
Role of Chitinase 3-Like 1 Protein in the Pathogenesis of Hepatic Insulin Resistance in Nonalcoholic Fatty Liver Disease

 ,
,
Abstract
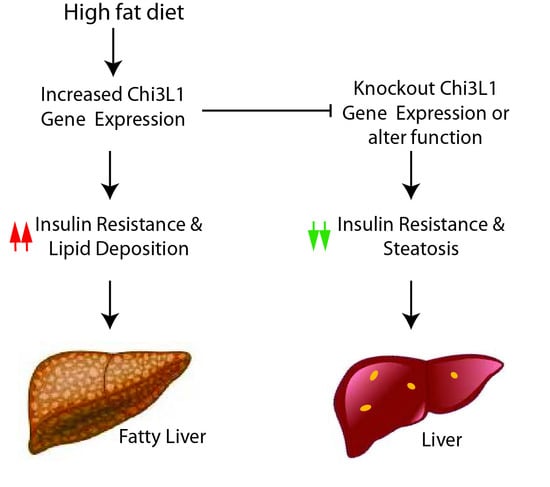
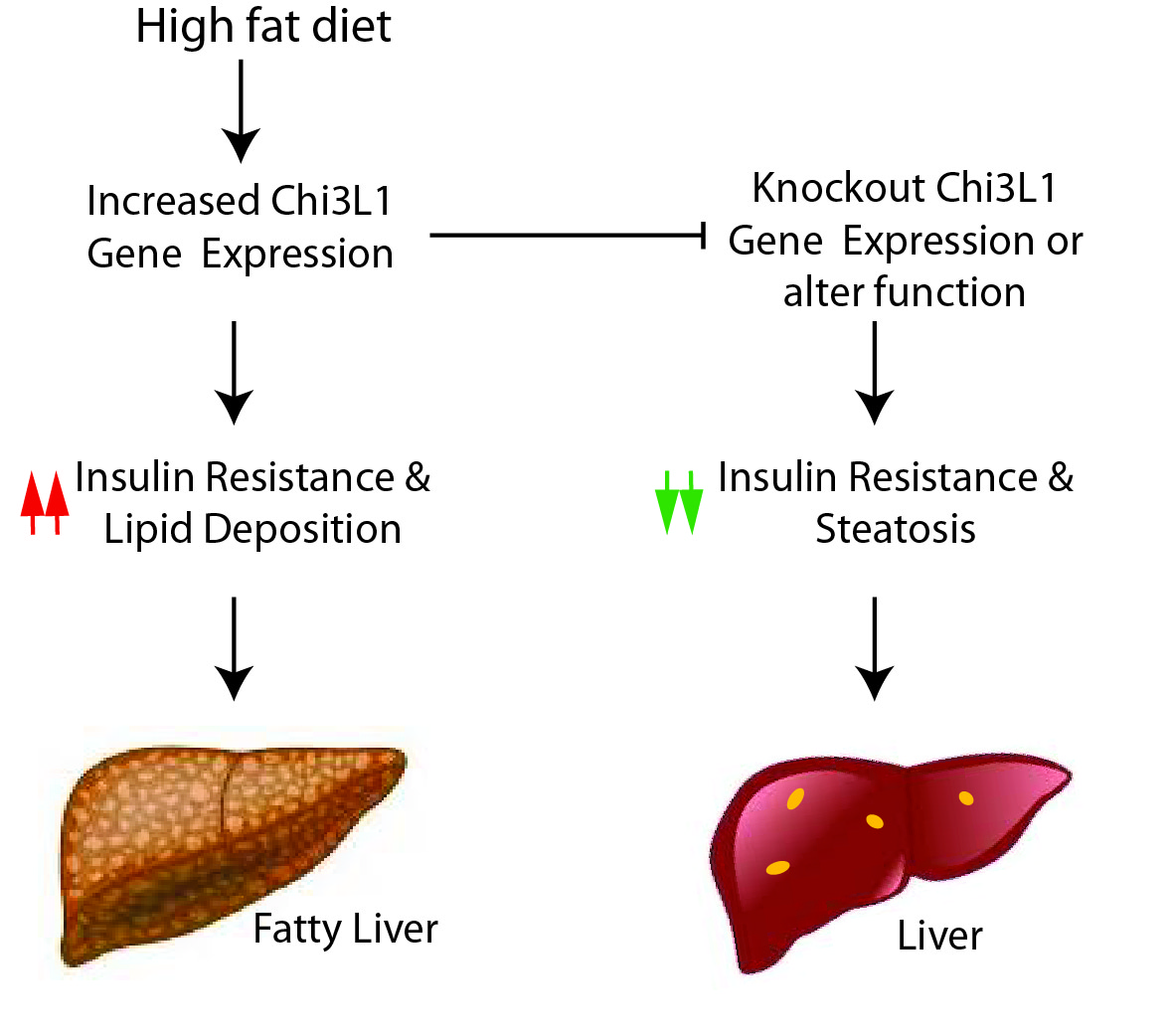
:
1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Human Liver Biopsy Specimens
2.3. Preparation of Monoclonal Anti-Chi3L1 Antibodies
2.4. Insulin Stimulation
2.5. Measurement of Chi3L1 Gene Expression
2.6. Protein Extraction
2.7. Western Blot Analysis
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Hematoxylin & Eosin Staining
2.10. Oil Red O (ORO) Staining
2.11. Quantitative Liver Triglyceride Analysis
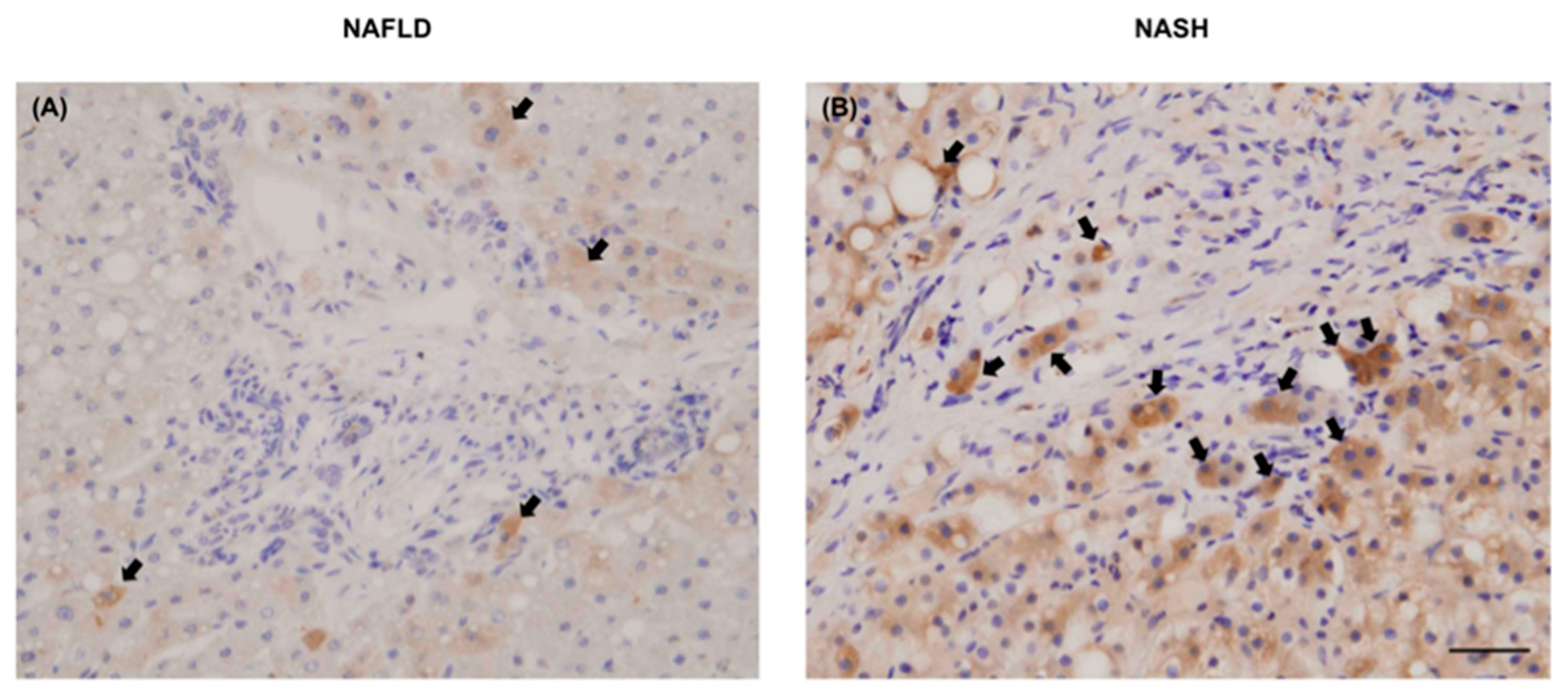
2.12. Immunohistochemical Staining of Human Liver Biopsies for Chi3L1 Protein Expression
2.13. Statistical Analysis
3. Results
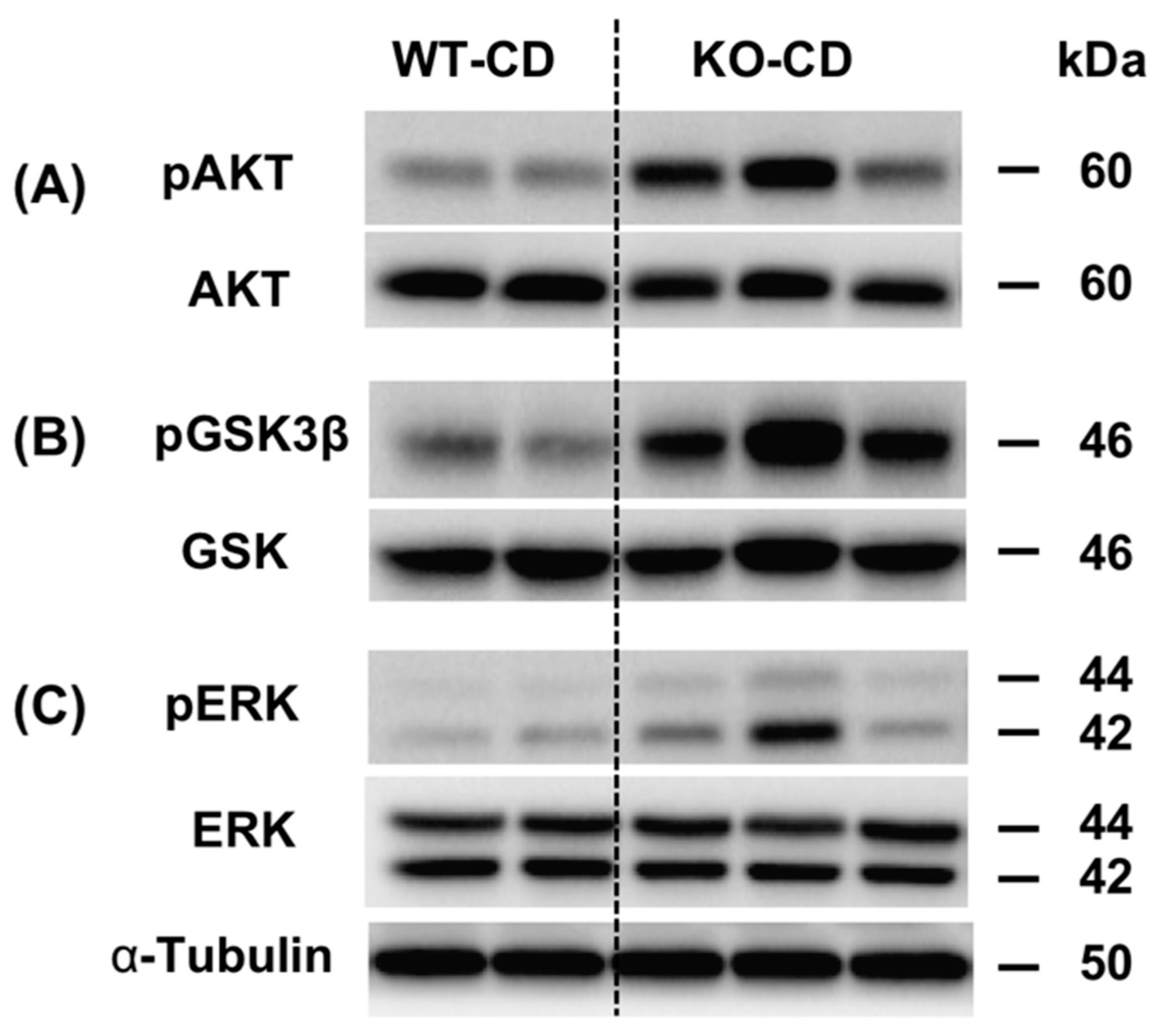
3.1. Knockout of the Chi3L1 Gene Enhanced Hepatic Insulin Signal Transduction
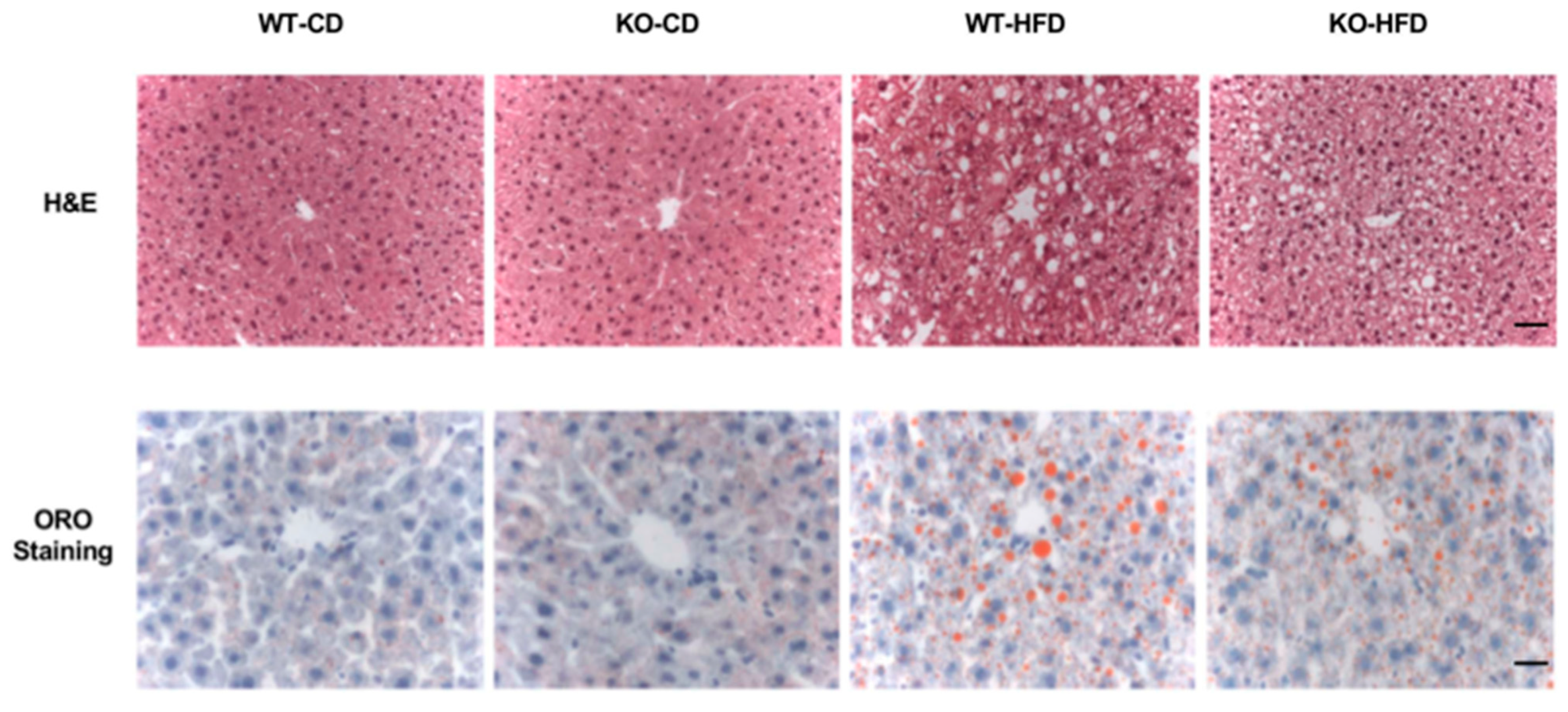
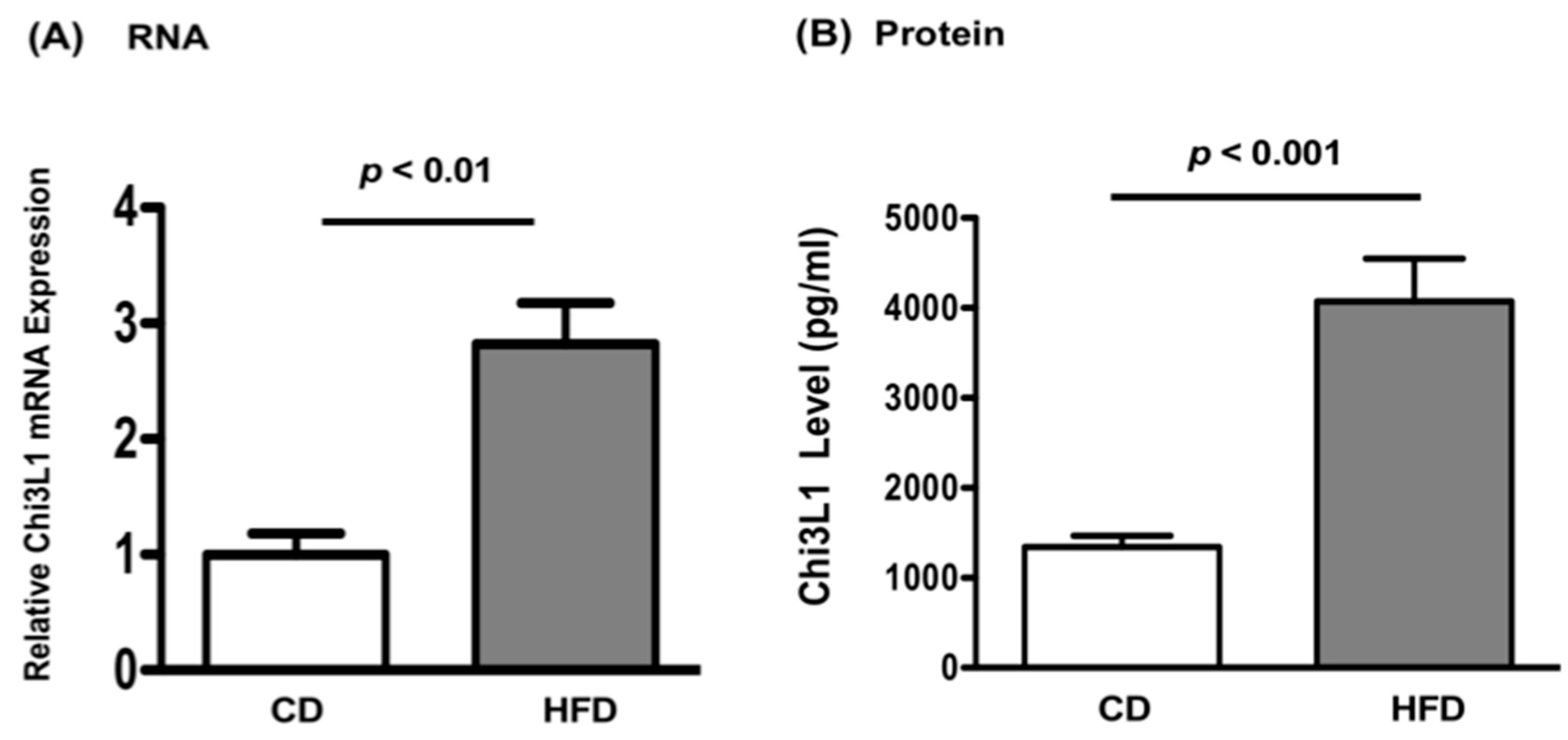
3.2. High Fat Diet Increased Chi3L1 Gene and Protein Expression in the Liver of Wild Type C57BL/6 Mice
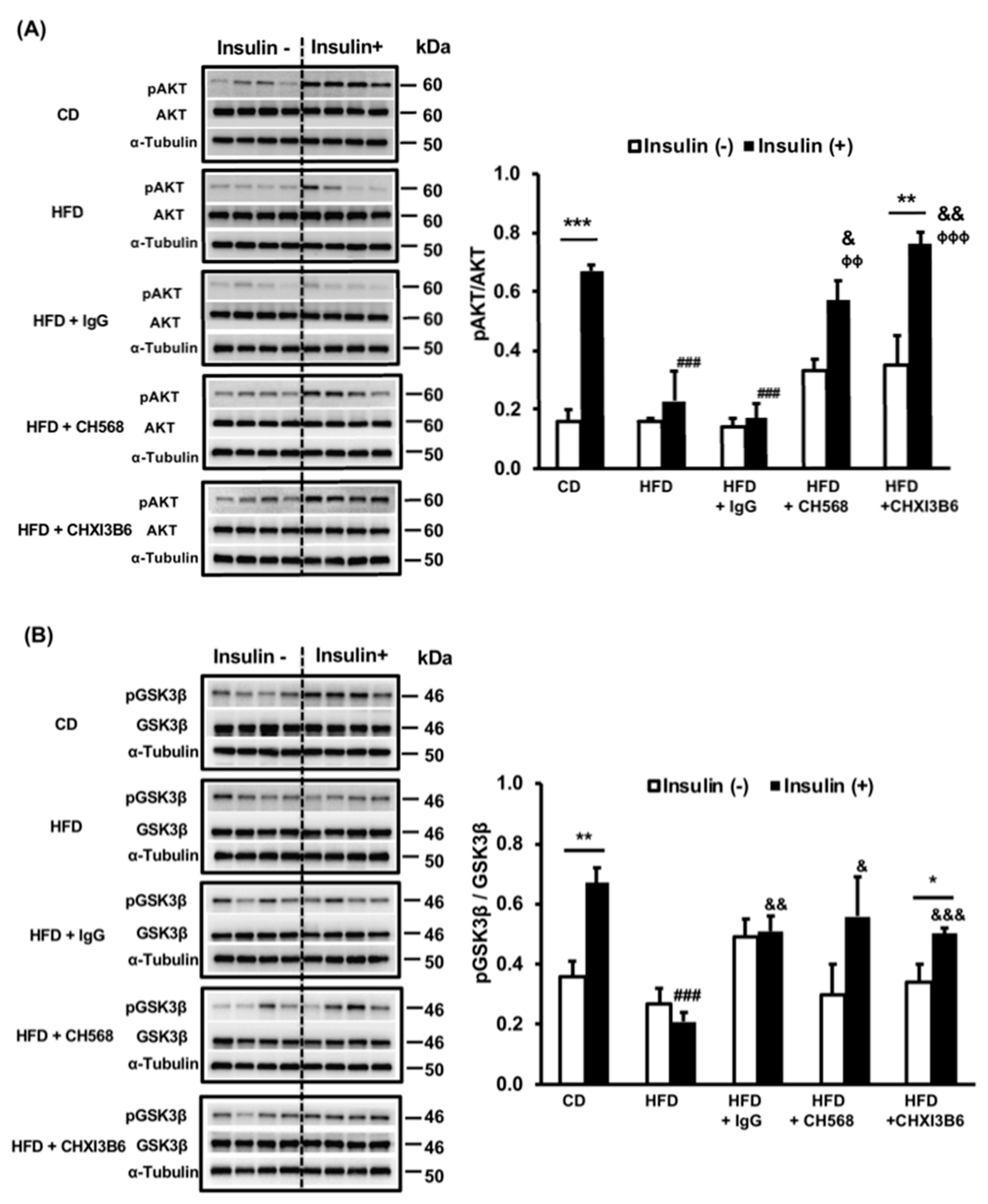
3.3. Monoclonal Anti-Chi3L1 Antibodies’ Administration Improves Hepatic Insulin Signaling in HFD Fed Mice
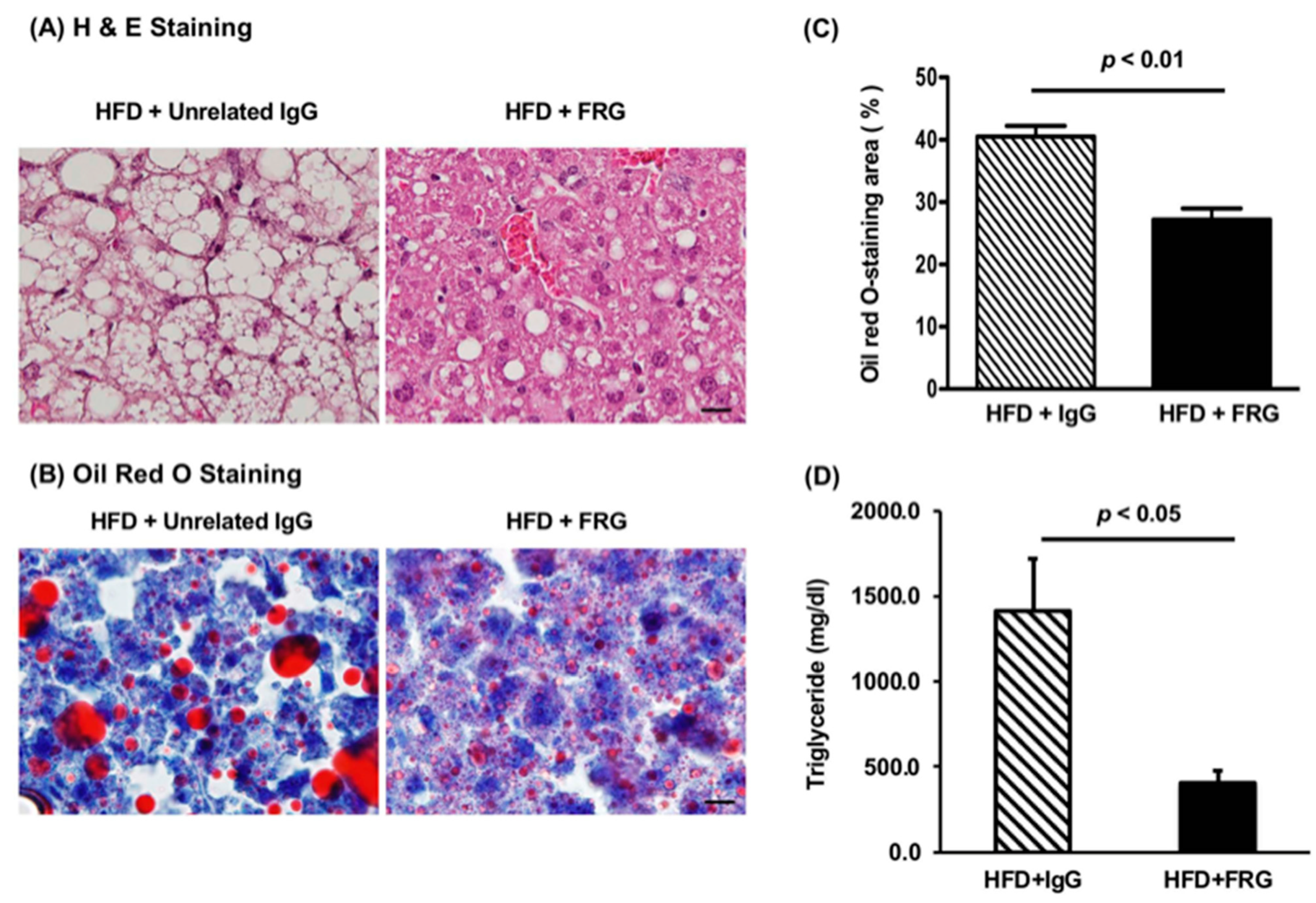
3.3.1. Effects of FRG on the Hepatic Insulin Signaling Cascade
3.3.2. Effects of Anti-Chi3L1 mAbs (CH568 and CHXI3B6) on Hepatic Insulin Resistance
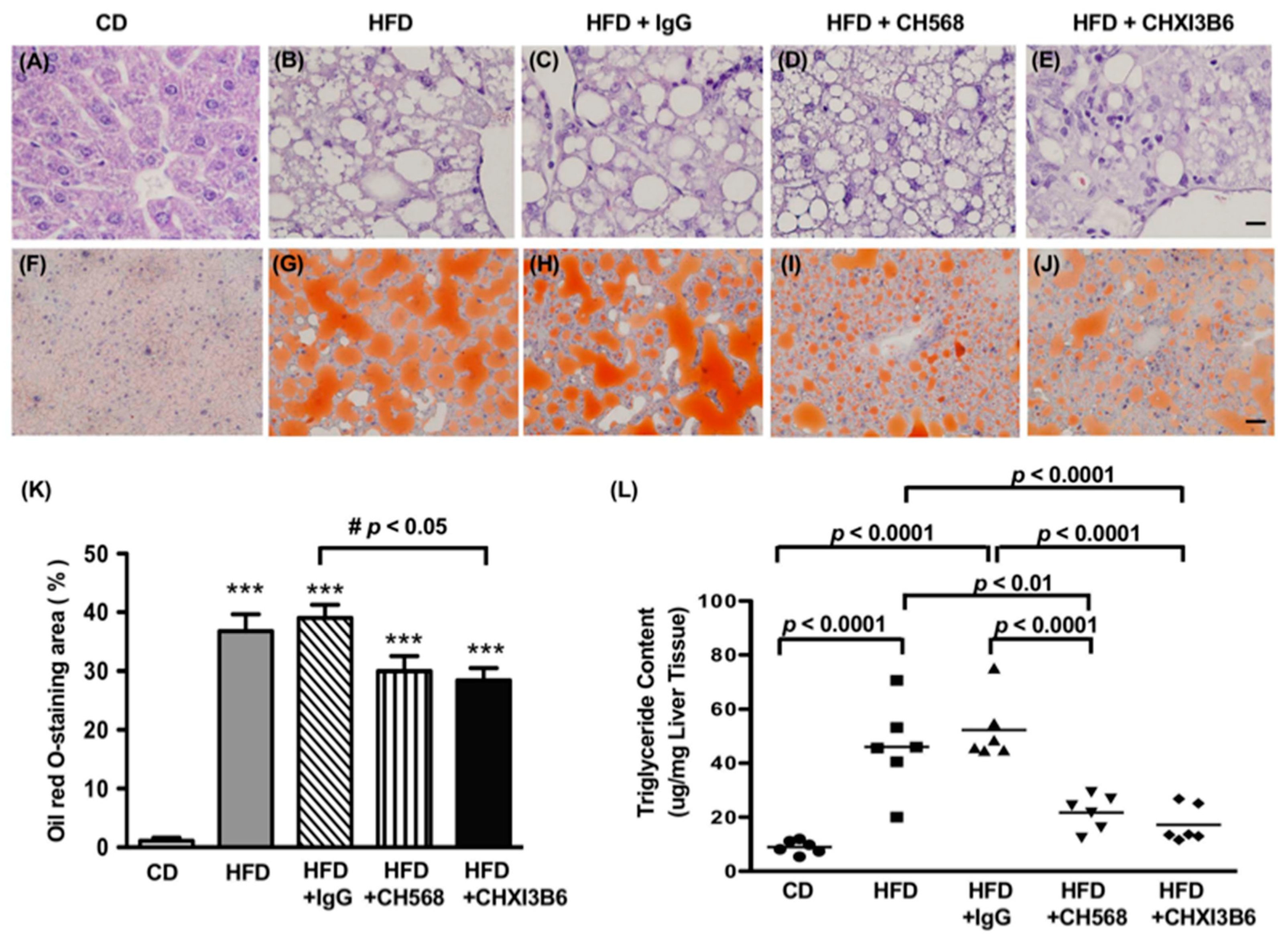
3.4. The Anti-Chi3L1 mAb Treatment Significantly Reduced Lipid Accumulation in HFD Fed Murine Liver
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef]
- Brenner, D.A. Of Mice and Men and Nonalcoholic Steatohepatitis. Hepatology 2018, 68, 2059–2061. [Google Scholar] [CrossRef] [Green Version]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; Thiel, D.H.; Lewis, P.J.; Ramadori, G. Insulin Production and Resistance in Different Models of Diet-Induced Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2017, 18, 285. [Google Scholar] [CrossRef] [Green Version]
- Alwahsh, S.M.; Xu, M.; Schultze, F.C.; Wilting, J.; Mihm, S.; Raddatz, D.; Ramadori, G. Combination of alcohol and fructose exacerbates metabolic imbalance in terms of hepatic damage, dyslipidemia, and insulin resistance in rats. PLoS ONE 2014, 9, e104220. [Google Scholar] [CrossRef]
- Pang, M.; de la Monte, S.M.; Longato, L.; Tong, M.; He, J.; Chaudhry, R.; Duan, K.; Ouh, J.; Wands, J.R. PPARdelta agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. J. Hepatol. 2009, 50, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Hayashi, N.; Ito, T.; Fusamoto, H.; Kamada, T.; Wands, J.R. Influence of ethanol on insulin receptor substrate-1-mediated signal transduction during rat liver regeneration. Alcohol. Alcohol. Suppl. 1994, 29, 99–106. [Google Scholar]
- Sasaki, Y.; Zhang, X.F.; Nishiyama, M.; Avruch, J.; Wands, J.R. Expression and phosphorylation of insulin receptor substrate 1 during rat liver regeneration. J. Biol. Chem. 1993, 268, 3805–3808. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Gratchev, A.; Goerdt, S. Human chitinases and chitinase-like proteins as indicators for inflammation and cancer. Biomark. Insights 2007, 2, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Da Silva, C.A.; Dela Cruz, C.S.; Ahangari, F.; Ma, B.; Kang, M.J.; He, C.H.; Takyar, S.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 2011, 73, 479–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Elias, J.A. Role of breast regression protein-39/YKL-40 in asthma and allergic responses. Allergy Asthma Immunol. Res. 2010, 2, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Hartl, D.; Lee, G.R.; Koller, B.; Matsuura, H.; Da Silva, C.A.; Sohn, M.H.; Cohn, L.; Homer, R.J.; Kozhich, A.A.; et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J. Exp. Med. 2009, 206, 1149–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Cruz, C.S.; Liu, W.; He, C.H.; Jacoby, A.; Gornitzky, A.; Ma, B.; Flavell, R.; Lee, C.G.; Elias, J.A. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe 2012, 12, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.H.; Lee, C.G.; Dela Cruz, C.S.; Lee, C.M.; Zhou, Y.; Ahangari, F.; Ma, B.; Herzog, E.L.; Rosenberg, S.A.; Li, Y.; et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep. 2013, 4, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; He, C.H.; Nour, A.M.; Zhou, Y.; Ma, B.; Park, J.W.; Kim, K.H.; Cruz, C.D.; Sharma, L.; Nasr, M.L.; et al. IL-13Ralpha2 uses TMEM219 in chitinase 3-like-1-induced signalling and effector responses. Nat. Commun. 2016, 7, 12752. [Google Scholar] [CrossRef]
- Matsuura, H.; Hartl, D.; Kang, M.J.; Dela Cruz, C.S.; Koller, B.; Chupp, G.L.; Homer, R.J.; Zhou, Y.; Cho, W.K.; Elias, J.A.; et al. Role of breast regression protein-39 in the pathogenesis of cigarette smoke-induced inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 44, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Ahangari, F.; Sood, A.; Ma, B.; Takyar, S.; Schuyler, M.; Qualls, C.; Dela Cruz, C.S.; Chupp, G.L.; Lee, C.G.; Elias, J.A. Chitinase 3-like-1 regulates both visceral fat accumulation and asthma-like Th2 inflammation. Am. J. Respir. Crit. Care Med. 2015, 191, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Kjaergaard, A.D.; Johansen, J.S.; Bojesen, S.E.; Nordestgaard, B.G. Role of inflammatory marker YKL-40 in the diagnosis, prognosis and cause of cardiovascular and liver diseases. Crit. Rev. Clin. Lab. Sci. 2016, 53, 396–408. [Google Scholar] [CrossRef]
- Kyrgios, I.; Galli-Tsinopoulou, A.; Stylianou, C.; Papakonstantinou, E.; Arvanitidou, M.; Haidich, A.B. Elevated circulating levels of the serum acute-phase protein YKL-40 (chitinase 3-like protein 1) are a marker of obesity and insulin resistance in prepubertal children. Metabolism 2012, 61, 562–568. [Google Scholar] [CrossRef]
- Huang, H.; Wu, T.; Mao, J.; Fang, Y.; Zhang, J.; Wu, L.; Zheng, S.; Lin, B.; Pan, H. CHI3L1 Is a Liver-Enriched, Noninvasive Biomarker That Can Be Used to Stage and Diagnose Substantial Hepatic Fibrosis. OMICS 2015, 19, 339–345. [Google Scholar] [CrossRef]
- Kumagai, E.; Mano, Y.; Yoshio, S.; Shoji, H.; Sugiyama, M.; Korenaga, M.; Ishida, T.; Arai, T.; Itokawa, N.; Atsukawa, M.; et al. Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci. Rep. 2016, 6, 35282. [Google Scholar] [CrossRef]
- Harrison, S.A.; Castiblanco, J.E.; Park, S.; Handler, T.E.; Barstow, K.; He, W.; Harper, N.; Ahuja, S. Whole genome RNA sequencing (RNA-seq) identifies CHI3L1 gene expression as a stronger discriminator of isolated steatosis vs. NASH compared with established biomarker scoring systems. Hepatology 2014, 60, 1290A. [Google Scholar]
- Tomita, K.; Higashiyama, M.; Furuhashi, H.; Shirakabe, K.; Maruta, K.; Okada, Y.; Kurihara, C.; Watanabe, C.; Komoto, S.; Hokari, R. Chitinase 3-like 1 plays a key role in the progression of nonalcholic steatohepatitis in mice. Hepatology 2017, 66, 1039A–1040A. [Google Scholar]
- Carr, R.M.; Oranu, A.; Khungar, V. Nonalcoholic Fatty Liver Disease: Pathophysiology and Management. Gastroenterol. Clin. N. Am. 2016, 45, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef]
- Di Rosa, M.; Malaguarnera, L. Chitinase 3 Like-1: An Emerging Molecule Involved in Diabetes and Diabetic Complications. Pathobiology 2016, 83, 228–242. [Google Scholar] [CrossRef]
- Gorgens, S.W.; Eckardt, K.; Elsen, M.; Tennagels, N.; Eckel, J. Chitinase-3-like protein 1 protects skeletal muscle from TNFalpha-induced inflammation and insulin resistance. Biochem. J. 2014, 459, 479–488. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Pang, M.; Chaudhry, R.; Duan, K.; Longato, L.; Carter, J.; Ouh, J.; Wands, J.R. Peroxisome proliferator-activated receptor agonist treatment of alcohol-induced hepatic insulin resistance. Hepatol. Res. 2011, 41, 386–398. [Google Scholar] [CrossRef] [Green Version]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High fat diet induced hepatic steatosis and insulin resistance: Role of dysregulated ceramide metabolism. Hepatol. Res. 2012, 42, 412–427. [Google Scholar] [CrossRef] [Green Version]
- Wands, J.R.; Zurawski, V.R., Jr. High affinity monoclonal antibodies to hepatitis B surface antigen (HBsAg) produced by somatic cell hybrids. Gastroenterology 1981, 80, 225–232. [Google Scholar] [CrossRef]
- He, J.; de la Monte, S.; Wands, J.R. Acute ethanol exposure inhibits insulin signaling in the liver. Hepatology 2007, 46, 1791–1800. [Google Scholar] [CrossRef]
- Ito, T.K.; Ishii, G.; Chiba, H.; Ochiai, A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene 2007, 26, 7194–7203. [Google Scholar] [CrossRef] [Green Version]
- Sandler, N.G.; Mentink-Kane, M.M.; Cheever, A.W.; Wynn, T.A. Global gene expression profiles during acute pathogen-induced pulmonary inflammation reveal divergent roles for Th1 and Th2 responses in tissue repair. J. Immunol. 2003, 171, 3655–3667. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [Green Version]
- Nandi, A.; Kitamura, Y.; Kahn, C.R.; Accili, D. Mouse models of insulin resistance. Physiol. Rev. 2004, 84, 623–647. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Roberts-Toler, C.; O’Neill, B.T.; Cypess, A.M. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity (Silver Spring) 2015, 23, 1765–1770. [Google Scholar] [CrossRef]
- Wang, C.Y.; Liao, J.K. A mouse model of diet-induced obesity and insulin resistance. Methods Mol. Biol. 2012, 821, 421–433. [Google Scholar]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Su, Z.; Li, Y.; Zhang, X.; You, Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal Transduct. Target. Ther. 2020, 5, 201. [Google Scholar] [CrossRef]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., Koch, C., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Meshkani, R.; Adeli, K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin. Biochem. 2009, 42, 1331–1346. [Google Scholar] [CrossRef]
- Bazotte, R.B.; Silva, L.G.; Schiavon, F.P. Insulin resistance in the liver: Deficiency or excess of insulin? Cell Cycle 2014, 13, 2494–2500. [Google Scholar] [CrossRef] [Green Version]
- Fengler, V.H.; Macheiner, T.; Kessler, S.M.; Czepukojc, B.; Gemperlein, K.; Muller, R.; Kiemer, A.K.; Magnes, C.; Haybaeck, J.; Lackner, C. Susceptibility of Different Mouse Wild Type Strains to Develop Diet-Induced NAFLD/AFLD-Associated Liver Disease. PLoS ONE 2016, 11, e0155163. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Dittmann, A.; Kennedy, N.J.; Soltero, N.L.; Morshed, N.; Mana, M.D.; Yilmaz, O.H.; Davis, R.J.; White, F.M. High-fat diet in a mouse insulin-resistant model induces widespread rewiring of the phosphotyrosine signaling network. Mol. Syst. Biol. 2019, 15, e8849. [Google Scholar] [CrossRef]
- Chen, C.C.; Llado, V.; Eurich, K.; Tran, H.T.; Mizoguchi, E. Carbohydrate-binding motif in chitinase 3-like 1 (CHI3L1/YKL-40) specifically activates Akt signaling pathway in colonic epithelial cells. Clin. Immunol. 2011, 140, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Higashiyama, M.; Tomita, K.; Sugihara, N.; Nakashima, H.; Furuhashi, H.; Nishikawa, M.; Inaba, K.; Wada, A.; Horiuchi, K.; Hanawa, Y.; et al. Chitinase 3-like 1 deficiency ameliorates liver fibrosis by promoting hepatic macrophage apoptosis. Hepatol. Res. 2019, 49, 1316–1328. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.N.; Lee, K.E.; Hong, J.Y.; Heo, W.I.; Kim, K.W.; Kim, K.E.; Sohn, M.H. Involvement of the MAPK and PI3K pathways in chitinase 3-like 1-regulated hyperoxia-induced airway epithelial cell death. Biochem. Biophys. Res. Commun. 2012, 421, 790–796. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | N | Diet | Treatment | Insulin Stimulation |
|---|---|---|---|---|
| Group 1 | 8 | Chow Diet | None | + (4 mice) |
| − (4 mice) | ||||
| Group 2 | 8 | High Fat Diet | None | + (4 mice) |
| − (4 mice) | ||||
| Group 3 | 8 | High Fat Diet | Unrelated IgG 200 µg/mouse, twice a week | + (4 mice) |
| − (4 mice) | ||||
| Group 4 | 8 | High Fat Diet | Chi3L1 IgG Ab. (CH568), 200 µg/mouse, twice a week | + (4 mice) |
| − (4 mice) | ||||
| Group 5 | 8 | High Fat Diet | Chi3L1 IgG Ab. (CHXI3B6), 200 µg/mouse, twice a week | + (4 mice) |
| − (4 mice) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Sousa, A.; Lin, M.; Iwano, A.; Jain, R.; Ma, B.; Lee, C.M.; Park, J.W.; Kamle, S.; Carlson, R.; et al. Role of Chitinase 3-Like 1 Protein in the Pathogenesis of Hepatic Insulin Resistance in Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 201. https://doi.org/10.3390/cells10020201
Zhang S, Sousa A, Lin M, Iwano A, Jain R, Ma B, Lee CM, Park JW, Kamle S, Carlson R, et al. Role of Chitinase 3-Like 1 Protein in the Pathogenesis of Hepatic Insulin Resistance in Nonalcoholic Fatty Liver Disease. Cells. 2021; 10(2):201. https://doi.org/10.3390/cells10020201
Chicago/Turabian StyleZhang, Songhua, Aryanna Sousa, Mengqui Lin, Ayako Iwano, Rishubh Jain, Bing Ma, Chang Min Lee, Jin Wook Park, Suchitra Kamle, Rolf Carlson, and et al. 2021. "Role of Chitinase 3-Like 1 Protein in the Pathogenesis of Hepatic Insulin Resistance in Nonalcoholic Fatty Liver Disease" Cells 10, no. 2: 201. https://doi.org/10.3390/cells10020201