
First-Principles Modeling of Direct versus Oxygen-Assisted Water Dissociation on Fe(100) Surfaces

Abstract
:1. Introduction
2. Results and Discussion
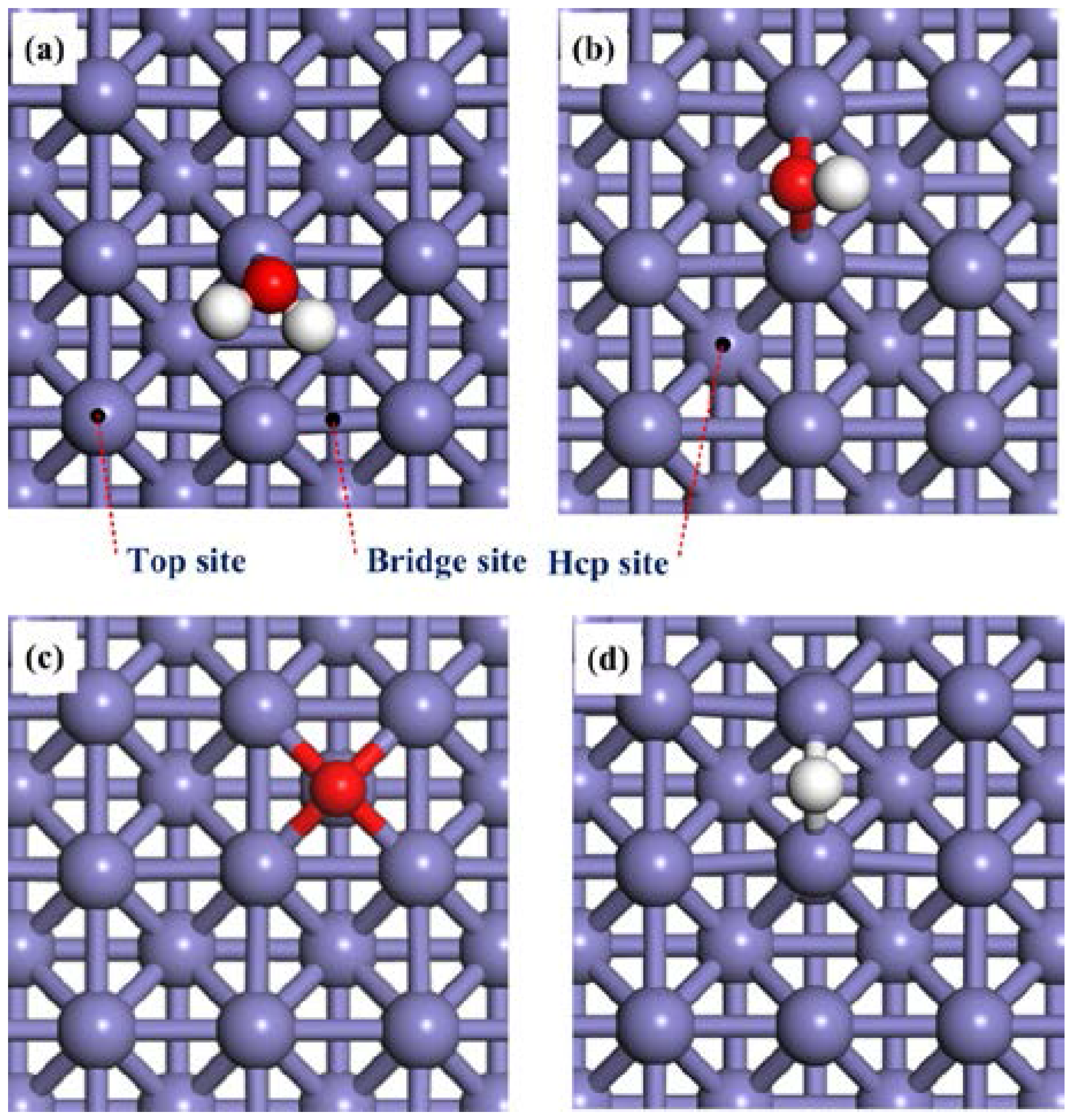
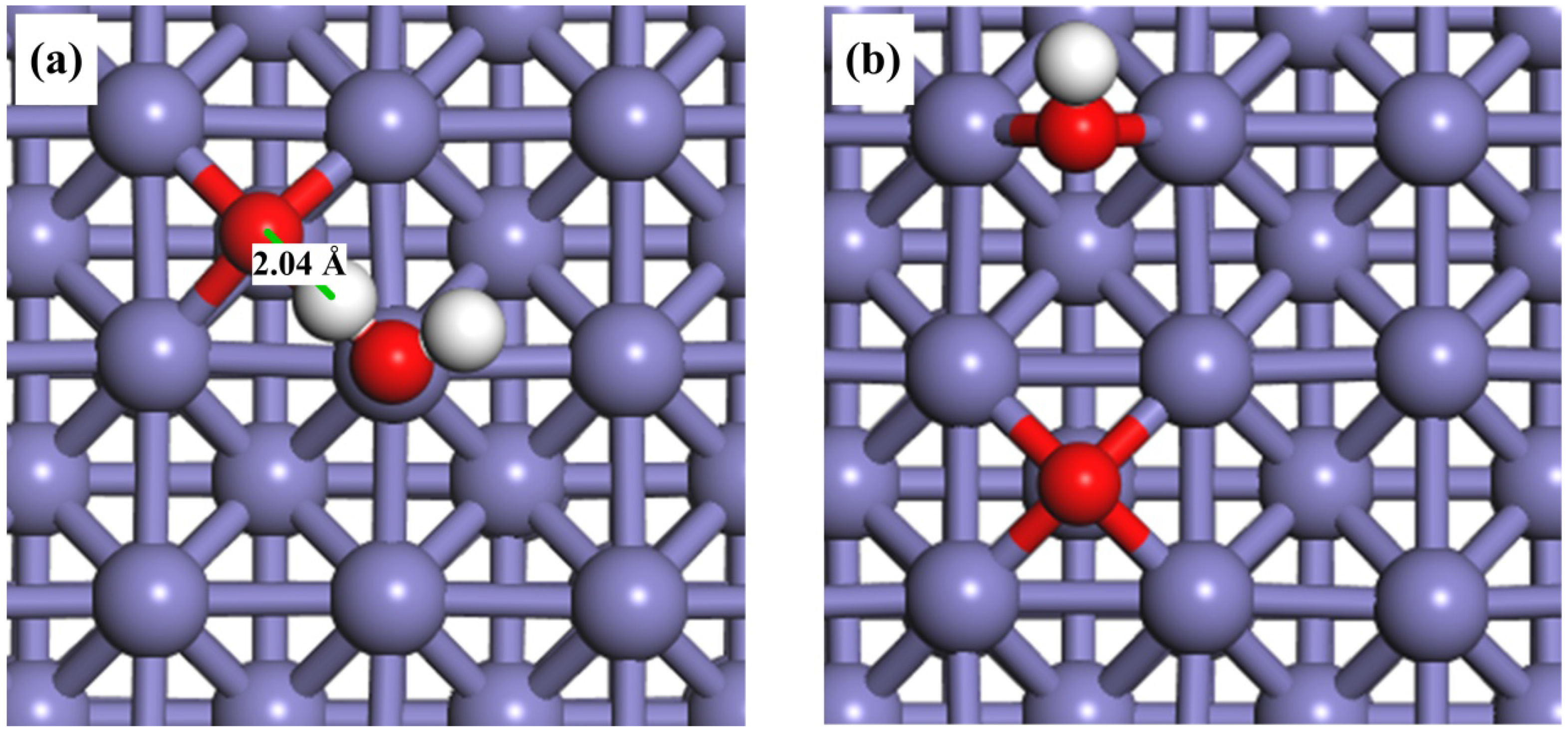
2.1. Adsorption of H2O, OH, O and H on the Clean Fe(100) Surface
2.2. Adsorption of H2O and OH on the O-Pre-adsorbed Fe(100) Surface
2.3. Reaction Mechanisms
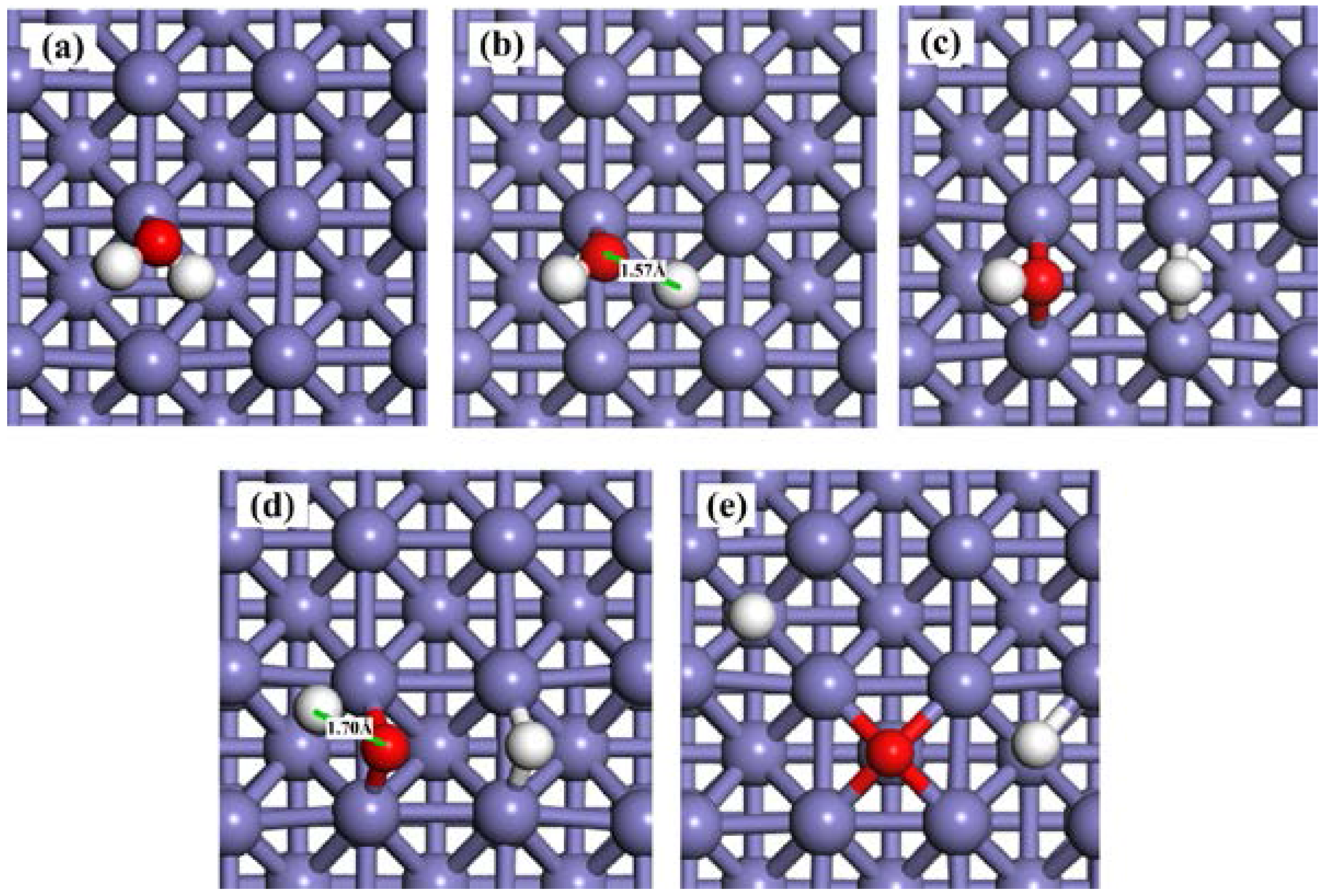
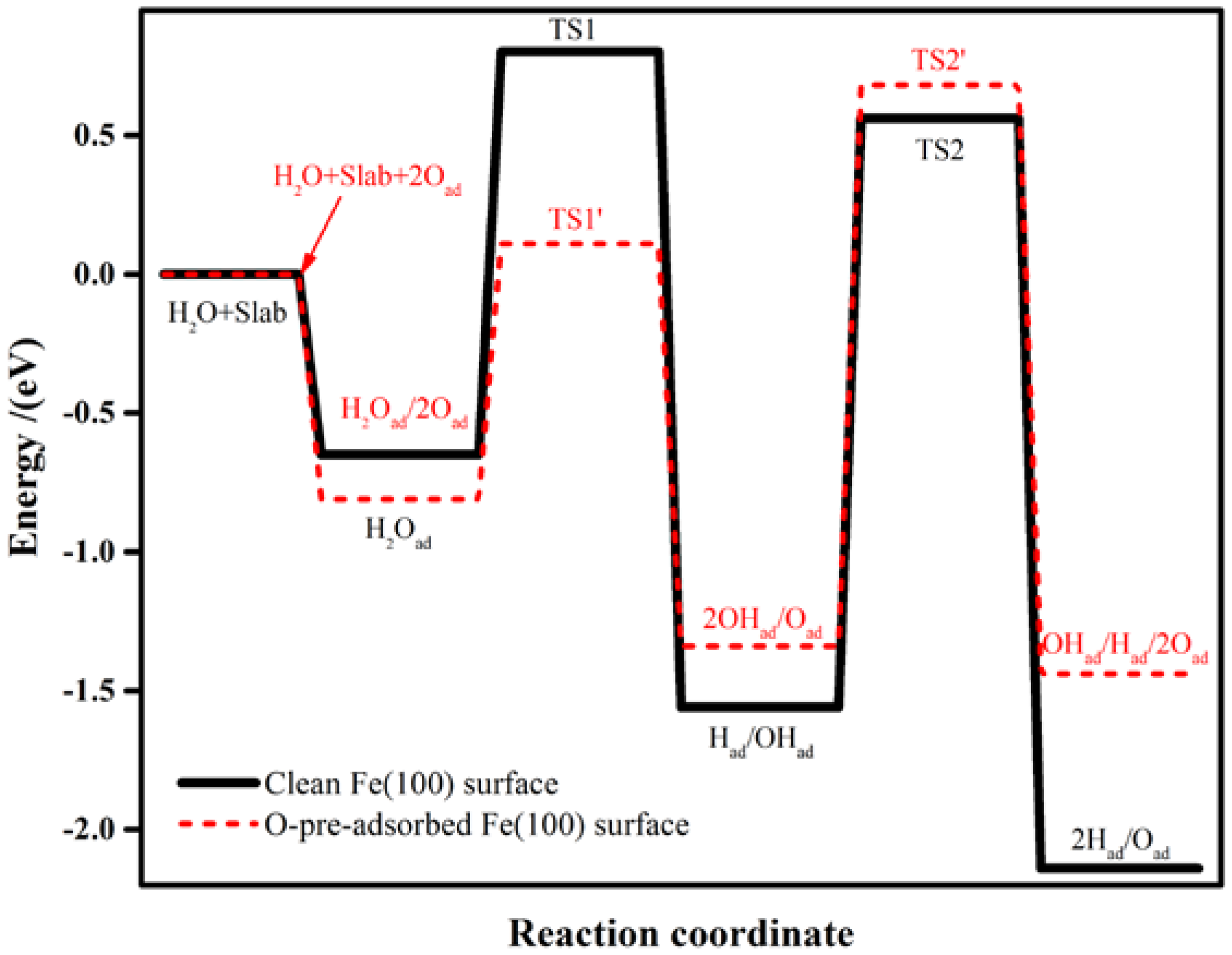
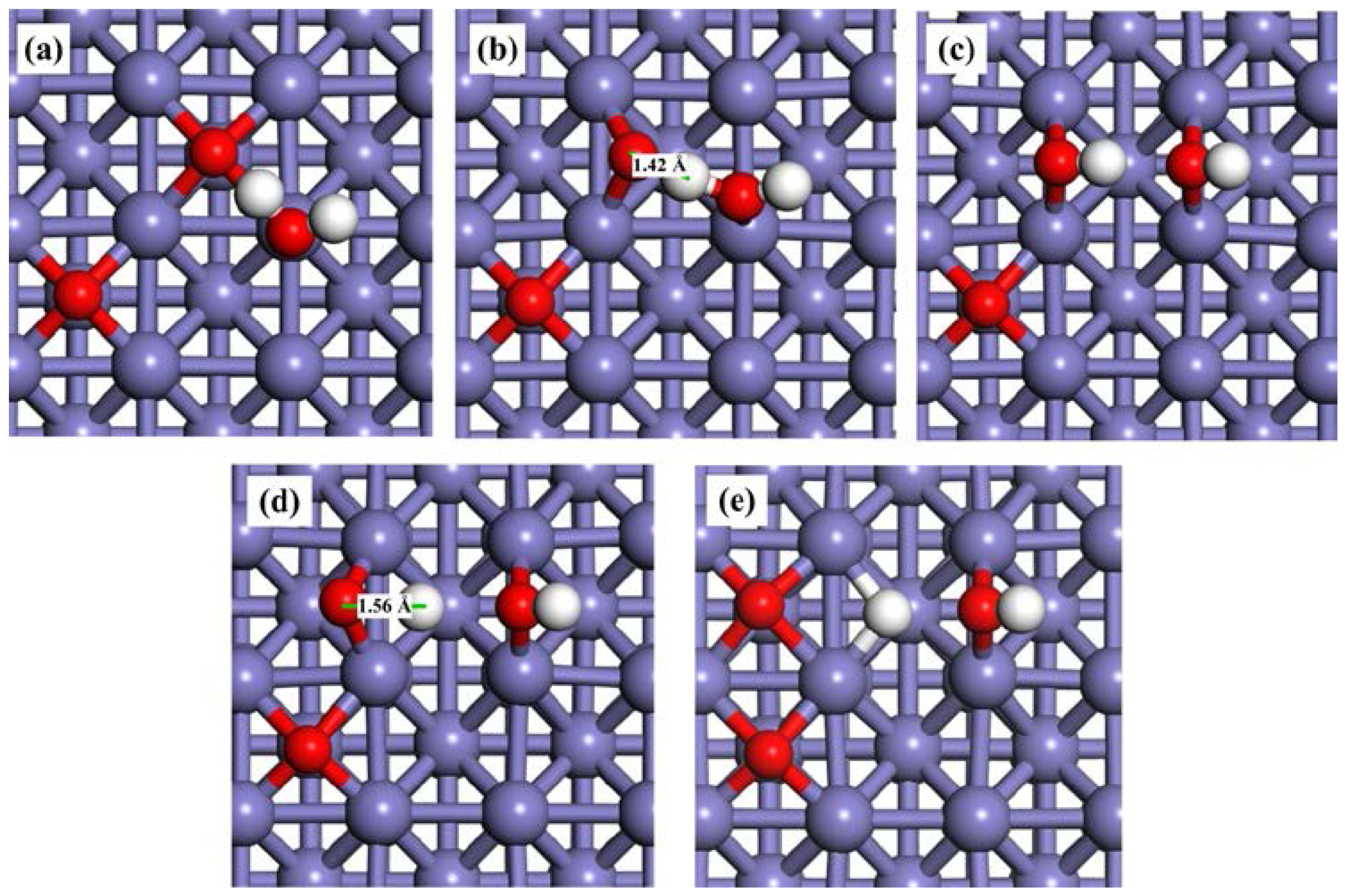
2.3.1. H2O Dissociation on the Clean Fe(100) Surface
2.3.2. H2O Dissociation on the O-Preadsorbed Fe(100) Surface
3. Computational Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Henniker, J.C. The depth of the surface zone of a liquid. Rev. Mod. Phys. 1949, 21, 322–341. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Veziroglu, T.N. Estimates of the price of hydrogen as a medium for wind and solar sources. Int. J. Hydrogen Energy 2007, 32, 1605–1610. [Google Scholar] [CrossRef]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.W. Hydrogen storage in metal-organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef] [PubMed]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and physical solutions for hydrogen storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef] [PubMed]
- Dicks, A.L. Hydrogen generation from natural gas for the fuel cell systems of tomorrow. J. Power Sources 1996, 61, 113–124. [Google Scholar] [CrossRef]
- Dybkjaer, I. Tubular reforming and autothermal reforming of natural gas—An overview of available processes. Fuel Process. Technol. 1995, 42, 85–107. [Google Scholar] [CrossRef]
- Takezawa, N.; Iwasa, N. Steam reforming and dehydrogenation of methanol: Difference in the catalytic functions of copper and group VIII metals. Catal. Today 1997, 36, 45–56. [Google Scholar] [CrossRef]
- Haryanto, A.; Fernando, S.; Murali, N.; Adhikari, S. Current status of hydrogen production techniques by steam reforming of ethanol: A review. Energy Fuels 2005, 19, 2098–2106. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Reaction pathways and site requirements for the activation and chemical conversion of methane on Ru-based catalysts. J. Phys. Chem. B 2004, 108, 7253–7262. [Google Scholar] [CrossRef]
- Rostrupnielsen, J.R.; Hansen, J.H.B. CO2-reforming of methane over transition metal. J. Catal. 1993, 144, 38–49. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Structural requirements and reaction pathways in methane activation and chemical conversion catalyzed by rhodium. J. Catal. 2004, 225, 116–127. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Isotopic and kinetic assessment of the mechanism of methane reforming and decomposition reactions on supported iridium catalysts. Phys. Chem. Chem. Phys. 2004, 6, 3754–3759. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Structural and mechanistic requirements for methane activation and chemical conversion on supported iridium clusters. Angew. Chem. Int. Ed. 2004, 43, 3685–3688. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.M.; Iglesia, E. Mechanism and site requirements for activation and chemical conversion of methane on supported Pt clusters and turnover rate comparisons among noble metals. J. Phys. Chem. B 2004, 108, 4094–4103. [Google Scholar] [CrossRef]
- Sehested, J. Four challenges for nickel steam-reforming catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Murakhtina, T.; Site, L.D.; Sebastiani, D. Vibrational frequencies of water adsorbed on (111) and (221) nickel surfaces from first principle calculations. Chem. Phys. Chem. 2006, 7, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Catapan, R.C.; Oliveira, A.A.M.; Chen, Y.; Vlachos, D.G. DFT study of the water–gas shift reaction and coke formation on Ni(111) and Ni(211) surfaces. J. Phys. Chem. C 2012, 116, 20281–20291. [Google Scholar] [CrossRef]
- Lindström, B.; Pettersson, L.J. Hydrogen generation by steam reforming of methanol over copper-based catalysts for fuel cell applications. Int. J. Hydrogen Energy 2001, 26, 923–933. [Google Scholar] [CrossRef]
- Andersson, K.; Ketteler, G.; Bluhm, H.; Yamamoto, S.; Ogasawara, H.; Pettersson, L.G.M.; Salmeron, M.; Nilsson, A. Autocatalytic water dissociation on Cu(110) at near ambient conditions. J. Am. Chem. Soc. 2008, 130, 2793–2797. [Google Scholar] [CrossRef] [PubMed]
- Llorca, J.; Homs, N.; Sales, J.; de la Piscina, P.R. Efficient production of hydrogen over supported cobalt catalysts from ethanol steam reforming. J. Catal. 2002, 209, 306–317. [Google Scholar] [CrossRef]
- Xu, L.S.; Ma, Y.S.; Zhang, Y.L.; Chen, B.H.; Wu, Z.F.; Jiang, Z.Q.; Huang, W.X. Water adsorption on a Co(0001) surface. J. Phys. Chem. C 2010, 114, 17023–17029. [Google Scholar] [CrossRef]
- Murata, K.; Wang, L.S.; Saito, M.; Inaba, M.; Takahara, I.; Mimura, N. Hydrogen production from steam reforming of hydrocarbons over alkaline-earth metal-modified Fe- or Ni-based catalysts. Energy Fuels 2004, 18, 122–126. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Bakandritsos, A.; Tzitzios, V.; Fierro, J.L.G.; Efstathiou, A.M. Absorption-enhanced reforming of phenol by steam over supported Fe catalysts. J. Catal. 2006, 241, 132–148. [Google Scholar] [CrossRef]
- Liang, C.H.; Ma, Z.Q.; Lin, H.Y.; Ding, L.; Qiu, J.S.; Frandsen, W.; Su, D.S. Template preparation of nanoscale CexFe1−xO2 solid solutions and their catalytic properties for ethanol steam reforming. J. Mater. Chem. 2009, 19, 1417–1424. [Google Scholar] [CrossRef]
- Noichi, H.; Uddin, A.; Sasaoka, E. Steam reforming of naphthalene as model biomass tar over iron-aluminum and iron-zirconium oxide catalyst catalysts. Fuel Process. Technol. 2010, 91, 1609–1616. [Google Scholar] [CrossRef]
- Mawdsley, J.R.; Krause, T.R. Rare earth-first-row transition metal perovskites as catalysts for the autothermal reforming of hydrocarbon fuels to generate hydrogen. Appl. Catal. A 2008, 334, 311–320. [Google Scholar] [CrossRef]
- Sato, K.; Nagaoka, K.; Nishiguchi, H.; Takita, Y. n-C4H10 autothermal reforming over MgO-supported base metal catalysts. Int. J. Hydrogen Energy 2009, 34, 333–342. [Google Scholar] [CrossRef]
- Anderson, A.B. Reactions and structures of water on clean and oxygen covered Pt(111) and Fe(100). Surf. Sci. 1981, 105, 159–176. [Google Scholar] [CrossRef]
- Baró1, A.M.; Erley1, W. The adsorption of H2O on Fe(100) studied by EELS. J. Vac. Sci. Technol. 1982, 20, 580–583. [Google Scholar] [CrossRef]
- Lu, J.P.; Albert, M.R.; Bernasek, S.L. The adsorption of oxygen on the Fe(100) surface. Surf. Sci. 1989, 215, 348–362. [Google Scholar] [CrossRef]
- Hung, W.H.; Schwartz, J.; Bernasek, S.L. Sequential oxidation of Fe(100) by water adsorption: formation of an ordered hydroxylated surface. Surf. Sci. 1991, 248, 332–342. [Google Scholar] [CrossRef]
- Freitas, R.R.Q.; Rivelino, R.; de Brito Mota, F.; de Castilho, C.M.C. Dissociative adsorption and aggregation of water on the Fe(100) surface: A DFT study. J. Phys. Chem. C 2012, 116, 20306–20314. [Google Scholar] [CrossRef]
- Eder, M.; Terakura, K. Initial stages of oxidation of (100) and (110) surfaces of iron caused by water. Phys. Rev. B 2001, 64, 115426:1–115426:7. [Google Scholar] [CrossRef]
- Jung, S.C.; Kang, M.H. Adsorption of a water molecule on Fe(100): Density-functional calculations. Phys. Rev. B 2010, 81, 115460:1–115460:7. [Google Scholar] [CrossRef]
- Govender, A.; Ferré, D.C.; Niemantsverdriet, J.W. The surface chemistry of water on Fe(100): A density functional theory study. Chem. Phys. Chem. 2012, 13, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Lazar, P.; Otyepka, M. Dissociation of water at iron surfaces: Generalized gradient functional and range-separated hybrid functional study. J. Phys. Chem. C 2012, 116, 25470–25477. [Google Scholar] [CrossRef]
- Liu, S.; Tian, X.; Wang, T.; Wen, X.; Li, Y.W.; Wang, J.; Jiao, H. High coverage water aggregation and dissociation on Fe(100): A computational analysis. J. Phys. Chem. C 2014, 118, 26139–26154. [Google Scholar] [CrossRef]
- Michaelides, A.; Ranea, V.A.; de Andres, P.L.; King, D.A. General model for water monomer adsorption on close-packed transition and noble metal surfaces. Phys. Rev. Lett. 2003, 90, 216102:1–216102:4. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, A.; Ranea, V.A.; de Andres, P.L.; King, D.A. First-principles study of H2O diffusion on a metal surface: H2O on Al{100}. Phys. Rev. B 2004, 69, 075409:1–075409:4. [Google Scholar] [CrossRef]
- Wang, S.; Cao, Y.; Rikvold, P.A. First-principles calculations for the adsorption of water molecules on the Cu(100) surface. Phys. Rev. B 2004, 70, 205410:1–205410:4. [Google Scholar] [CrossRef]
- Li, J.B.; Zhu, S.L.; Li, Y.; Wang, F.H. Water adsorption on Pd {100} from first principles. Phys. Rev. B 2007, 76, 235433:1–235433:8. [Google Scholar] [CrossRef]
- Thiel, P.A.; Madey, T.E. The interaction of water with solid surfaces: fundamental aspects. Surf. Sci. Rep. 1987, 7, 211–385. [Google Scholar] [CrossRef]
- Henderson, M.A. The interaction of water with solid surfaces: fundamental aspects revisited. Surf. Sci. Rep. 2002, 46, 1–308. [Google Scholar] [CrossRef]
- Heras, J.M.; Estiu, G.; Viscido, L. The interaction of water with clean palladium films: A thermal desorption and work function study. Appl. Surf. Sci. 1997, 108, 455–464. [Google Scholar] [CrossRef]
- Nyberg, C.; Tengstal, C.G. Adsorption and reaction of water, oxygen, and hydrogen on Pd(100): Identification of adsorbed hydroxyl and implications for the catalytic H2–O2 reaction. J. Chem. Phys. 1984, 80, 3463–3468. [Google Scholar] [CrossRef]
- Wolf, M.; Nettesheim, S.; White, J.M.; Hasselbrink, E.; Ertl, G. Ultraviolet-laser induced dissociation and desorption of water adsorbed on Pd(111). J. Chem. Phys. 1990, 92, 1509–1510. [Google Scholar] [CrossRef]
- Wolf, M.; Nettesheim, S.; White, J.M.; Hasselbrink, E.; Ertl, G. Dynamics of the ultraviolet photochemistry of water adsorbed on Pd(111). J. Chem. Phys. 1991, 94, 4609–4619. [Google Scholar] [CrossRef]
- Zhu, X.Y.; White, J.M.; Wolf, M.; Hasselbrink, E.; Ertl, G. Photochemical pathways of water on palladium (111) at 6.4 eV. J. Phys. Chem. 1991, 95, 8393–8402. [Google Scholar] [CrossRef]
- Cao, Y.L.; Chen, Z.X. Theoretical studies on the adsorption and decomposition of H2O on Pd(111) surface. Surf. Sci. 2006, 600, 4572–4583. [Google Scholar] [CrossRef]
- Shavorskiy, A.; Eralp, T.; Gladys, M.J.; Held, G. A stable pure hydroxyl layer on Pt{110}-(1×2). J. Phys. Chem. C 2009, 113, 21755–21764. [Google Scholar] [CrossRef]
- Liu, S.L.; Tian, X.X.; Wang, T.; Wen, X.D.; Li, Y.W.; Wang, J.G.; Jiao, H.J. Coverage dependent water dissociative adsorption on the cleanand O-precovered Fe(111) surfaces. J. Phys. Chem. C 2015, 119, 11714–11724. [Google Scholar] [CrossRef]
- Hung, W.H.; Schwartz, J.; Bernasek, S.L. Adsorption of H2O on oxidized Fe(100) surfaces: comparison between the oxidation of iron by H2O and O2. Surf. Sci. 1993, 294, 21–32. [Google Scholar] [CrossRef]
- Pozzo, M.; Carlini, G.; Rosei, R.; Alfè, D. Comparative study of water dissociation on Rh(111) and Ni(111) studied with first principles calculations. J. Chem. Phys. 2007, 126, 164706:1–164706:11. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Wang, G.P. A theoretical study of water adsorption and dissociation on Ni(111) surface during oxidative steam reforming and water gas shift processes. J. Energy Inst. 2015, 88, 112–117. [Google Scholar] [CrossRef]
- Mohsenzadeh, A.; Bolton, K.; Richards, T. DFT study of the adsorption and dissociation of water on Ni(111), Ni(110) and Ni(100) surfaces. Surf. Sci. 2014, 627, 1–10. [Google Scholar] [CrossRef]
- Tang, Q.L.; Chen, Z.X.; He, X. A theoretical study of the water gas shift reaction mechanism on Cu(111) model system. Surf. Sci. 2009, 603, 2138–2144. [Google Scholar] [CrossRef]
- Wang, W.J.; Wang, G.P. Theoretical study of direct versus oxygen-assisted water dissociation on the Cu(110) surface. Appl. Surf. Sci. 2015, 351, 846–852. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Illas, F.; Gomes, J.R.B. Descriptors controlling the catalytic activity of metallic surfaces towardwater splitting. J. Catal. 2010, 276, 92–100. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Illas, F.; Gomes, J.R.B. Generalized Brønsted-Evans-Polanyi relationships and descriptors for O–H bond cleavage of organic molecules on transition metal surfaces. J. Catal. 2014, 313, 24–33. [Google Scholar] [CrossRef]
- Błoński, P.; Kiejna, A.; Hafner, J. Theoretical study of oxygen adsorption at the Fe(110) and (100) surfaces. Surf. Sci. 2005, 590, 88–100. [Google Scholar] [CrossRef]
- Sorescu, D.C. First principles calculations of the adsorption and diffusion of hydrogen on Fe(100) surface and in the bulk. Catal. Today 2005, 105, 44–65. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Yan, L.F.; Wang, G.C. Oxygen-assisted water partial dissociation on copper: a model study. Phys. Chem. Chem. Phys. 2015, 17, 8231–8238. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, M.M.; Yan, T.; Fang, T. Decomposition of H2O on clean and oxygen-covered Au(100) surface: A DFT study. Appl. Surf. Sci. 2014, 315, 16–21. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, Z. Slab model studies of water adsorption and decomposition on clean and X- (X = C, N and O) contaminated Pd(111) surfaces. Phys. Chem. Chem. Phys. 2007, 9, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Kurth, S.; Perdew, J.P.; Blaha, P. Molecular and solid-state tests of density functional approximations: LSD, GGAs, and meta-GGAs. Int. J. Quantum Chem. 1999, 75, 889–909. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley: New York, NY, USA, 2005. [Google Scholar]
- Kohlhaas, R.; Donner, P.; Schmitz-Pranghe, N. The temperature-dependence of the lattice parameters of iron, cobalt, and nickel in the high temperature range. Z. Angew. Phys. 1967, 23, 245. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics, 7th ed.; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Jiang, D.E.; Carter, E.A. Diffusion of interstitial hydrogen into and through bcc Fe from first principles. Phys. Rev. B 2004, 70, 064102:1–064102:9. [Google Scholar] [CrossRef]
- Halgren, T.A. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comp. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Clean Fe(100) | O-pre-adsorbed Fe(100) | |||
|---|---|---|---|---|---|
| Top | Bridge | Hcp | Top | Bridge | |
| H2O | −0.65 | - | - | −1.13 | - |
| OH | - | −3.93 | −3.84 | - | −4.02 |
| O | - | −3.00 | −3.67 | - | - |
| H | - | −3.99 | −3.86 | - | - |
| Species | Clean Fe(100) | O-pre-adsorbed Fe(100) | ||||
|---|---|---|---|---|---|---|
| dO–H (Å) | AH–O–H (°) | dO(/H)–Fe (Å) a | dO–H (Å) | AH–O–H (°) | dO–Fe (Å) a | |
| H2O | 0.99/0.98 (0.98/0.98) b | 105.4 (104.4) b | 2.17 | 0.99/0.98 | 106.4 | 2.20 |
| OH | 0.98 (0.99) b | - | 1.99/1.99 | 0.98 | - | 1.98/1.98 |
| O | - | - | 2.04/2.04/2.04/2.04 | - | - | - |
| H | - | - | 1.70/1.70 | - | - | - |
| Surfaces | Reactions | ΔE (eV) | Ea (eV) |
|---|---|---|---|
| Clean Fe(100) | Equation (1) | −1.02 | 1.45 (TS1) |
| Equation (2) | −0.58 | 2.12 (TS2) | |
| O-pre-adsorbed Fe(100) | Equation (3) | −0.53 | 0.92 (TS1’) |
| Equation (4) | −0.10 | 2.02 (TS2’) |
| Species | DFT(GGA-PBE) | DFT + U |
|---|---|---|
| H2O (top) | −0.65 | −0.67 |
| OH (brg) | −3.93 | −3.89 |
| O (hcp) | −3.67 | −3.54 |
| H (brg) | −3.99 | −3.66 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, G.; Shao, M. First-Principles Modeling of Direct versus Oxygen-Assisted Water Dissociation on Fe(100) Surfaces. Catalysts 2016, 6, 29. https://doi.org/10.3390/catal6020029
Wang W, Wang G, Shao M. First-Principles Modeling of Direct versus Oxygen-Assisted Water Dissociation on Fe(100) Surfaces. Catalysts. 2016; 6(2):29. https://doi.org/10.3390/catal6020029
Chicago/Turabian StyleWang, Wenju, Guoping Wang, and Minhua Shao. 2016. "First-Principles Modeling of Direct versus Oxygen-Assisted Water Dissociation on Fe(100) Surfaces" Catalysts 6, no. 2: 29. https://doi.org/10.3390/catal6020029