
Enhancer RNA Transcription Is Essential for a Novel CSF1 Enhancer in Triple-Negative Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Global Run-On Sequencing (GRO-Seq) and ChIP-Seq
2.3. RT-PCR of Patient Tumors
2.4. Quantitative RT-PCR
2.5. Expression of CSF1 across Breast Cancer or All TCGA Cancer Types
2.6. TCGA Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing (ATAC-Seq) Data Analysis
2.7. CSF1 Enhancer Knockout with CRISPR-Cas9
2.8. Cell Proliferation Assays
2.9. Cell Migration Assays
2.10. Colony Formation Assay
2.11. Engineering MDA-MB-436 and OVCAR3 dCas9-KRAB Expressing Cells
2.12. CRISPRi sgRNA Design and Vector Cloning
2.13. CRISPR Interference (CRISPRi) Targeted Perturbation
2.14. RNA-Sequencing and Analysis
2.15. Kaplan Meier Survival Analyses in Breast Cancer and Ovarian Cancer
2.16. Engineering PspCas13b Stable Expression Cell Lines
2.17. PspCas13b eRNA Knockdown Transfections
2.18. Engineering RfxCas13d Stable Expression Cell Lines
2.19. RNA Interference (RNAi)
2.20. Apoptosis Assay of MDA-MB-436 CSF1 Enhancer Knockout Cells
3. Results
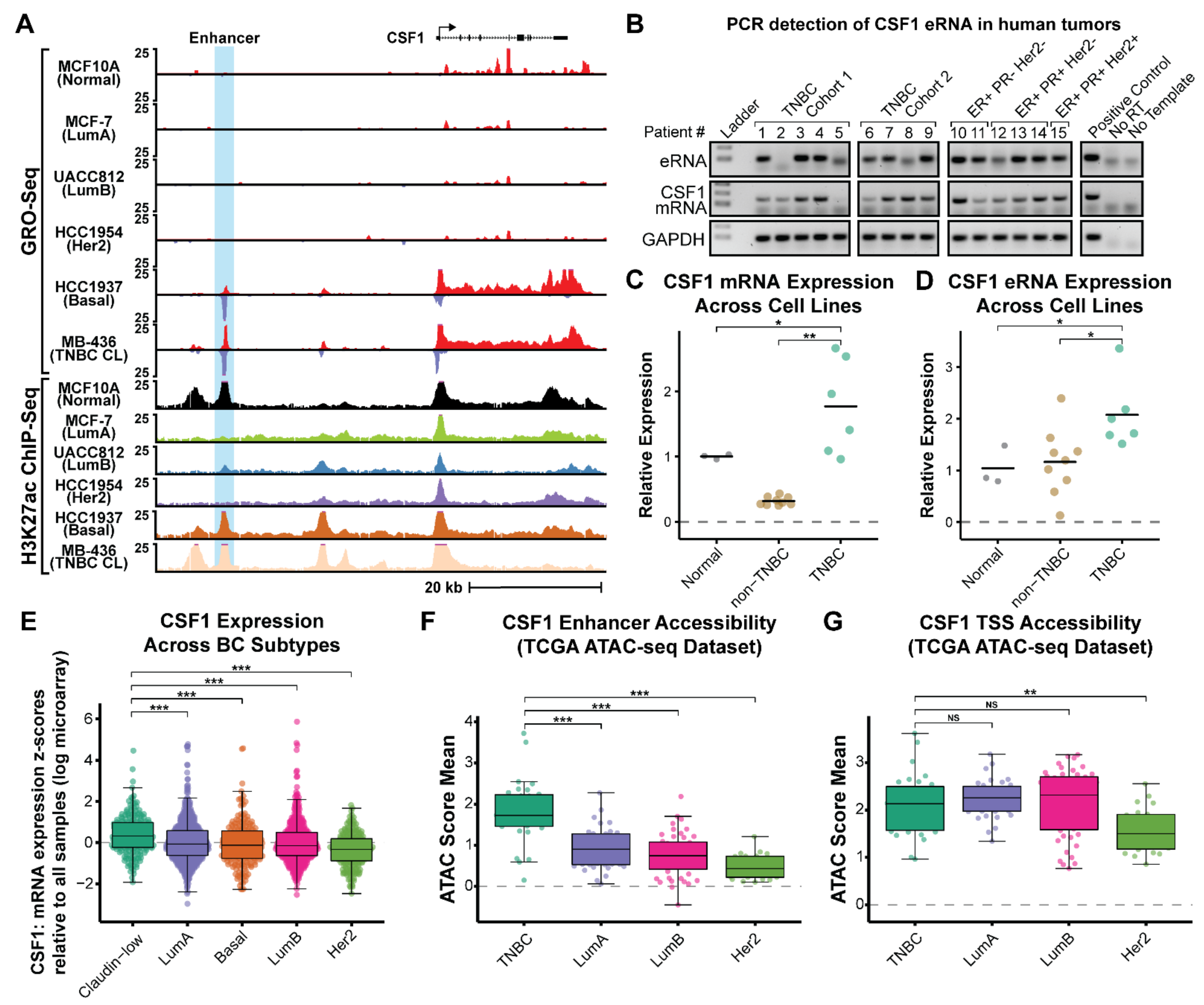
3.1. Enhancer RNA Production Highlights a Novel Enhancer of CSF1 in Triple-Negative Breast Cancer (TNBC)
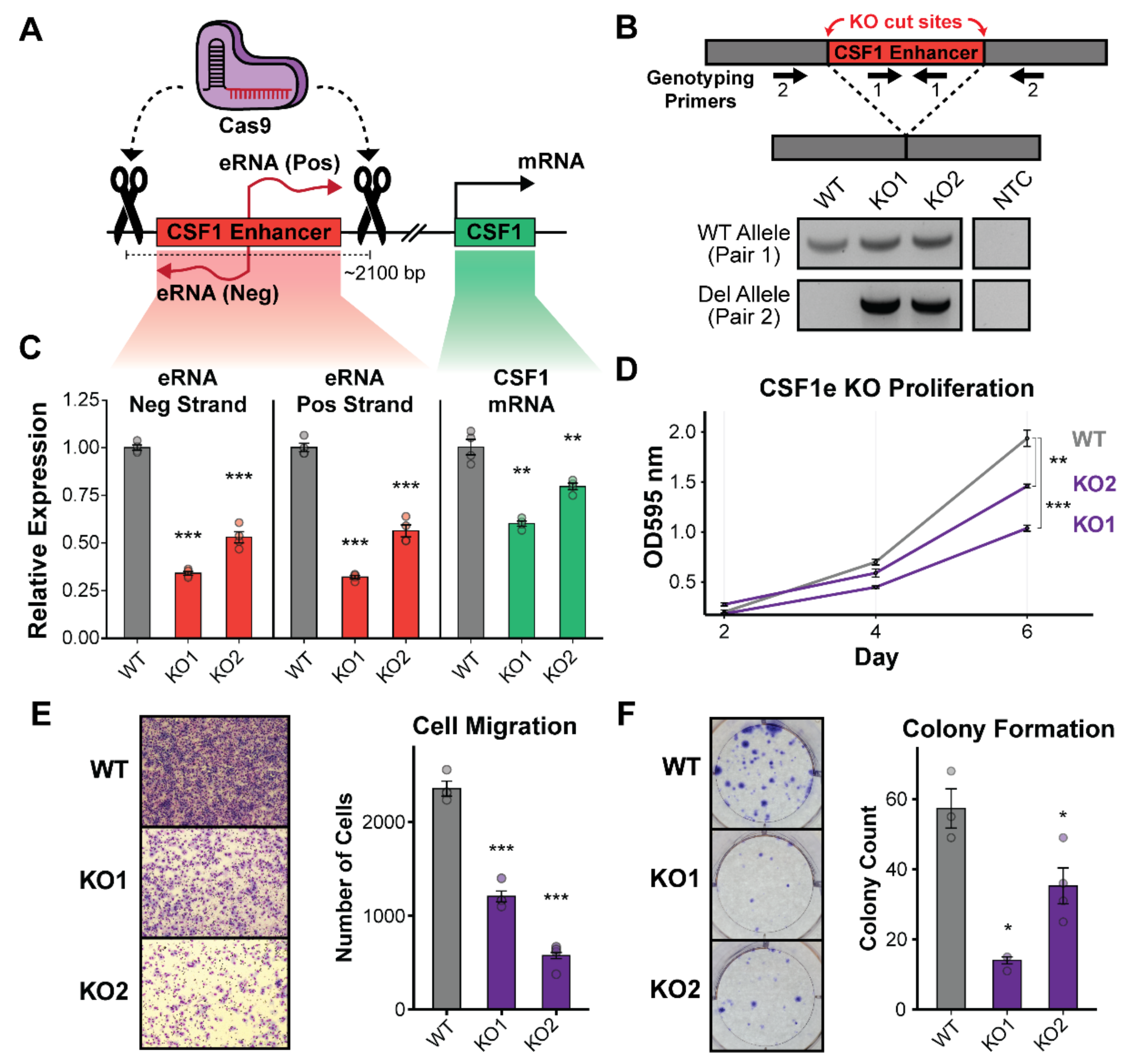
3.2. Deletion of the CSF1 Enhancer Reduces CSF1 mRNA and Leads to Growth Defects in TNBC Cells
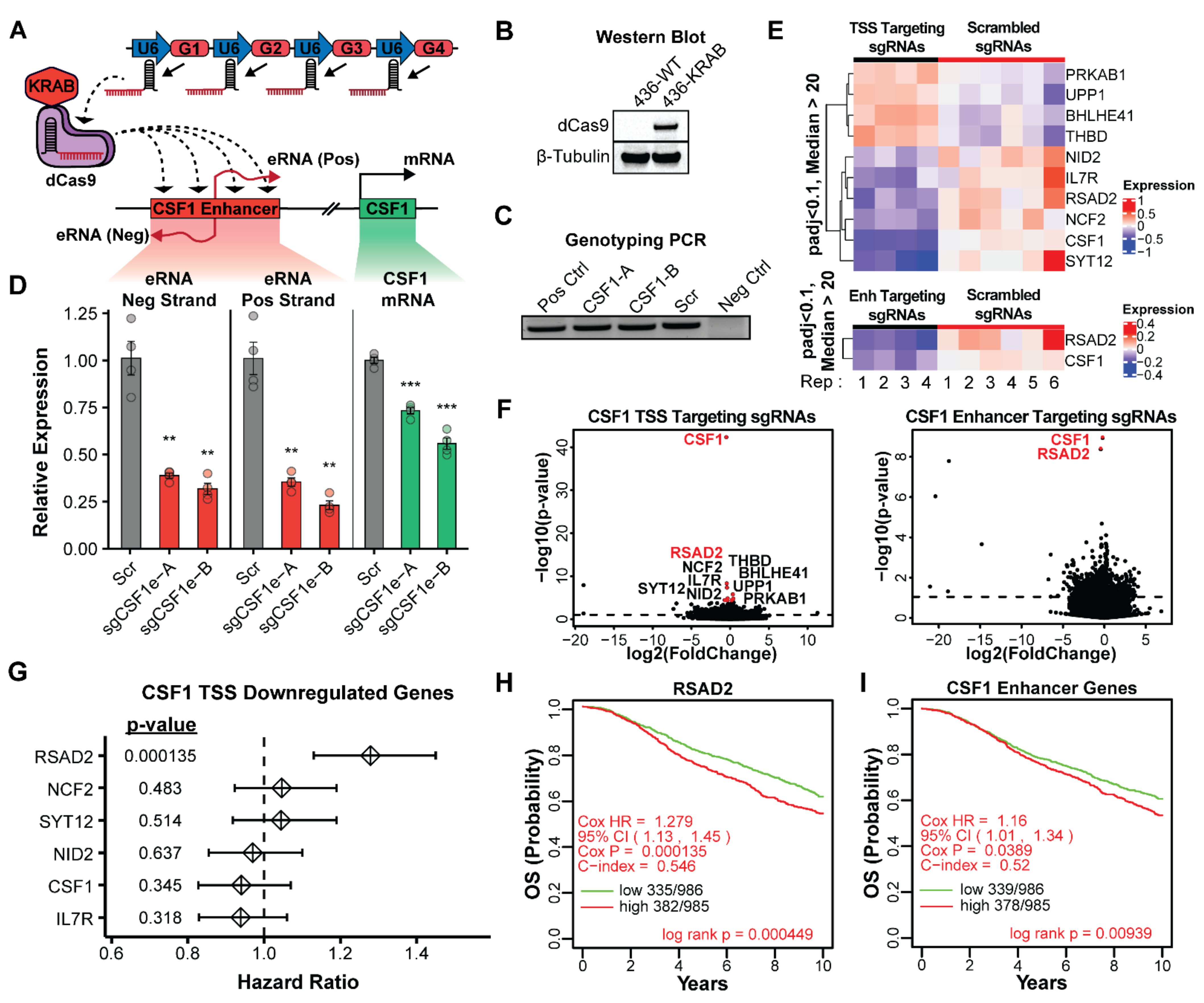
3.3. Expression and Accessibility of the CSF1 Enhancer Is Critical for Its Activity
3.4. Activity of the CSF1 Enhancer Is Correlated with Reduced Patient Survival
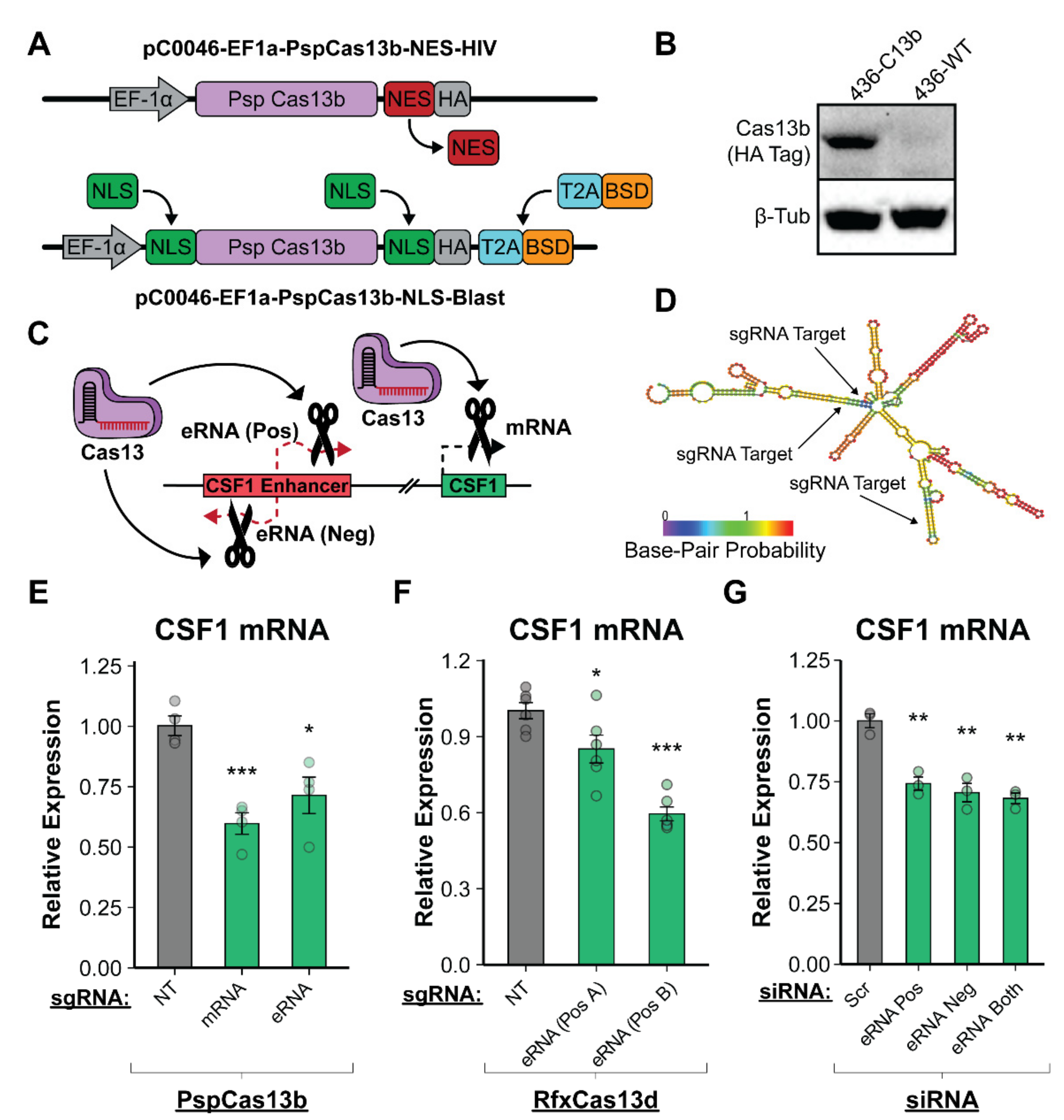
3.5. Enhancer RNA Transcripts Are Necessary for CSF1 Upregulation in a Post-Transcriptional Mechanism
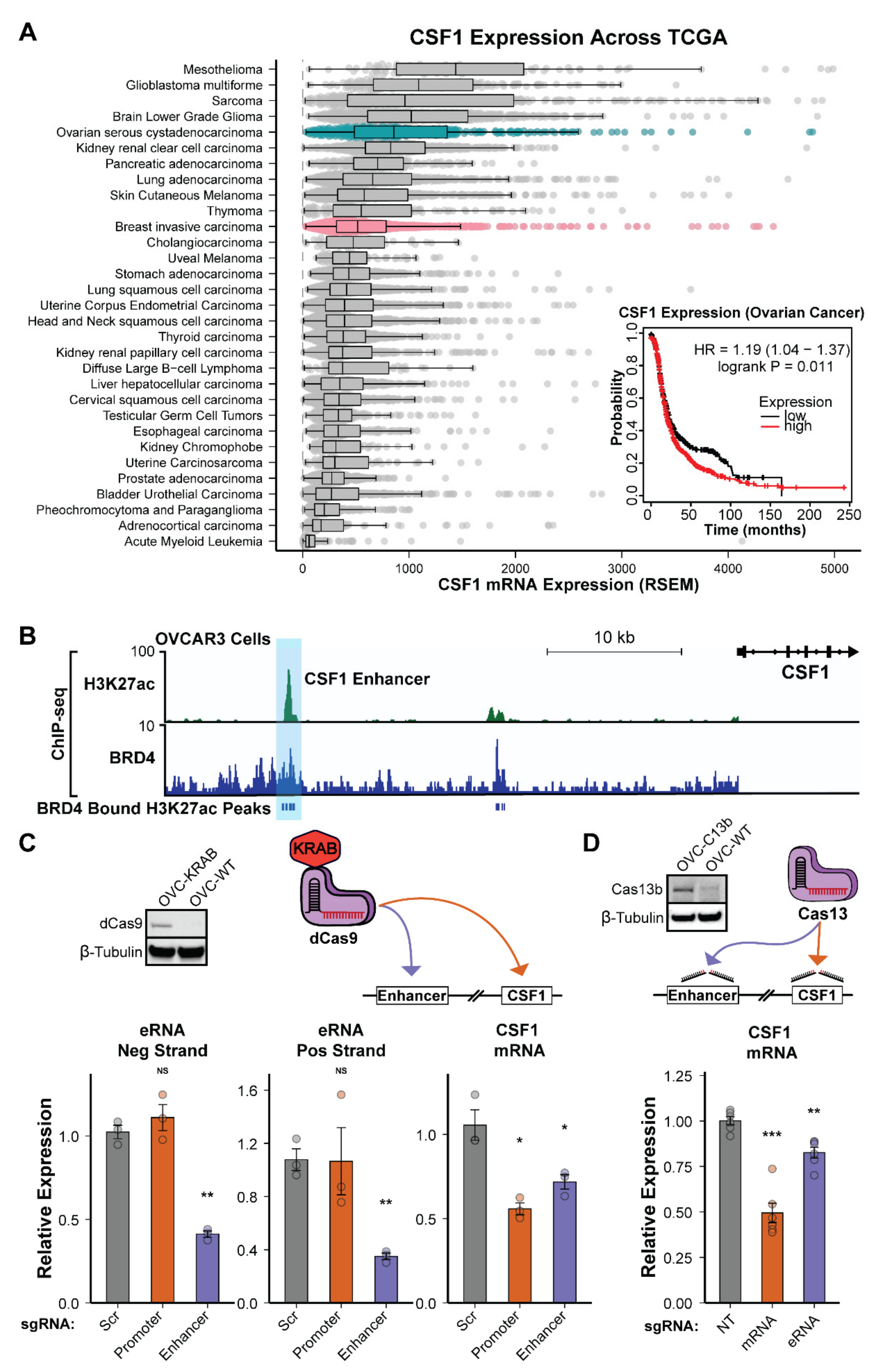
3.6. The CSF1 Enhancer Is Active in Ovarian Cancer and eRNA Transcription Is Essential for Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Provenzano, E.; Caldas, C. Triple negative breast cancers: Clinical and prognostic implications. Eur. J. Cancer 2009, 45 (Suppl. S1), 27–40. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.L.; Updike, K.L.; Factor, R.E.; Henry, N.L.; Boucher, K.M.; Bernard, P.S.; Varley, K.E. A Multigene Assay Determines Risk of Recurrence in Patients with Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3466–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.U.; Vanderplas, A.; Hughes, M.E.; Theriault, R.L.; Edge, S.B.; Wong, Y.N.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; Weeks, J.C. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Bulut, N.; Kilickap, S.; Sari, E.; Altundag, K. Response to taxanes in triple negative breast cancer. Cancer Chemother. Pharmacol. 2008, 63, 189. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16 (Suppl. S1), 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Cancer Genome Atlas Research, N.; Liang, H. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386–399.e312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, H.L.; Nagari, A.; Malladi, V.S.; Li, W.; Xi, Y.; Richardson, D.; Allton, K.L.; Tanaka, K.; Li, J.; Murakami, S.; et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome. Res. 2018, 28, 159–170. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Kadonaga, J.T. Going the distance: A current view of enhancer action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 2010, 20, R754–R763. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Seumois, G.; Chavez, L.; Gerasimova, A.; Lienhard, M.; Omran, N.; Kalinke, L.; Vedanayagam, M.; Ganesan, A.P.; Chawla, A.; Djukanovic, R.; et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat. Immunol. 2014, 15, 777–788. [Google Scholar] [CrossRef]
- Verzi, M.P.; Shin, H.; He, H.H.; Sulahian, R.; Meyer, C.A.; Montgomery, R.K.; Fleet, J.C.; Brown, M.; Liu, X.S.; Shivdasani, R.A. Differentiation-specific histone modifications reveal dynamic chromatin interactions and partners for the intestinal transcription factor CDX2. Dev. Cell 2010, 19, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Lee, S.; Yun, J.; Kim, A. Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the beta-globin locus. Biosci. Rep. 2015, 35, e00179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Satpathy, A.T.; Boyle, E.A.; Dai, C.; Gowen, B.G.; Cho, S.W.; Nguyen, M.L.; Rubin, A.J.; Granja, J.M.; Kazane, K.R.; et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017, 49, 1602–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lin, F.; Xing, K.; He, X. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Q.; Zhang, W.; Lin, J.R.; Cai, Y.; Mitra, J.; Zhang, Z.D. HEDD: Human Enhancer Disease Database. Nucleic Acids Res. 2018, 46, D113–D120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Rickels, R.; Shilatifard, A. Enhancer Logic and Mechanics in Development and Disease. Trends. Cell Biol. 2018, 28, 608–630. [Google Scholar] [CrossRef] [PubMed]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017, 547, 55–60. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.L.; Natoli, G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010, 8, e1000384. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.T.; Cho, H.; Lesch, H.P.; Gosselin, D.; Heinz, S.; Tanaka-Oishi, Y.; Benner, C.; Kaikkonen, M.U.; Kim, A.S.; Kosaka, M.; et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013, 498, 511–515. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013, 498, 516–520. [Google Scholar] [CrossRef]
- Rahnamoun, H.; Lee, J.; Sun, Z.; Lu, H.; Ramsey, K.M.; Komives, E.A.; Lauberth, S.M. RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat. Struct. Mol. Biol. 2018, 25, 687–697. [Google Scholar] [CrossRef]
- Tsai, P.F.; Dell’ Orso, S.; Rodriguez, J.; Vivanco, K.O.; Ko, K.D.; Jiang, K.; Juan, A.H.; Sarshad, A.A.; Vian, L.; Tran, M.; et al. A Muscle-Specific Enhancer RNA Mediates Cohesin Recruitment and Regulates Transcription In trans. Mol. Cell 2018, 71, 129–141.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaukowitch, K.; Joo, J.Y.; Liu, X.; Watts, J.K.; Martinez, C.; Kim, T.K. Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 2014, 56, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Ivaldi, M.S.; Diaz, L.F.; Chakalova, L.; Lee, J.; Krivega, I.; Dean, A. Fetal gamma-globin genes are regulated by the BGLT3 long noncoding RNA locus. Blood 2018, 132, 1963–1973. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.W.; Li, S.; Franco, H.L. Transcriptional control by enhancers and enhancer RNAs. Transcription 2019, 10, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arner, E.; Daub, C.O.; Vitting-Seerup, K.; Andersson, R.; Lilje, B.; Drablos, F.; Lennartsson, A.; Ronnerblad, M.; Hrydziuszko, O.; Vitezic, M.; et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015, 347, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Hah, N.; Murakami, S.; Nagari, A.; Danko, C.G.; Kraus, W.L. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013, 23, 1210–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.S.; Chen, S.; et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324. [Google Scholar] [CrossRef] [Green Version]
- Bose, D.A.; Donahue, G.; Reinberg, D.; Shiekhattar, R.; Bonasio, R.; Berger, S.L. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 2017, 168, 135–149.e22. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, K.; Zare, H.; Dell’ orso, S.; Grontved, L.; Gutierrez-Cruz, G.; Derfoul, A.; Hager, G.L.; Sartorelli, V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 2013, 51, 606–617. [Google Scholar] [CrossRef] [Green Version]
- Sigova, A.A.; Abraham, B.J.; Ji, X.; Molinie, B.; Hannett, N.M.; Guo, Y.E.; Jangi, M.; Giallourakis, C.C.; Sharp, P.A.; Young, R.A. Transcription factor trapping by RNA in gene regulatory elements. Science 2015, 350, 978–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Xi, Y.; Shi, J.; Li, W.; Tanaka, K.; Allton, K.L.; Richardson, D.; Li, J.; Franco, H.L.; Nagari, A.; Malladi, V.S.; et al. Histone modification profiling in breast cancer cell lines highlights commonalities and differences among subtypes. BMC Genom. 2018, 19, 150. [Google Scholar] [CrossRef] [Green Version]
- Kalender Atak, Z.; Imrichova, H.; Svetlichnyy, D.; Hulselmans, G.; Christiaens, V.; Reumers, J.; Ceulemans, H.; Aerts, S. Identification of cis-regulatory mutations generating de novo edges in personalized cancer gene regulatory networks. Genome Med. 2017, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, O.M.; Sammut, S.J.; Seoane, J.A.; Chin, S.F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic. Acids. Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Duan, J.; Li, B.; Zhou, P.; Hon, G.C. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 2017, 66, 285–299.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawhorn, I.E.; Ferreira, J.P.; Wang, C.L. Evaluation of sgRNA target sites for CRISPR-mediated repression of TP53. PLoS ONE 2014, 9, e113232. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Lánczky, A.; Szállási, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessels, H.-H.; Méndez-Mancilla, A.; Guo, X.; Legut, M.; Daniloski, Z.; Sanjana, N.E. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 2020, 38, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sun, L.; Chen, Z.; Whitaker, J.W.; Wang, T.; Wang, W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013, 41, 10032–10043. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.H.; Pan, D.Z.; Tsai, Z.T.; Tsai, H.K. Genome-wide analysis of enhancer RNA in gene regulation across 12 mouse tissues. Sci. Rep. 2015, 5, 12648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Hatake, K.; Terado, Y.; Tsukino, H.; Okegawa, T.; Nutahara, K.; Higashihara, E.; Horie, S. Serum level of macrophage colony-stimulating factor is increased in prostate cancer patients with bone metastasis. Hum. Cell 2008, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pixley, F.J.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, A.; Barbetti, V.; Riverso, M.; Dello Sbarba, P.; Rovida, E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS ONE 2011, 6, e27450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Guo, C.; Hu, P.; Chen, J.; Liu, Q.; Wu, X.; Cao, Y.; Wu, J. CSF1 is involved in breast cancer progression through inducing monocyte differentiation and homing. Int. J. Oncol. 2016, 49, 2064–2074. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Pollard, J.W. Role of infiltrated leucocytes in tumour growth and spread. Br. J. Cancer 2004, 90, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, D.W.; Chan, A.; Dirix, L.Y.; Cortes, J.; Pivot, X.; Tomczak, P.; Delozier, T.; Sohn, J.H.; Provencher, L.; Puglisi, F.; et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2010, 28, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Begg, G.E.; Harper, S.L.; Friedman, J.R.; Speicher, D.W.; Rauscher, F.J., 3rd. Biochemical analysis of the Kruppel-associated box (KRAB) transcriptional repression domain. J. Biol. Chem. 2000, 275, 18000–18010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J., 3rd. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlan, T.S.; Gifford, W.D.; Agarwal, S.; Driscoll, S.; Lettieri, K.; Wang, J.; Andrews, S.E.; Franco, L.; Rosenfeld, M.G.; Ren, B.; et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011, 25, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Ayyanathan, K.; Lechner, M.S.; Bell, P.; Maul, G.G.; Schultz, D.C.; Yamada, Y.; Tanaka, K.; Torigoe, K.; Rauscher, F.J., 3rd. Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: A mammalian cell culture model of gene variegation. Genes Dev. 2003, 17, 1855–1869. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.S.; Begg, G.E.; Speicher, D.W.; Rauscher, F.J., 3rd. Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: Direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell Biol. 2000, 20, 6449–6465. [Google Scholar] [CrossRef]
- Rowe, H.M.; Friedli, M.; Offner, S.; Verp, S.; Mesnard, D.; Marquis, J.; Aktas, T.; Trono, D. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 2013, 140, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Wiznerowicz, M.; Jakobsson, J.; Szulc, J.; Liao, S.; Quazzola, A.; Beermann, F.; Aebischer, P.; Trono, D. The Kruppel-associated box repressor domain can trigger de novo promoter methylation during mouse early embryogenesis. J. Biol. Chem. 2007, 282, 34535–34541. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Racko, D.; Benedetti, F.; Dorier, J.; Stasiak, A. Transcription-induced supercoiling as the driving force of chromatin loop extrusion during formation of TADs in interphase chromosomes. Nucleic Acids Res. 2018, 46, 1648–1660. [Google Scholar] [CrossRef] [Green Version]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Oude Vrielink, J.A.; Elkon, R.; Melo, S.A.; Léveillé, N.; Kalluri, R.; et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pnueli, L.; Rudnizky, S.; Yosefzon, Y.; Melamed, P. RNA transcribed from a distal enhancer is required for activating the chromatin at the promoter of the gonadotropin α-subunit gene. Proc. Natl. Acad. Sci. USA 2015, 112, 4369–4374. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Dominguez, J.R.; Hu, W.; Yuan, B.; Shi, J.; Park, S.S.; Gromatzky, A.A.; van Oudenaarden, A.; Lodish, H.F. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood 2014, 123, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.R.; Kim, Y.J.; Kim, T.H. A novel virus-inducible enhancer of the interferon-β gene with tightly linked promoter and enhancer activities. Nucleic Acids Res. 2014, 42, 12537–12554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, A.; Mimura, J.; Itoh, K. Non-coding RNA derived from the region adjacent to the human HO-1 E2 enhancer selectively regulates HO-1 gene induction by modulating Pol II binding. Nucleic Acids Res. 2014, 42, 13599–13614. [Google Scholar] [CrossRef] [Green Version]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Onodera, C.S.; Underwood, J.G.; Katzman, S.; Jacobs, F.; Greenberg, D.; Salama, S.R.; Haussler, D. Gene isoform specificity through enhancer-associated antisense transcription. PLoS ONE 2012, 7, e43511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ounzain, S.; Pezzuto, I.; Micheletti, R.; Burdet, F.; Sheta, R.; Nemir, M.; Gonzales, C.; Sarre, A.; Alexanian, M.; Blow, M.J.; et al. Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. J. Mol. Cell Cardiol. 2014, 76, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shechner, D.M.; Hacisuleyman, E.; Younger, S.T.; Rinn, J.L. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods 2015, 12, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, S.; Maurer, K.; Zhang, Z.; Petri, M.; Sullivan, K.E. Enhancer RNA and NFkappaB-dependent P300 regulation of ADAMDEC1. Mol. Immunol. 2018, 103, 312–321. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, J.S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell 2015, 58, 1028–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhyasen, G.W.; Yao, Y.; Zhang, J.; Dulak, A.; Castriotta, L.; Jacques, K.; Zhao, W.; Gharahdaghi, F.; Hattersley, M.M.; Lyne, P.D.; et al. BRD4 amplification facilitates an oncogenic gene expression program in high-grade serous ovarian cancer and confers sensitivity to BET inhibitors. PLoS ONE 2018, 13, e0200826. [Google Scholar] [CrossRef] [Green Version]
- Qing, T.; Mohsen, H.; Marczyk, M.; Ye, Y.; O’Meara, T.; Zhao, H.; Townsend, J.P.; Gerstein, M.; Hatzis, C.; Kluger, Y.; et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat. Commun. 2020, 11, 2438. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Marty, R.; Hofree, M.; Gross, A.M.; Jensen, J.; Fisch, K.M.; Wu, X.; DeBoever, C.; Van Nostrand, E.L.; Song, Y.; et al. Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer. Cancer Discov. 2017, 7, 410–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Kitchen-Smith, I.; Xiong, L.; Stracquadanio, G.; Brown, K.; Richter, P.H.; Wallace, M.D.; Bond, E.; Sahgal, N.; Moore, S.; et al. Germline and Somatic Genetic Variants in the p53 Pathway Interact to Affect Cancer Risk, Progression, and Drug Response. Cancer Res. 2021, 81, 1667–1680. [Google Scholar] [CrossRef]
- Claringbould, A.; Zaugg, J.B. Enhancers in disease: Molecular basis and emerging treatment strategies. Trends Mol. Med. 2021, 27, 1060–1073. [Google Scholar] [CrossRef]
- Mirzadeh Azad, F.; Atlasi, Y. Deregulation of Transcriptional Enhancers in Cancer. Cancers 2021, 13, 3532. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smargon, A.A.; Cox, D.B.T.; Pyzocha, N.K.; Zheng, K.; Slaymaker, I.M.; Gootenberg, J.S.; Abudayyeh, O.A.; Essletzbichler, P.; Shmakov, S.; Makarova, K.S.; et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol. Cell 2017, 65, 618–630.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.X.; Chong, S.; Zhang, H.; Makarova, K.S.; Koonin, E.V.; Cheng, D.R.; Scott, D.A. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 2018, 70, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Zorca, C.E.; Traboulsi, T.; Noutahi, E.; Krause, M.R.; Mader, S.; Zenklusen, D. Single-cell profiling reveals that eRNA accumulation at enhancer-promoter loops is not required to sustain transcription. Nucleic Acids Res. 2017, 45, 3017–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Almada, A.E.; Zamudio, J.R.; Sharp, P.A. Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc. Natl. Acad. Sci. USA 2011, 108, 10460–10465. [Google Scholar] [CrossRef] [Green Version]
- Lubas, M.; Andersen, P.R.; Schein, A.; Dziembowski, A.; Kudla, G.; Jensen, T.H. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep. 2015, 10, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.H.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 4562. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Keating, M.J.; Gandhi, V.; Plunkett, W. Transcription inhibition by flavopiridol: Mechanism of chronic lymphocytic leukemia cell death. Blood 2005, 106, 2513–2519. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Refsing Andersen, P.; Valen, E.; Core, L.J.; Bornholdt, J.; Boyd, M.; Heick Jensen, T.; Sandelin, A. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun. 2014, 5, 5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaCava, J.; Houseley, J.; Saveanu, C.; Petfalski, E.; Thompson, E.; Jacquier, A.; Tollervey, D. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 2005, 121, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Kazadi, D.; Sun, J.; Federation, A.; Chao, J.; Elliott, O.; Liu, Z.P.; et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 2015, 161, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, H.L.; Kraus, W.L. No driver behind the wheel? Targeting transcription in cancer. Cell 2015, 163, 28–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, M.W.; Wisniewska, K.; King, C.M.; Li, S.; Coffey, A.; Kelly, M.R.; Regner, M.J.; Franco, H.L. Enhancer RNA Transcription Is Essential for a Novel CSF1 Enhancer in Triple-Negative Breast Cancer. Cancers 2022, 14, 1852. https://doi.org/10.3390/cancers14071852
Lewis MW, Wisniewska K, King CM, Li S, Coffey A, Kelly MR, Regner MJ, Franco HL. Enhancer RNA Transcription Is Essential for a Novel CSF1 Enhancer in Triple-Negative Breast Cancer. Cancers. 2022; 14(7):1852. https://doi.org/10.3390/cancers14071852
Chicago/Turabian StyleLewis, Michael W., Kamila Wisniewska, Caitlin M. King, Shen Li, Alisha Coffey, Michael R. Kelly, Matthew J. Regner, and Hector L. Franco. 2022. "Enhancer RNA Transcription Is Essential for a Novel CSF1 Enhancer in Triple-Negative Breast Cancer" Cancers 14, no. 7: 1852. https://doi.org/10.3390/cancers14071852