Inst. 2012, 104, 240–253. [Google Scholar] [CrossRef] [Green Version]
- Amos, C.I.; Wang, L.-E.; Lee, J.E.; Gershenwald, J.E.; Chen, W.V.; Fang, S.; Kosoy, R.; Zhang, M.; Qureshi, A.A.; Vattathil, S.; et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet. 2011, 20, 5012–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; De Mariano, M.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinog 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozen, W.; Timofeeva, M.N.; Li, D.; Diepstra, A.; Hazelett, D.J.; Delahayesourdeix, M.; Edlund, C.K.; Franke, L.; Rostgaard, K.; Berg, D.J.V.D.; et al. A meta-analysis of Hodgkin lymphoma reveals 19p13.3 TCF3 as a novel susceptibility locus. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genotypes and Phenotypes Database. Available online: https://wwwncbinlmnihgov/gap (accessed on 10 February 2020).
- Online Platform for MR Analyses. Available online: http://wwwmrbaseorg (accessed on 10 February 2020).
- Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int. J. Epidemiol. 2014, 43, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Dudbridge, F.; Thompson, S.G. Combining information on multiple instrumental variables in Mendelian randomization: Comparison of allele score and summarized data methods. Stat. Med. 2016, 35, 1880–1906. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Bowden, J.K.; Smith, G.D.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, F.P.; Smith, G.D.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magno, R.; Maia, A.-T. gwasrapidd: An R package to query, download and wrangle GWAS Catalog data. Bioinform 2019, 36, 649–650. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Baird, D.; Borges, M.-C.; Bowden, J.; Hemani, G.; Haycock, P.; Evans, D.M.; Smith, G.D. Recent Developments in Mendelian Randomization Studies. Curr. Epidemiol. Rep. 2017, 4, 330–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.-E.; Kimm, H.; Jee, S.H. Combined Effects of Smoking and Bilirubin Levels on the Risk of Lung Cancer in Korea: The Severance Cohort Study. PLoS ONE 2014, 9, e103972. [Google Scholar] [CrossRef]
- Wen, C.-P.; Zhang, F.; Liang, D.; Wen, C.; Gu, J.; Skinner, H.D.; Chow, W.-H.; Ye, Y.; Pu, X.; Hildebrandt, M.A.; et al. The Ability of Bilirubin in Identifying Smokers with Higher Risk of Lung Cancer: A Large Cohort Study in Conjunction with Global Metabolomic Profiling. Clin. Cancer Res. 2015, 21, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Freisling, H.; Khoei, N.S.; Viallon, V.; Wagner, K.-H. Gilbert’s syndrome, circulating bilirubin and lung cancer: A genetic advantage? Thorax 2020, 75, 916–917. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A. Epstein Barr Virus-Associated Hodgkin Lymphoma. Cancers 2018, 10, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Wilson, A.T.; Luxon, B.A.; Brown, K.E.; Mathahs, M.M.; Bandyopadhyay, S.; McCaffrey, A.P.; Schmidt, W.N. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: Mechanism for the antiviral effects of heme oxygenase? Hepatology (Baltimore Md) 2010, 52, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Li, H.; Luo, X.; Li, Y.; Bode, A.; Cao, Y. The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int. J. Cancer 2017, 141, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Qiu, L.-X.; Yu, L.; Yang, Z.; Yu, X.-J.; Zhong, Y.; Yu, L. The association between TA-repeat polymorphism in the promoter region of UGT1A1 and breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2010, 122, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tracy, T.S.; Remmel, R.P. Correlation between Bilirubin Glucuronidation and Estradiol-3-Gluronidation in the Presence of Model UDP-Glucuronosyltransferase 1A1 Substrates/Inhibitors. Drug Metab. Dispos. 2010, 39, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Saed, G.M.; Fletcher, N.M.; Jiang, Z.L.; Abu-Soud, H.M.; Diamond, M.P. Dichloroacetate Induces Apoptosis of Epithelial Ovarian Cancer Cells Through a Mechanism Involving Modulation of Oxidative Stress. Reprod. Sci. 2011, 18, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Khoei, N.S.; Jenab, M.; Murphy, N.; Banbury, B.L.; Carreras-Torres, R.; Viallon, V.; Kühn, T.; Bueno-de-Mesquita, B.; Aleksandrova, K.; Cross, A.J.; et al. Circulating bilirubin levels and risk of colorectal cancer: Serological and Mendelian randomization analyses. BMC Med. 2020, 18, 229. [Google Scholar]
- Haycock, P.C.; Burgess, S.; Wade, K.H.; Bowden, J.; Relton, C.L.; Smith, G.D. Best (but oft-forgotten) practices: The design, analysis, and interpretation of Mendelian randomization studies. Am. J. Clin. Nutr. 2016, 103, 965–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, P.G.; Mackenzie, P.I.; Chowdhury, J.R.; Guillemette, C.; Gregory, P.A.; Ishii, Y.; Hansen, A.J.; Kessler, F.K.; Kim, P.M.; Chowdhury, N.R.; et al. Glucuronidation and the UDP-glucuronosyltransferases in health and disease. Drug Metab. Dispos. Biol. Fate Chem. 2004, 32, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Innocenti, F.; Ratain, M.J. Linkage disequilibrium across the UGT1A locus should not be ignored in association studies of cancer susceptibility. Clin. Cancer Res. 2005, 11, 1348. [Google Scholar]
- Schooling, C.M. Biases in GWAS–the dog that did not bark. bioRxiv 2019, 709063. [Google Scholar] [CrossRef]


{kind=link}
| Cancer Type | Subtype | N Cases | N Controls | SNP Set | Minimum Detectable OR |
|---|---|---|---|---|---|
| Pancreatic cancer | overall | 7110 | 7264 | UGT1A1 SNP | 1.12/0.89 |
| Non-UGT1A1 SNPs (n = 113) | 1.30/0.77 | ||||
| men | 3861 | 4056 | UGT1A1 SNP | 1.17/0.86 | |
| Non-UGT1A1 SNPs (n = 113) | 1.43/0.70 | ||||
| women | 3252 | 3268 | UGT1A1 SNP | 1.18/0.84 | |
| Non-UGT1A1 SNPs (n = 113) | 1.48/0.67 | ||||
| Renal cell cancer | overall | 10,784 | 20,406 | UGT1A1 SNP | 1.08/0.92 |
| Non-UGT1A1 SNPs (n = 111) | 1.21/0.83 | ||||
| men | 3227 | 4916 | UGT1A1 SNP | 1.17/0.86 | |
| Non-UGT1A1 SNPs (n = 109) | 1.43/0.70 | ||||
| women | 1992 | 3095 | UGT1A1 SNP | 1.22/0.82 | |
| Non-UGT1A1 SNPs (n = 109) | 1.58/0.63 | ||||
| Lung cancer | overall | 29,266 | 56,450 | UGT1A1 SNP | 1.05/0.95 |
| Non-UGT1A1 SNPs (n = 109) | 1.12/0.89 | ||||
| ever smokers | 23,223 | 16,964 | UGT1A1 SNP | 1.07/0.93 | |
| Non-UGT1A1 SNPs (n = 109) | 1.17/0.85 | ||||
| never smokers | 2355 | 7504 | UGT1A1 SNP | 1.17/0.85 | |
| Non-UGT1A1 SNPs (n = 109) | 1.46/0.69 | ||||
| adenocarcinoma | 11,273 | 55,483 | UGT1A1 SNP | 1.07/0.93 | |
| Non-UGT1A1 SNPs (n = 109) | 1.18/0.85 | ||||
| squamous cell | 7426 | 55,627 | UGT1A1 SNP | 1.09/0.92 | |
| Non-UGT1A1 SNPs (n = 109) | 1.22/0.82 | ||||
| small cell | 2664 | 21,444 | UGT1A1 SNP | 1.15/0.87 | |
| Non-UGT1A1 SNPs (n = 109) | 1.39/0.72 | ||||
| Ovarian cancer | overall | 25,509 | 40,941 | UGT1A1 SNP | 1.06/0.95 |
| Non-UGT1A1 SNPs (n = 111) | 1.14/0.88 | ||||
| serous | 16,003 | 40,941 | UGT1A1 SNP | 1.07/0.94 | |
| Non-UGT1A1 SNPs (n = 111) | 1.16/0.86 | ||||
| Breast cancer | overall | 122,977 | 105,974 | UGT1A1 SNP | 1.03/0.97 |
| Non-UGT1A1 SNPs (n = 112) | 1.07/0.94 | ||||
| Endometrial cancer | overall | 12,906 | 108,979 | UGT1A1 SNP | 1.07/0.94 |
| Non-UGT1A1 SNPs (n = 110) | 1.16/0.86 | ||||
| Prostate cancer | overall | 79,194 | 61,112 | UGT1A1 SNP | 1.04/0.96 |
| Non-UGT1A1 SNPs (n = 107) | 1.09/0.92 | ||||
| Hodgkin’s lymphoma | overall | 1200 | 6417 | UGT1A1 SNP | 1.24/0.81 |
| Non-UGT1A1 SNPs (n = 91) | 1.65/0.61 | ||||
| Melanoma | overall | 1804 | 1026 | UGT1A1 SNP | 1.31/0.77 |
| Non-UGT1A1 SNPs (n = 75) | 1.86/0.54 | ||||
| Neuroblastoma | overall | 1627 | 3254 | UGT1A1 SNP | 1.23/0.81 |
| Non-UGT1A1 SNPs (n = 57) | 1.62/0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seyed Khoei, N.; Carreras-Torres, R.; Murphy, N.; Gunter, M.J.; Brennan, P.; Smith-Byrne, K.; Mariosa, D.; Mckay, J.; O’Mara, T.A., ECAC group; Jarrett, R.; et al. Genetically Raised Circulating Bilirubin Levels and Risk of Ten Cancers: A Mendelian Randomization Study. Cells 2021, 10, 394. https://doi.org/10.3390/cells10020394
Seyed Khoei N, Carreras-Torres R, Murphy N, Gunter MJ, Brennan P, Smith-Byrne K, Mariosa D, Mckay J, O’Mara TA ECAC group, Jarrett R, et al. Genetically Raised Circulating Bilirubin Levels and Risk of Ten Cancers: A Mendelian Randomization Study. Cells. 2021; 10(2):394. https://doi.org/10.3390/cells10020394
Chicago/Turabian StyleSeyed Khoei, Nazlisadat, Robert Carreras-Torres, Neil Murphy, Marc J. Gunter, Paul Brennan, Karl Smith-Byrne, Daniela Mariosa, James Mckay, Tracy A. O’Mara ECAC group, Ruth Jarrett, and et al. 2021. "Genetically Raised Circulating Bilirubin Levels and Risk of Ten Cancers: A Mendelian Randomization Study" Cells 10, no. 2: 394. https://doi.org/10.3390/cells10020394