Counteracting Rapid Catalyst Deactivation by Concomitant Temperature Increase during Catalytic Upgrading of Biomass Pyrolysis Vapors Using Solid Acid Catalysts

 , ,
, , 
Abstract
:1. Introduction
2. Results
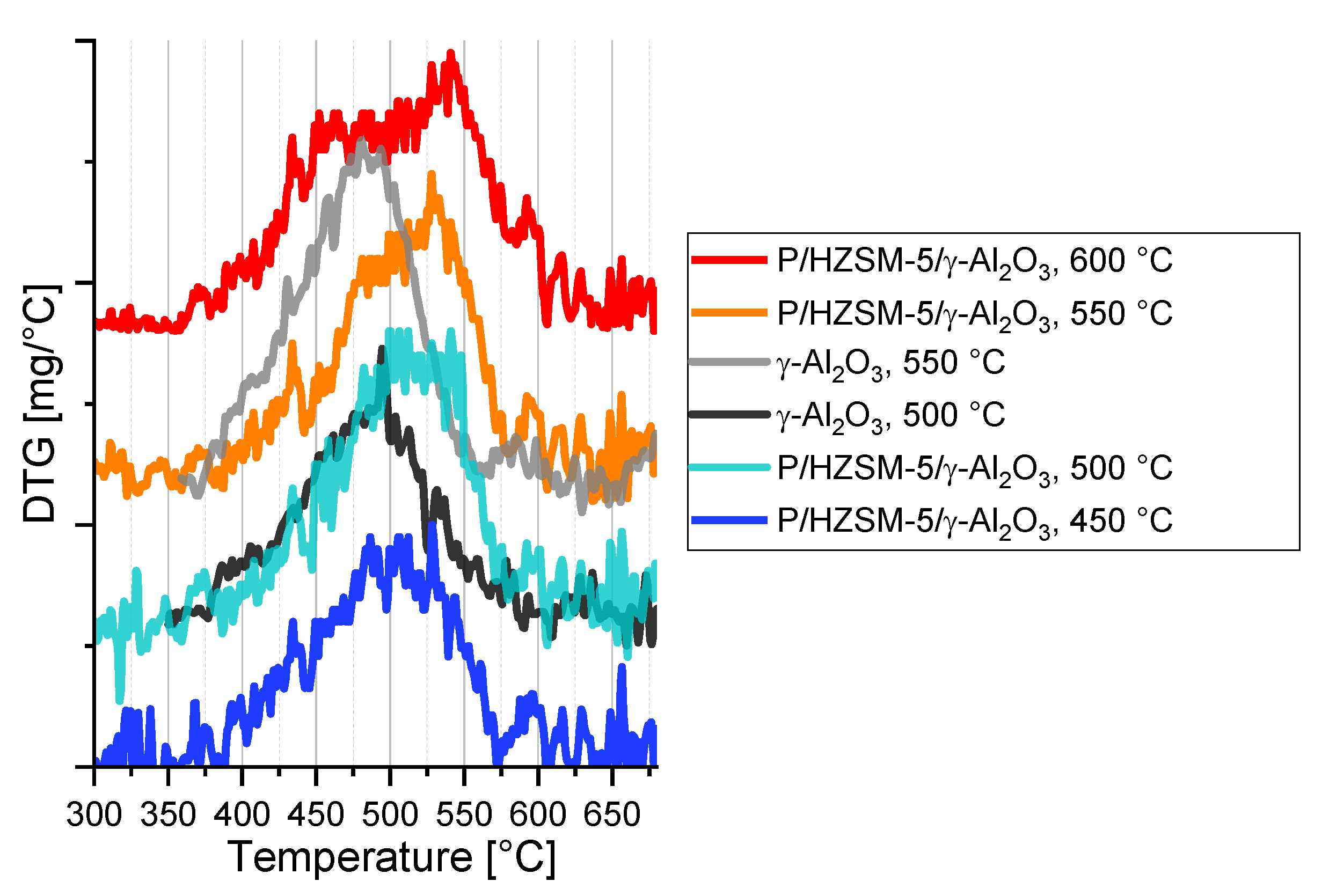
2.1. Catalyst Properties
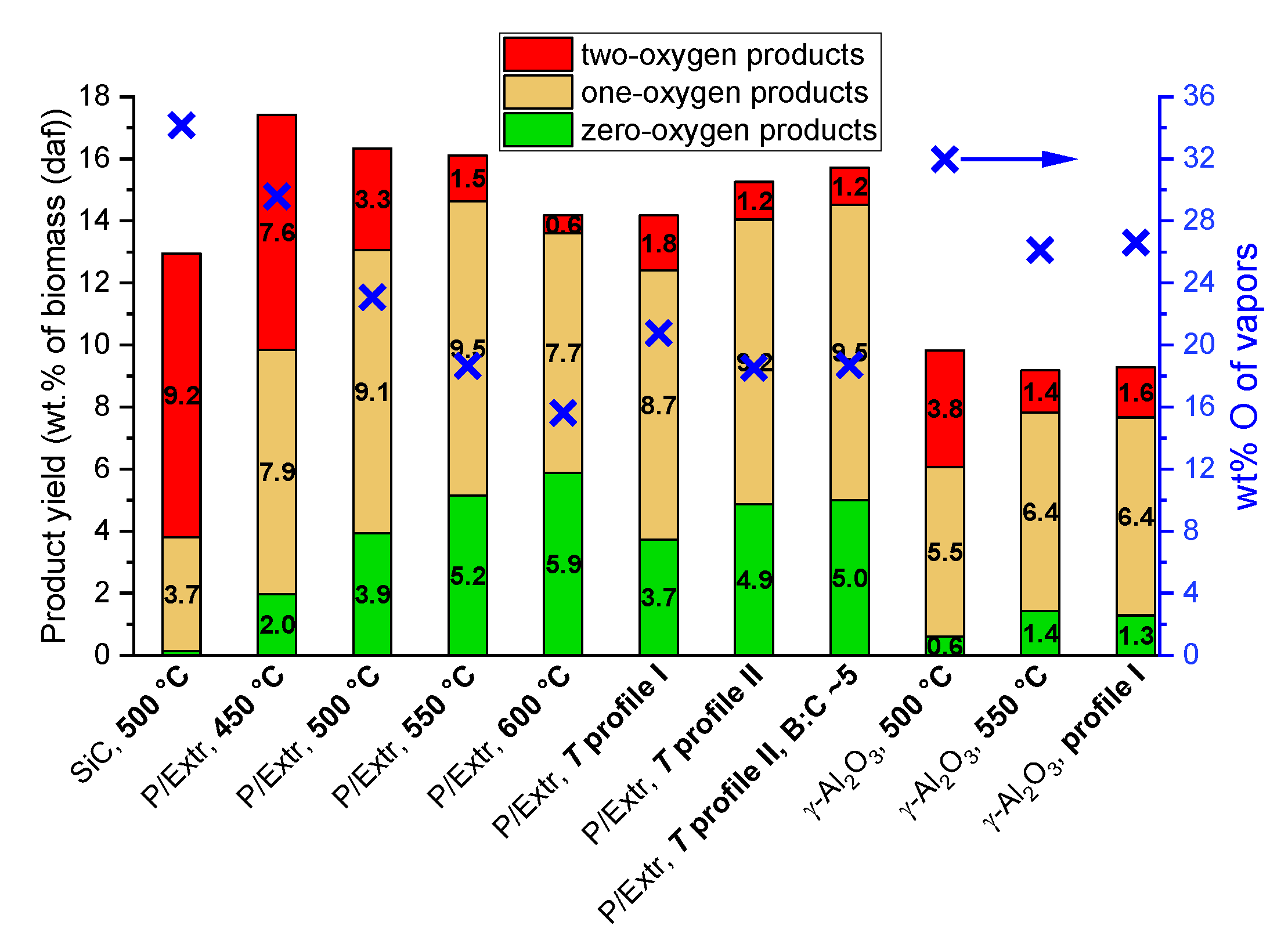
2.2. Product Yields
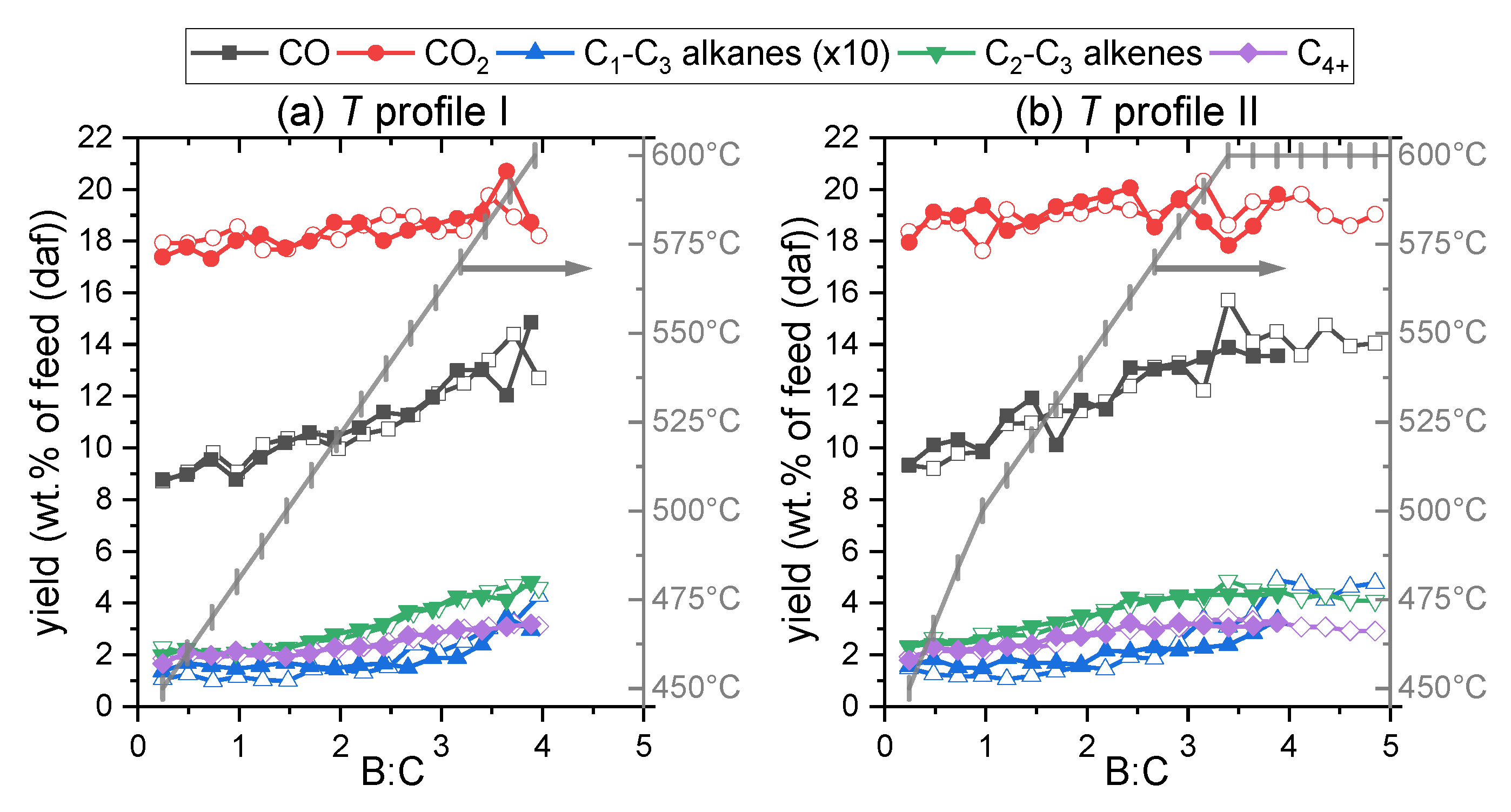
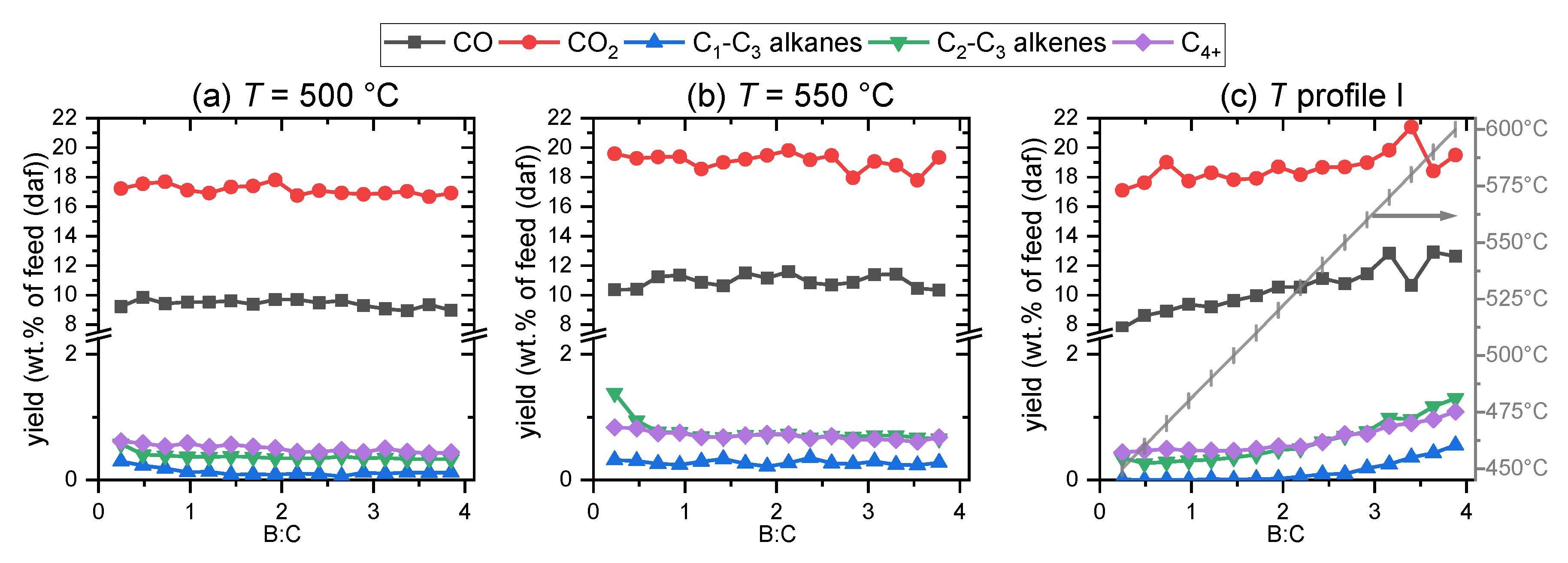
2.2.1. Light Gases
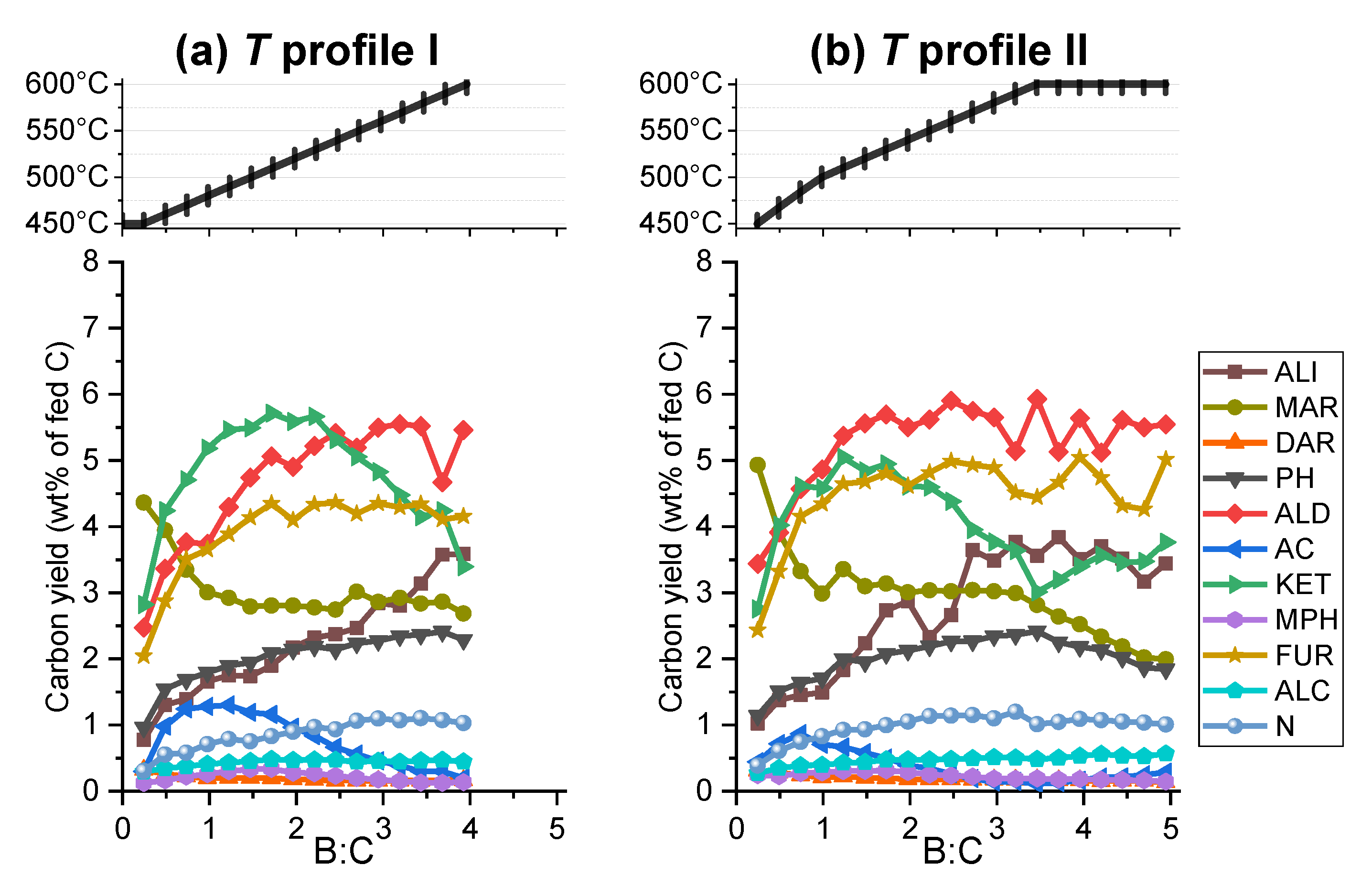
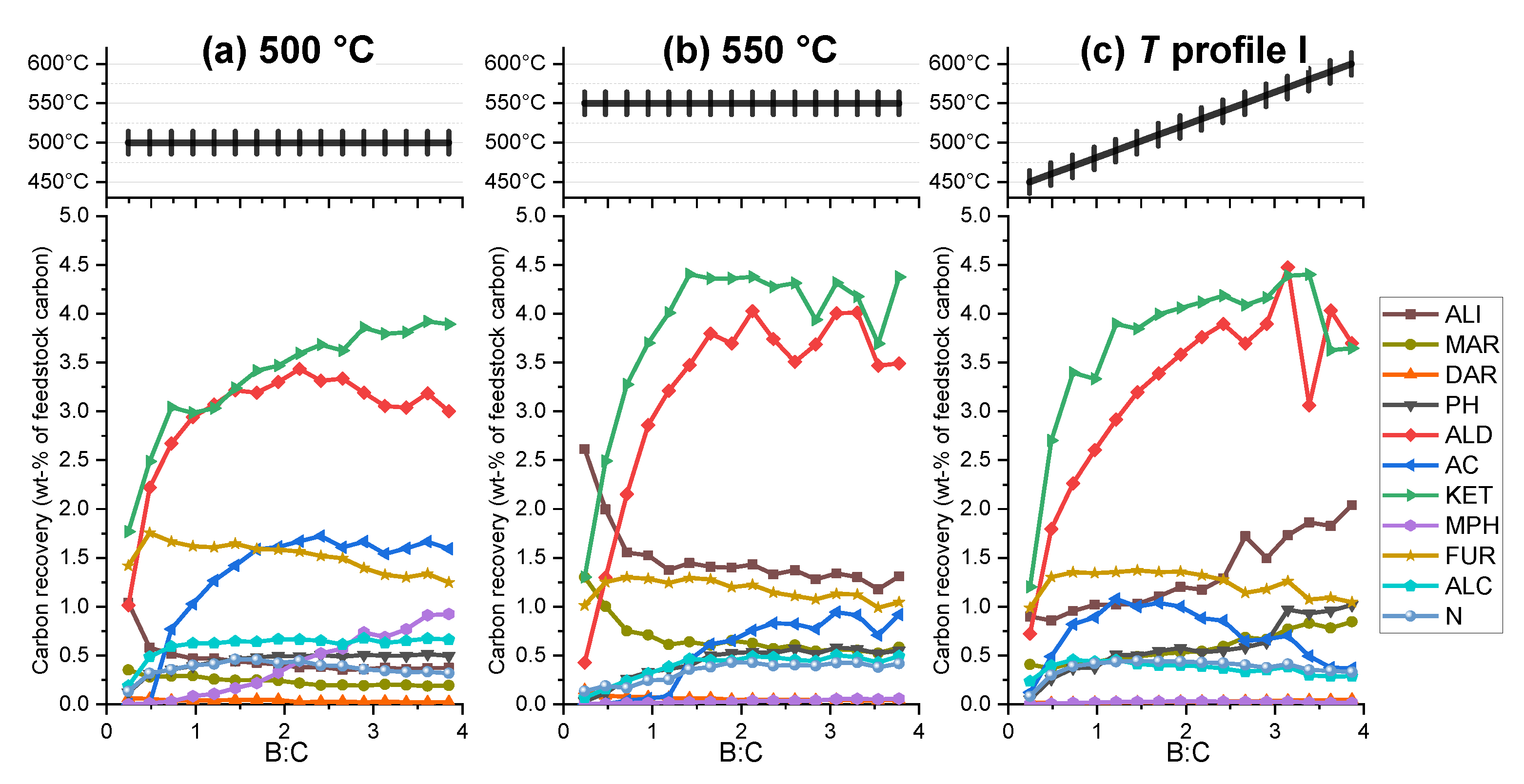
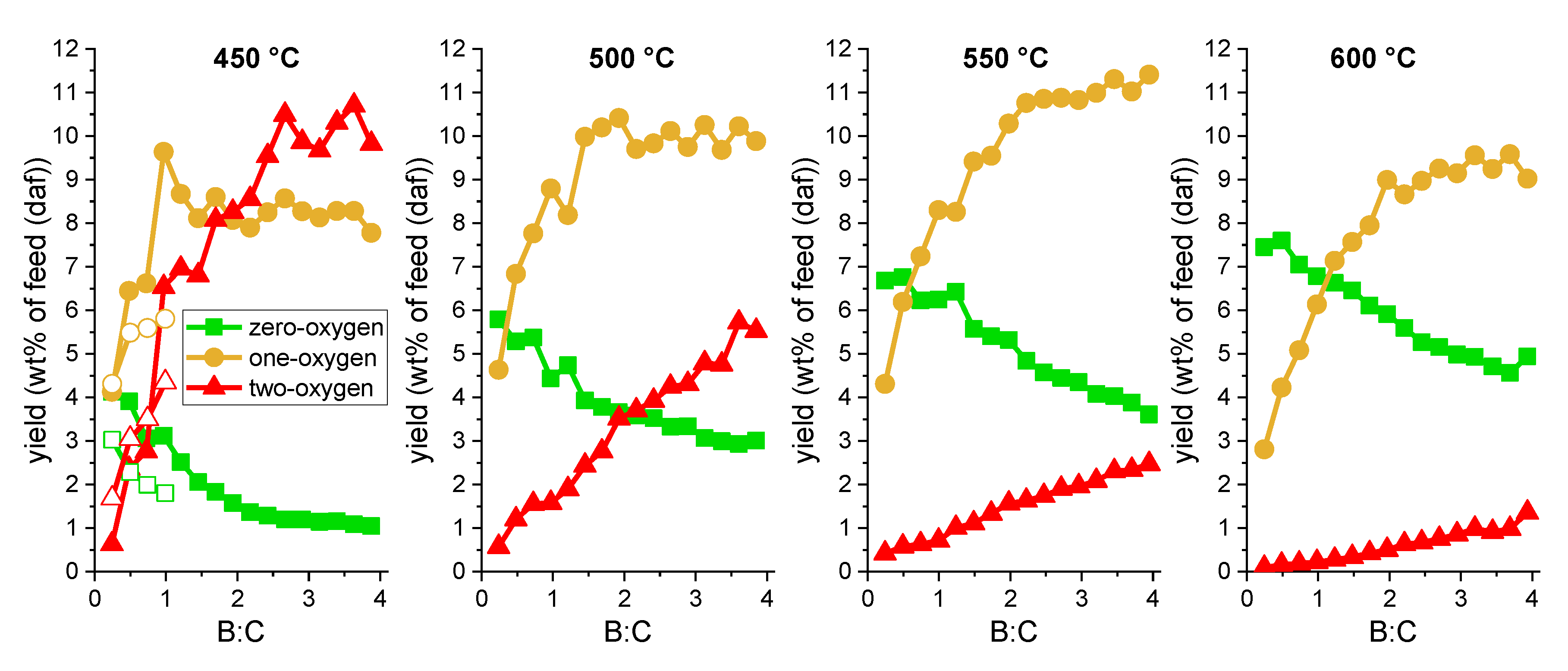
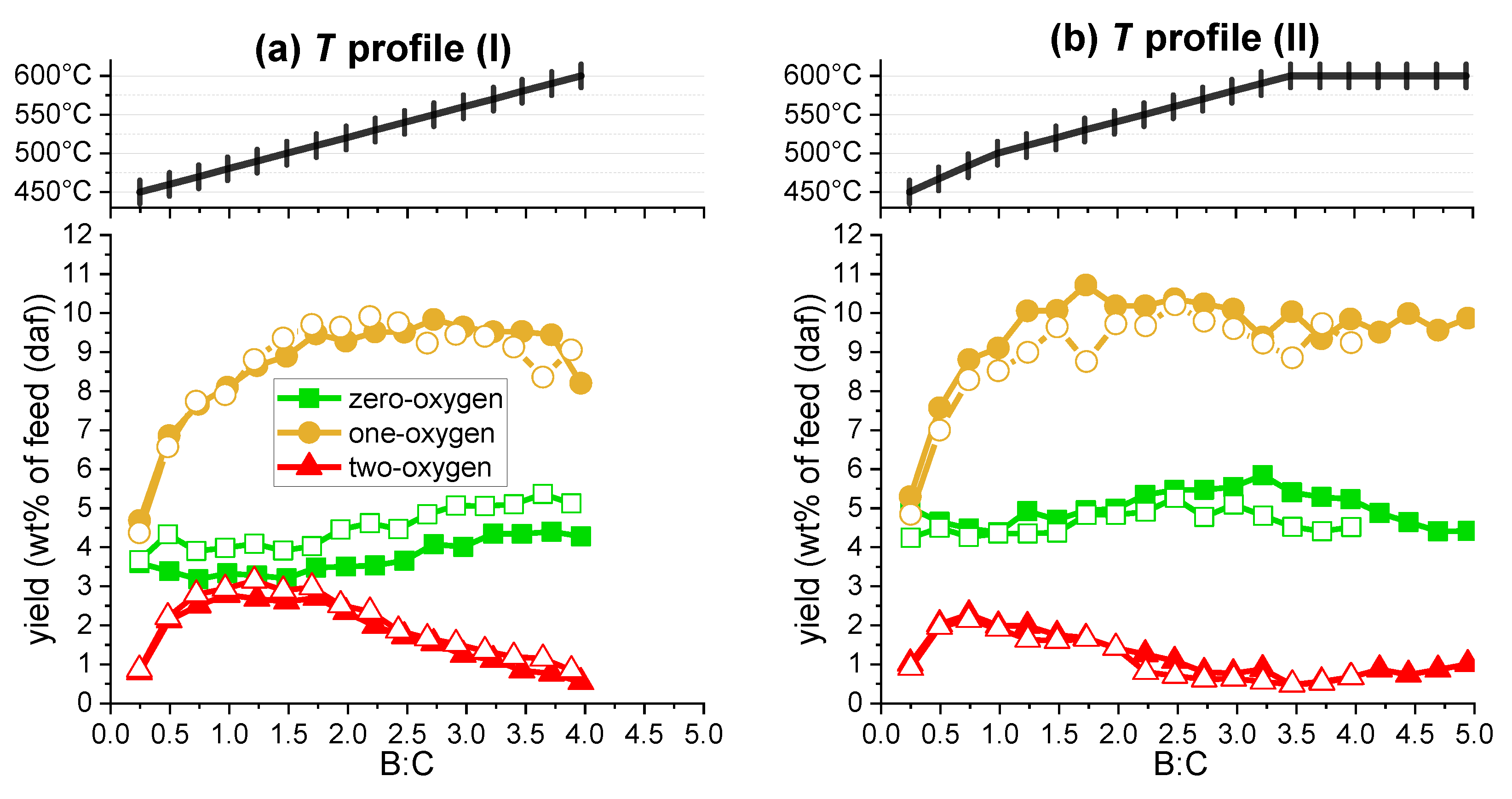
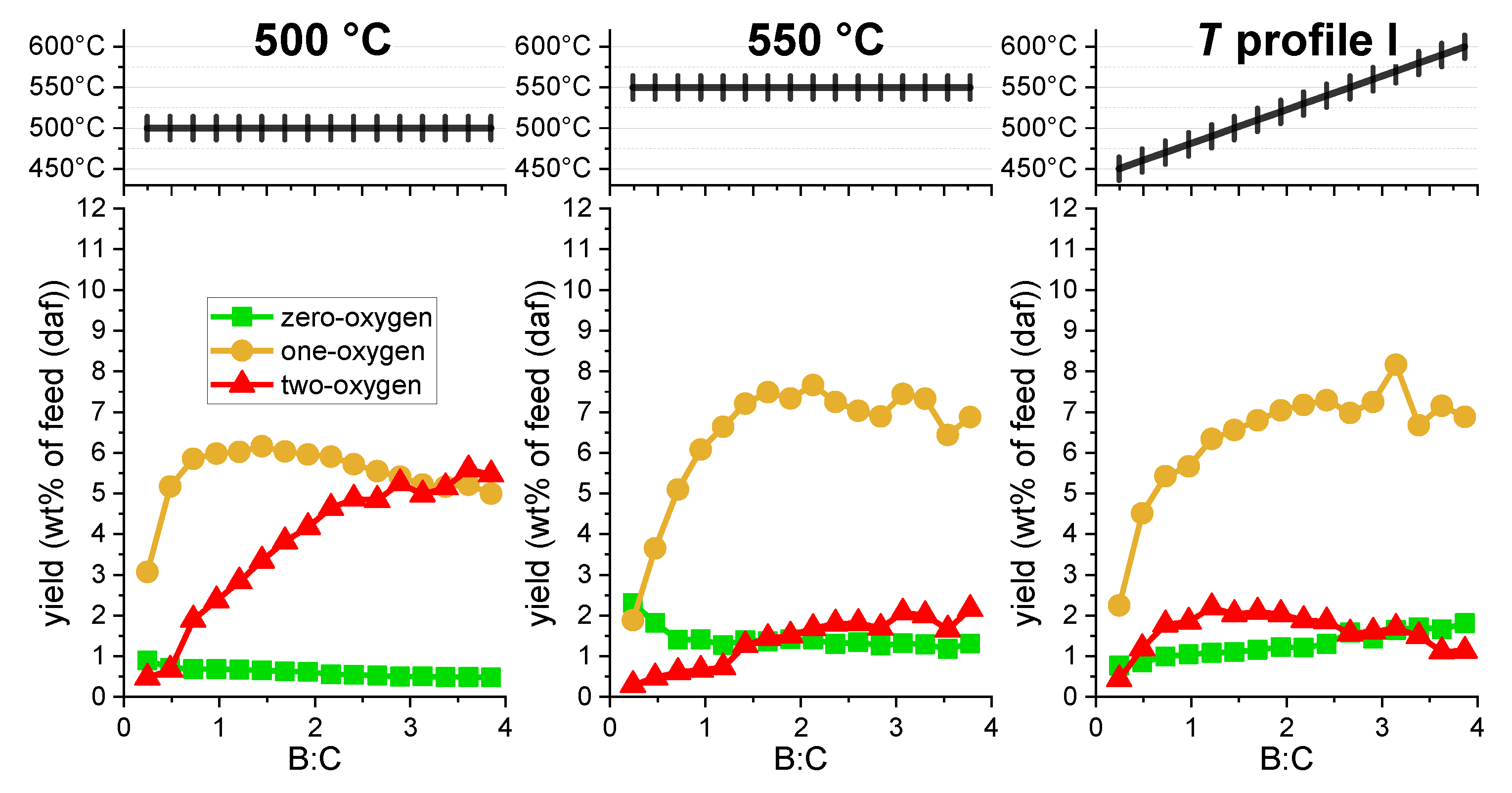
2.2.2. Vapors
2.2.3. Coke
2.2.4. Cumulative Product Yields
2.3. Product Quality
3. Discussion
4. Materials and Methods
4.1. Biomass
4.2. Catalyst Preparation
4.3. Catalyst Characterization
4.4. Micro-Pyrolyzer
4.5. Test Conditions
- T profile I: Starting from a temperature of 450 °C, the catalyst temperature was increased by 10 °C in between each injection (corresponding to delta B:C ~0.25) until reaching 600 °C at the 16th injection (at B:C ~4).
- T profile II: Starting from a temperature of 450 °C, the catalyst temperature was increased by 16.7 °C per injection for the first three injections (until reaching 500 °C), followed by a 10 °C increase per injection for the next ten injections and holding the temperature at 600 °C for the remaining injections. An additional test was completed with continued biomass feeding until reaching B:C ~5 while holding the temperature at 600 °C.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass and Bioenergy 2011, 38, 68–94. [Google Scholar] [CrossRef]
- Sharifzadeh, M.; Sadeqzadeh, M.; Guo, M.; Borhani, T.N.; Konda, N.V.S.N.M.; Garcia, M.C.; Wang, L.; Hallett, J.; Shah, N. The multi-scale challenges of biomass fast pyrolysis and bio-oil upgrading: Review of the state of art and future research directions. Prog. Energy Combust. Sci. 2019, 71, 1–80. [Google Scholar] [CrossRef]
- Krutof, A.; Hawboldt, K.A. Upgrading of biomass sourced pyrolysis oil review: Focus on co-pyrolysis and vapour upgrading during pyrolysis. Biomass Convers. Biorefin. 2018, 8, 775–787. [Google Scholar] [CrossRef]
- Ruddy, D.A.; Schaidle, J.A.; Ferrell, J.R., III; Wang, J.; Moens, L.; Hensley, J.E. Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: Catalyst development through the study of model compounds. Green Chem. 2014, 16, 454–490. [Google Scholar] [CrossRef]
- Stefanidis, S.D.; Kalogiannis, K.G.; Iliopoulou, E.F.; Lappas, A.A.; Pilavachi, P.A. In-situ upgrading of biomass pyrolysis vapors: Catalyst screening on a fixed bed reactor. Bioresour. Technol. 2011, 102, 8261–8267. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulou, E.F.; Stefanidis, S.D.; Kalogiannis, K.; Psarras, A.C.; Delimitis, A.; Triantafyllidis, K.S.; Lappas, A.A. Pilot-scale validation of Co-ZSM-5 catalyst performance in the catalytic upgrading of biomass pyrolysis vapours. Green Chem. 2014, 16, 662–674. [Google Scholar] [CrossRef]
- Williams, P.T.; Horne, P.A. The influence of catalyst regeneration on the composition of zeolite-upgraded biomass pyrolysis oils. Fuel 1995, 74, 1839–1851. [Google Scholar] [CrossRef]
- Iisa, K.; French, R.J.; Orton, K.A.; Dutta, A.; Schaidle, J.A. Production of low-oxygen bio-oil via ex situ catalytic fast pyrolysis and hydrotreating. Fuel 2017, 207, 413–422. [Google Scholar] [CrossRef]
- Williams, P.T.; Nugranad, N. Comparison of products from the pyrolysis and catalytic pyrolysis of rice husks. Energy 2000, 25, 493–513. [Google Scholar] [CrossRef]
- Hernando, H.; Fermoso, J.; Ochoa-Hernández, C.; Opanasenko, M.; Pizarro, P.; Coronado, J.M.; Čejka, J.; Serrano, D.P. Performance of MCM-22 zeolite for the catalytic fast-pyrolysis of acid-washed wheat straw. Catal. Today 2018, 304, 30–38. [Google Scholar] [CrossRef]
- Hernando, H.; Hernández-Giménez, A.M.; Gutiérrez-Rubio, S.; Fakin, T.; Horvat, A.; Danisi, R.M.; Pizarro, P.; Fermoso, J.; Heracleous, E.; Bruijnincx, P.C.A.; et al. Scaling-Up of Bio-Oil Upgrading during Biomass Pyrolysis over ZrO2/ZSM-5-Attapulgite. ChemSusChem 2019, 12, 2428–2438. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, I.Y.; Kazi, F.K.; Yusup, S.; Alaba, P.A.; Sani, Y.M.; Abakr, Y.A. Catalytic intermediate pyrolysis of Napier grass in a fixed bed reactor with ZSM-5, HZSM-5 and zinc-exchanged zeolite-a as the catalyst. Energies 2016, 9, 246. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhang, X.; Chen, L.; Zhao, B.; Yang, S.; Xie, X. Comparision of catalytic fast pyrolysis of biomass to aromatic hydrocarbons over ZSM-5 and Fe/ZSM-5 catalysts. J. Anal. Appl. Pyrolysis 2016, 121, 342–346. [Google Scholar] [CrossRef]
- Du, S.; Sun, Y.; Gamliel, D.P.; Valla, J.A.; Bollas, G.M. Catalytic pyrolysis of miscanthus×giganteus in a spouted bed reactor. Bioresour. Technol. 2014, 169, 188–197. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbach, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- Carlson, T.R.; Tompsett, G.A.; Conner, W.C.; Huber, G.W. Aromatic production from catalytic fast pyrolysis of biomass-derived feedstocks. Top. Catal. 2009, 52, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Saraeian, A.; Nolte, M.W.; Shanks, B.H. Deoxygenation of biomass pyrolysis vapors: Improving clarity on the fate of carbon. Renew. Sustain. Energy Rev. 2019, 104, 262–280. [Google Scholar] [CrossRef]
- Stöcker, M. Biofuels and biomass-to-liquid fuels in the biorefinery: Catalytic conversion of lignocellulosic biomass using porous materials. Angew. Chemie Int. Ed. 2008, 47, 9200–9211. [Google Scholar] [CrossRef]
- Charoenwiangnuea, P.; Maihom, T.; Kongpracha, P.; Sirijaraensre, J.; Limtrakul, J. Adsorption and decarbonylation of furfural over H-ZSM-5 zeolite: A DFT study. RSC Adv. 2016, 6, 105888–105894. [Google Scholar] [CrossRef]
- Foster, A.J.; Jae, J.; Cheng, Y.; Huber, G.W.; Lobo, R.F. Optimizing the aromatic yield and distribution from catalytic fast pyrolysis of biomass over ZSM-5. Appl. Catal. A Gen. 2012, 423–424, 154–161. [Google Scholar] [CrossRef]
- Cheng, Y.T.; Huber, G.W. Production of targeted aromatics by using Diels-Alder classes of reactions with furans and olefins over ZSM-5. Green Chem. 2012, 14, 3114–3125. [Google Scholar] [CrossRef]
- Patel, H.; Hao, N.; Iisa, K.; French, R.J.; Orton, K.A.; Mukarakate, C.; Ragauskas, A.J.; Nimlos, M.R. Detailed Oil Compositional Analysis Enables Evaluation of Impact of Temperature and Biomass-to-Catalyst Ratio on ex Situ Catalytic Fast Pyrolysis of Pine Vapors over ZSM-5. ACS Sustain. Chem. Eng. 2020, 8, 1762–1773. [Google Scholar] [CrossRef]
- Carlson, T.R.; Jae, J.; Lin, Y.C.; Tompsett, G.A.; Huber, G.W. Catalytic fast pyrolysis of glucose with HZSM-5: The combined homogeneous and heterogeneous reactions. J. Catal. 2010, 270, 110–124. [Google Scholar] [CrossRef]
- Carlson, T.R.; Cheng, Y.; Jae, J.; Huber, G.W. Production of green aromatics and olefins by catalytic fast pyrolysis of wood sawdust. Energy Environ. Sci. 2011, 4, 145–161. [Google Scholar] [CrossRef] [Green Version]
- Lazaridis, P.A.; Fotopoulos, A.P.; Karakoulia, S.A. Catalytic Fast Pyrolysis of Kraft Lignin With Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Schaidle, J.A.; Humbird, D.; Baddour, F.G.; Sahir, A. Conceptual Process Design and Techno-Economic Assessment of Ex Situ Catalytic Fast Pyrolysis of Biomass: A Fixed Bed Reactor Implementation Scenario for Future Feasibility. Top. Catal. 2016, 59, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Eschenbacher, A.; Jensen, P.A.; Henriksen, U.B.; Ahrenfeldt, J.; Li, C.; Duus, J.Ø.; Mentzel, U.V.; Jensen, A.D. Impact of ZSM-5 deactivation on bio-oil quality during upgrading of straw derived pyrolysis vapors. Energy Fuels 2019, 33, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Eschenbacher, A.; Jensen, P.A.; Henriksen, U.B.; Ahrenfeldt, J.; Li, C.; Duus, J.Ø.; Mentzel, U.V.; Jensen, A.D. Deoxygenation of wheat straw fast pyrolysis vapors using HZSM-5, Al2O3, HZSM-5/Al2O3 extrudates, and desilicated HZSM-5/Al2O3 extrudates. Energy Fuels 2019, 33, 6405–6420. [Google Scholar] [CrossRef]
- Kalogiannis, K.G.; Stefanidis, S.D.; Lappas, A.A. Catalyst deactivation, ash accumulation and bio-oil deoxygenation during ex situ catalytic fast pyrolysis of biomass in a cascade thermal-catalytic reactor system. Fuel Process. Technol. 2019, 186, 99–109. [Google Scholar] [CrossRef]
- Horne, P.A.; Williams, P.T. The effect of zeolite ZSM-5 catalyst deactivation during the upgrading of biomass-derived pyrolysis vapours. J. Anal. Appl. Pyrolysis 1995, 34, 65–85. [Google Scholar] [CrossRef]
- Diebold, J.P.; Scahill, J.W. Biomass To Gasoline (Btg): Upgrading Pyrolysis Vapors To Aromatic Gasoline With Zeolite Catalysis At Atmospheric Pressure. ACS Div. Fuel Chem. Prepr. 1987, 32, 297–307. [Google Scholar] [CrossRef]
- Scahill, J.W.; Diebold, J.P. Engineering Aspects of Upgrading Pyrolysis oil Using Zeolites; Elsevier: Amsterdam, The Netherlands, 1988; pp. 927–940. [Google Scholar]
- Xu, M.; Mukarakate, C.; Iisa, K.; Budhi, S.; Menart, M.; Davidson, M.; Robichaud, D.J.; Nimlos, M.R.; Trewyn, B.G.; Richards, R.M. Deactivation of Multilayered MFI Nanosheet Zeolite during Upgrading of Biomass Pyrolysis Vapors; ACS Publications: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Andersen, J.A.; Jensen, A.D. Catalytic conversion of acetol over HZSM-5 catalysts—influence of Si/Al ratio and introduction of mesoporosity. Catal. Today 2020. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Jensen, P.A.; Henriksen, U.B.; Ahrenfeldt, J.; Ndoni, S.; Li, C.; Duus, J.Ø.; Mentzel, U.V.; Jensen, A.D. Catalytic deoxygenation of vapors obtained from ablative fast pyrolysis of wheat straw using mesoporous HZSM-5. Fuel Process. Technol. 2019, 194, 106119. [Google Scholar] [CrossRef]
- Perkins, G.; Bhaskar, T.; Konarova, M. Process development status of fast pyrolysis technologies for the manufacture of renewable transport fuels from biomass. Renew. Sustain. Energy Rev. 2018, 90, 292–315. [Google Scholar] [CrossRef]
- Menon, P.G. Coke on catalysts-harmful, harmless, invisible and beneficial types. J. Mol. Catal. 1990, 59, 207–220. [Google Scholar] [CrossRef]
- Tamm, P.W.; Harnsberger, H.F.; Bridge, A.G. Effects of Feed Metals on Catalyst Aging in Hydroprocessing Residuum. Ind. Eng. Chem. Process Des. Dev. 1981, 20, 262–273. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Saraeian, A.; Shanks, B.H.; Mentzel, U.V.; Jensen, P.A.; Henriksen, U.B.; Ahrenfeldt, J.; Jensen, A.D. Performance-screening of metal-impregnated industrial HZSM-5/Al2O3 extrudates for deoxygenation and hydrodeoxygenation of fast pyrolysis vapors. J. Anal. Appl. Pyrolysis. 2020. under review. [Google Scholar]
- Blasco, T.; Corma, A.; Martínez-Triguero, J. Hydrothermal stabilization of ZSM-5 catalytic-cracking additives by phosphorus addition. J. Catal. 2006, 237, 267–277. [Google Scholar] [CrossRef]
- Huang, L.; Li, Q. Enhanced Acidity and Thermal Stability of Mesoporous Materials with Post-treatment with Phosphoric Acid. Chem. Lett. 1999, 28, 829–830. [Google Scholar] [CrossRef]
- Van der Bij, H.E.; Meirer, F.; Kalirai, S.; Wang, J.; Weckhuysen, B.M. Hexane cracking over steamed phosphated zeolite H-ZSM-5: Promotional effect on catalyst performance and stability. Chem. A Eur. J. 2014, 20, 16922–16932. [Google Scholar] [CrossRef]
- Caeiro, G.; Magnoux, P.; Lopes, J.M.; Ribeiro, F.R.; Menezes, S.M.C.; Costa, A.F.; Cerqueira, H.S. Stabilization effect of phosphorus on steamed H-MFI zeolites. Appl. Catal. A Gen. 2006, 314, 160–171. [Google Scholar] [CrossRef]
- Corma, A.; Mengual, J.; Miguel, P.J. Stabilization of ZSM-5 zeolite catalysts for steam catalytic cracking of naphtha for production of propene and ethene. Appl. Catal. A Gen. 2012, 421–422, 121–134. [Google Scholar] [CrossRef]
- Mante, O.D.; Agblevor, F.A.; Oyama, S.T.; McClung, R. The effect of hydrothermal treatment of FCC catalysts and ZSM-5 additives in catalytic conversion of biomass. Appl. Catal. A Gen. 2012, 445–446, 312–320. [Google Scholar] [CrossRef]
- Yao, W.; Li, J.; Feng, Y.; Wang, W.; Zhang, X.; Chen, Q.; Komarneni, S.; Wang, Y. Thermally stable phosphorus and nickel modified ZSM-5 zeolites for catalytic co-pyrolysis of biomass and plastics. RSC Adv. 2015, 5, 1–4. [Google Scholar] [CrossRef]
- Cerqueira, H.S.; Caeiro, G.; Costa, L.; Ribeiro, F.R. Deactivation of FCC catalysts. J. Mol. Catal. A Chem. 2008, 292, 1–13. [Google Scholar] [CrossRef]
- Van der Bij, H.E.; Weckhuysen, B.M. Local silico-aluminophosphate interfaces within phosphated H-ZSM-5 zeolites. Phys. Chem. Chem. Phys. 2014, 16, 9892. [Google Scholar] [CrossRef] [Green Version]
- Van der Bij, H.E.; Weckhuysen, B.M. Phosphorus promotion and poisoning in zeolite-based materials: Synthesis, characterisation and catalysis. Chem. Soc. Rev. 2015, 44, 7406–7428. [Google Scholar] [CrossRef] [Green Version]
- Bjørgen, M.; Svelle, S.; Joensen, F.; Nerlov, J.; Kolboe, S.; Bonino, F.; Palumbo, L.; Bordiga, S.; Olsbye, U. Conversion of methanol to hydrocarbons over zeolite H-ZSM-5: On the origin of the olefinic species. J. Catal. 2007, 249, 195–207. [Google Scholar] [CrossRef]
- Knöll, J.; Singh, U.; Nicolich, J.; Gonzalez, R.; Ziebarth, M.; Fougret, C.; Brandt, S. Unit cell volume as a measure of dealumination of ZSM-5 in fluid catalytic cracking catalyst. Ind. Eng. Chem. Res. 2014, 53, 16270–16274. [Google Scholar] [CrossRef]
- Degnan, T.F. Recent Progress in the Development of Zeolitic Catalysts for the Retroleum Refining and Petrochemical Manufacturing Industries; Elsevier: Amsterdam, The Netherlands, 2007; Volume 170, pp. 54–65. [Google Scholar] [CrossRef]
- Gao, X.; Tang, Z.; Ji, D.; Zhang, H. Modification of ZSM-5 zeolite for maximizing propylene in fluid catalytic cracking reaction. Catal. Commun. 2009, 10, 1787–1790. [Google Scholar] [CrossRef]
- Mukarakate, C.; McBrayer, J.D.; Evans, T.J.; Budhi, S.; Robichaud, D.J.; Iisa, K.; Dam, J.t.; Watson, M.J.; Baldwin, R.M.; Nimlos, M.R. Catalytic fast pyrolysis of biomass: The reactions of water and aromatic intermediates produces phenols. Green Chem. 2015, 17, 4217–4227. [Google Scholar] [CrossRef]
- Mullen, C.A.; Tarves, P.C.; Boateng, A.A. Role of Potassium Exchange in Catalytic Pyrolysis of Biomass over ZSM-5: Formation of Alkyl Phenols and Furans. ACS Sustain. Chem. Eng. 2017, 5, 2154–2162. [Google Scholar] [CrossRef]
- Stanton, A.; Iisa, K.; Mukarakate, C.; Nimlos, M.R. Role of Biopolymers in the Deactivation of ZSM-5 during Catalytic Fast Pyrolysis of Biomass. ACS Sustain. Chem. Eng. 2018, 6, 10030–10038. [Google Scholar] [CrossRef]
- Du, S.; Gamliel, D.P.; Valla, J.A.; Bollas, G.M. The effect of ZSM-5 catalyst support in catalytic pyrolysis of biomass and compounds abundant in pyrolysis bio-oils. J. Anal. Appl. Pyrolysis 2016, 122, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Eschenbacher, A.; Goodarzi, F.; Saraeian, A.; Kegnæs, S.; Shanks, B.H.; Jensen, A.D. Performance of Mesoporous HZSM-5 and Silicalite-1 Coated Mesoporous HZSM-5 Catalysts for Deoxygenation of Straw Fast Pyrolysis Vapors. J. Anal. Appl. Pyrolysis 2019, 145, 104712. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Saraeian, A.; Shanks, B.H.; Jensen, P.A.; Henriksen, U.B.; Ahrenfeldt, J.; Jensen, A.D. Insights into the scalability of catalytic upgrading of biomass pyrolysis vapors using micro and bench-scale reactors. Sustain. Energy Fuels 2020, 4, 3780–3796. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Griffin, M.B.; Iisa, K.; Wang, H.; Dutta, A.; Orton, K.A.; French, R.J.; Santosa, D.M.; Wilson, N.; Christensen, E.; Nash, C.; et al. Driving towards cost-competitive biofuels through catalytic fast pyrolysis by rethinking catalyst selection and reactor configuration. Energy Environ. Sci. 2018, 11, 2904–2918. [Google Scholar] [CrossRef]
- Hoff, T.C.; Thilakaratne, R.; Gardner, D.W.; Brown, R.C.; Tessonnier, J.P. Thermal Stability of Aluminum-Rich ZSM-5 Zeolites and Consequences on Aromatization Reactions. J. Phys. Chem. C 2016, 120, 20103–20113. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Rooney, D.W.; Halawy, S.A.; Mohamed, M.A.; Abdelkader, A. Effect of precursor on the performance of alumina for the dehydration of methanol to dimethyl ether. Appl. Catal. B Environ. 2012, 127, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Ong, L.H.; Dömök, M.; Olindo, R.; van Veen, A.C.; Lercher, J.A. Dealumination of HZSM-5 via steam-treatment. Microporous Mesoporous Mater. 2012, 164, 9–20. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Saraeian, A.; Shanks, B.H.; Jensen, P.A.; Li, C.; Duus, Ø. Enhancing bio-oil quality and energy recovery by atmospheric hydrodeoxygenation of wheat straw pyrolysis vapors using Pt and Mo-based catalysts. Sustain. Energy Fuels 2020, 4, 1991–2008. [Google Scholar] [CrossRef]
- Laumer, J.D.S.; Cicchetti, E.; Merle, P.; Egger, J. Quantification in Gas Chromatography Prediction of FID Response Factors from Combustion Enthalpies and Molecular Structures. Anal. Chem. 2010, 82, 6457–6462. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.Y.; Walsh, D.E.; Koenig, L.R. Fluidized Bed Upgrading of Wood Pyrolysis Liquids and Related Compounds. ACS Div. Fuel Chem. Prepr. 1987, 32, 264–275. [Google Scholar] [CrossRef]
- Channiwala, S.A.; Parikh, P.P. A unified correlation for estimating HHV of solid, liquid and gaseous fuels. Fuel 2002, 81, 1051–1063. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P Content (wt%) | BET (m2/g) | Vmeso (cm3/g) | Vtotal (cm3/g) | Acidity (mmol NH3/g) | |
|---|---|---|---|---|---|
| HZSM-5/γ-Al2O3 | - | 376 | 0.32 | 0.45 | 0.39 |
| P/HZSM-5/γ-Al2O3 | 0.41 | 381 | 0.28 | 0.41 | 0.40 |
| γ-Al2O3 | - | 235 | 0.53 | 0.52 | 0.31 |
| Catalyst | SiC * | P/HZSM-5/γ-Al2O3 | γ-Al2O3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature (°C) | 500 | 450 | 500 | 550 | 600 | profile (I) | profile (II) | 500 | 550 | profile (I) |
| Gas | 17.3 | 21.3 | 25.8 | 32.1 | 38.8 | 30.2 | 32.7 | 19.9 | 23.6 | 22.3 |
| CO | 6.4 | 7.2 | 8.6 | 10.4 | 12.4 | 9.7 | 10.6 | 8.4 | 9.7 | 9.3 |
| CO2 | 9.7 | 9.9 | 10.2 | 10.7 | 10.9 | 10.4 | 10.8 | 9.7 | 10.8 | 10.5 |
| C1-C3 alkanes | 0.13 | 0.04 | 0.21 | 0.31 | 0.66 | 0.30 | 0.34 | 0.21 | 0.44 | 0.21 |
| C2-C3 alkenes | 0.5 | 1.9 | 3.6 | 6.2 | 9.3 | 5.5 | 6.2 | 0.7 | 1.4 | 1.1 |
| C4+ | 0.6 | 2.2 | 3.4 | 4.5 | 5.6 | 4.3 | 4.8 | 0.9 | 1.2 | 1.1 |
| Vapors | 16.3 | 21.5 | 22.7 | 23.7 | 21.6 | 21.1 | 23.4 | 11.6 | 12.1 | 12.1 |
| ALI | 0.2 | 1.3 | 2.9 | 4.1 | 5.2 | 3.2 | 4.1 | 0.5 | 1.5 | 1.3 |
| Aromatics | 0.1 | 1.6 | 3.3 | 3.9 | 4.3 | 3.3 | 3.6 | 0.3 | 0.7 | 0.6 |
| Benzene | 0.0 | 0.1 | 0.3 | 0.4 | 0.5 | 0.3 | 0.4 | 0.1 | 0.1 | 0.1 |
| Toluene | 0.1 | 0.3 | 0.7 | 1.1 | 1.4 | 0.8 | 0.9 | 0.1 | 0.2 | 0.2 |
| Xylenes | 0.0 | 0.3 | 0.7 | 0.8 | 0.7 | 0.6 | 0.7 | 0.0 | 0.1 | 0.1 |
| Alkyl-benzenes | 0.1 | 0.3 | 0.6 | 0.4 | 0.3 | 0.4 | 0.5 | 0.0 | 0.1 | 0.1 |
| Alkenyl-benzenes | 0.0 | 0.2 | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0.0 | 0.0 | 0.1 |
| Indanes | 0.0 | 0.1 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 |
| Indenes | 0.0 | 0.2 | 0.3 | 0.4 | 0.4 | 0.3 | 0.3 | 0.0 | 0.2 | 0.1 |
| 2-ring aromatics | 0.0 | 0.1 | 0.2 | 0.4 | 0.4 | 0.3 | 0.3 | 0.0 | 0.1 | 0.0 |
| 3-ring aromatics | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 |
| PH | 0.6 | 1.4 | 1.9 | 2.3 | 2.2 | 2.2 | 2.6 | 0.4 | 0.4 | 0.6 |
| ALD | 3.1 | 2.5 | 3.1 | 3.3 | 2.9 | 2.9 | 3.2 | 2.9 | 3.2 | 3.2 |
| AC | 2.2 | 2.8 | 1.5 | 0.4 | 0.1 | 0.9 | 0.5 | 1.3 | 0.5 | 0.7 |
| KET | 6.3 | 6.4 | 5.2 | 4.8 | 3.0 | 4.4 | 4.5 | 3.3 | 3.8 | 3.7 |
| MPH | 1.3 | 1.2 | 0.6 | 0.1 | 0.0 | 0.1 | 0.0 | 0.4 | 0.0 | 0.0 |
| FUR | 1.4 | 3.1 | 2.9 | 3.2 | 2.7 | 2.8 | 3.1 | 1.5 | 1.2 | 1.2 |
| ALC | 0.7 | 0.5 | 0.4 | 0.4 | 0.3 | 0.5 | 0.4 | 0.6 | 0.4 | 0.4 |
| NIT | 0.0 | 0.7 | 0.9 | 1.1 | 1.0 | 0.9 | 1.1 | 0.4 | 0.3 | 0.4 |
| Coke | 0 | 5.9 | 6.9 | 6.6 | 8.2 | 7.7 | 6.9 | 5.7 | 7.2 | 8.6 |
| C-% closure † | 65 | 80 | 87 | 94 | 100 | 90 | 93 | 68 | 74 | 74 |
| Vapors | Gas | |||||||
|---|---|---|---|---|---|---|---|---|
| Catalyst | T (°C) | B:C | EHI | H/C | O/C | HHV (MJ/kg) | wt% O | Atom. CO/CO2 Ratio |
| SiC | 500 | 1 | 0.65 | 1.71 | 0.53 | 24.1 | 34.2 | 0.66 |
| HZSM-5/γ-Al2O3 | 500 | 4 | 0.97 | 1.46 | 0.24 | 31.7 | 22.2 | 1.22 |
| P/HZSM-5/γ-Al2O3 | 450 | 4 | 0.82 | 1.54 | 0.36 | 28.0 | 29.6 | 0.73 |
| 500 | 4 | 0.98 | 1.50 | 0.26 | 31.4 | 23.1 | 0.84 | |
| 550 | 4 | 1.07 | 1.47 | 0.20 | 33.7 | 18.7 | 0.97 | |
| 600 | 4 | 1.15 | 1.47 | 0.16 | 35.4 | 15.6 | 1.15 | |
| profile I * | 4 | 1.02 | 1.47 | 0.23 | 32.6 | 20.8 | 0.94 | |
| profile II * | 4 | 1.10 | 1.47 | 0.20 | 33.8 | 18.5 | 0.97 | |
| profile II * | 5 | 1.09 | 1.47 | 0.20 | 33.70 | 18.7 | 0.98 | |
| γ-Al2O3 | 500 | 4 | 0.89 | 1.71 | 0.41 | 27.3 | 32.0 | 0.86 |
| 550 | 4 | 1.08 | 1.69 | 0.31 | 30.6 | 26.1 | 0.90 | |
| profile I * | 4 | 1.05 | 1.68 | 0.32 | 30.2 | 26.6 | 0.88 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eschenbacher, A.; Saraeian, A.; Shanks, B.H.; Mentzel, U.V.; Ahrenfeldt, J.; Henriksen, U.B.; Jensen, A.D. Counteracting Rapid Catalyst Deactivation by Concomitant Temperature Increase during Catalytic Upgrading of Biomass Pyrolysis Vapors Using Solid Acid Catalysts. Catalysts 2020, 10, 748. https://doi.org/10.3390/catal10070748
Eschenbacher A, Saraeian A, Shanks BH, Mentzel UV, Ahrenfeldt J, Henriksen UB, Jensen AD. Counteracting Rapid Catalyst Deactivation by Concomitant Temperature Increase during Catalytic Upgrading of Biomass Pyrolysis Vapors Using Solid Acid Catalysts. Catalysts. 2020; 10(7):748. https://doi.org/10.3390/catal10070748
Chicago/Turabian StyleEschenbacher, Andreas, Alireza Saraeian, Brent H. Shanks, Uffe Vie Mentzel, Jesper Ahrenfeldt, Ulrik Birk Henriksen, and Anker Degn Jensen. 2020. "Counteracting Rapid Catalyst Deactivation by Concomitant Temperature Increase during Catalytic Upgrading of Biomass Pyrolysis Vapors Using Solid Acid Catalysts" Catalysts 10, no. 7: 748. https://doi.org/10.3390/catal10070748