
Interrogating the Genomic Landscape of Uterine Leiomyosarcoma: A Potential for Patient Benefit
by
Genevieve V. Dall
1,2,*, 
Anne Hamilton
2,3,4,
Gayanie Ratnayake
4,
Clare Scott
1,2,3,4 and
Holly Barker
1,2 1
Walter and Eliza Hall, Institute of Medical Research, Parkville, VIC 3052, Australia
2
Department of Medical Biology, University of Melbourne, Parkville, VIC 3010, Australia
3
Peter MacCallum Cancer Centre, Melbourne, VIC 3000, Australia
4
Royal Women’s Hospital, Parkville, VIC 3052, Australia
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(6), 1561; https://doi.org/10.3390/cancers14061561
Submission received: 11 February 2022
/
Revised: 10 March 2022
/
Accepted: 16 March 2022
/
Published: 18 March 2022
(This article belongs to the Special Issue Genomics of Gynaecological Cancer Defining Current and Future Treatment Options)
Abstract
:Simple Summary
Uterine leiomyosarcoma is an aggressive and rare cancer that is difficult to treat. There are a number of mutations that are common to uterine leiomyosarcoma that are currently not routinely targeted therapeutically in this cancer type. In this review, we summarise the studies being undertaken to investigate the effectiveness of targeting these mutations either pre-clinically in models of uterine leiomyosarcoma or in other cancers in the clinic. We hope this review will encourage the inclusion of uterine leiomyosarcoma in clinical trial design, which in turn will lead to improved survival outcomes for patients.
Abstract
Uterine leiomyosarcoma (uLMS) is a rare and aggressive gynaecological malignancy. Surgical removal and chemotherapy are commonly used to treat uLMS, but recurrence rates are high. Over the last few decades, clarification of the genomic landscape of uLMS has revealed a number of recurring mutations, including TP53, RB1, ATRX, PTEN, and MED12. Such genomic aberrations are difficult to target therapeutically or are actively targeted in other malignancies, and their potential as targets for the treatment of uLMS remains largely unexplored. Recent identification of deficiencies in homologous recombination in a minority of these tumours, however, has provided a rationale for investigation of PARP inhibitors in this sub-set. Here, we review these mutations and the evidence for therapeutic avenues that may be applied in uLMS. We also provide a comprehensive background on diagnosis and current therapeutic strategies as well as reviewing preclinical models of uLMS, which may be employed not only in testing emerging therapies but also in understanding this challenging and deadly disease.
1. Introduction
Uterine leiomyosarcoma (uLMS) is a rare gynaecological malignancy arising in the smooth muscle layer of the uterus. It accounts for less that 2% of uterine malignancy [1], affecting approximately 0.6–0.8/100,000 women each year [2,3], but the uterus is the most common primary site of leiomyosarcoma in women, and uLMS accounts for more than 10% of all soft tissue sarcomas [4], so that is usually well represented as a distinct subgroup in clinical trials of metastatic soft tissue sarcoma. The 5-year survival rate of uLMS is 42–76% [5,6], yet uLMS accounts for 70% of uterine sarcoma deaths [7]. This is due to initial diagnosis often being made when the disease is already advanced, and metastasis has occurred. Recurrence following optimal surgical de-bulking is also common [8]. Moreover, due to the rare nature of this disease, there are few large cohort trials testing emerging therapies, and thus, advances in therapeutic management of uLMS have been slow to evolve.
2. Disease Characteristics and Epidemiology
uLMS are generally large tumours, often presenting at greater than 5 cm in diameter [9]. uLMS is typically diagnosed in the 6th decade of life, with an average age of 53 reported at diagnosis [6,10,11]. Carriers of inherited TP53 and RB1 mutations carry an increased risk of uLMS [12,13], and occasional cases have been reported with tamoxifen use [14,15], but in the majority of cases, no risk factors have been identified. Age at diagnosis, tumour size, and disease stage all impact survival outcome, with increases in each of these prognostic categories leading to poorer outcomes for women with uLMS [6,10,11]. In addition, African-American patients have lower survival rates than their White counterparts irrespective of treatment modalities [6,10,16]. As a tumour of the uterus, hormone receptor expression is often recorded at diagnosis, with both oestrogen receptor (ER) and progesterone receptor (PR) levels expressed at similar rates (35−87% and 17−80%, respectively) [17,18,19,20,21,22]. PR expression has been associated with improved PFS (20.8 months in PR positive cases compared to 8.1 months in PR negative) irrespective of tumour grade, whilst an improvement in PFS has only been observed in ER positive cases when adjusting for tumour grade [19]. Histopathological assessment is the standard method of diagnosis, as currently there is no test or imaging technique that allows for preoperative diagnosis. uLMS is classified into three subtypes: spindle-cell type and the less common myxoid and epithelioid types [23]. Spindle-cell-type uLMS contains spindle cells with nuclear pleomorphism and show tumour type coagulative necrosis and >10 mitoses per 10 high-power fields [9]. Myxoid tumours contain an abundance of myxoid stroma [24], whilst epithelioid tumours are comprised of round or polygonal cells with eosinophilic cytoplasm [23]. As some of these features are not unique to uLMS, differential diagnosis includes benign smooth muscle tumours, such as symplastic leiomyomas, leiomyomas with intravenous extension, mitotically active leiomyomas, smooth muscle tumour of uncertain malignant potential (STUMPs), and other uterine sarcomas, such as high-grade endometrial stromal sarcoma (HGESS), undifferentiated uterine sarcoma, perivascular epithelioid cell tumour (PEComa), and inflammatory myofibroblastic tumour (IMT) [25]. Diagnosis requires assessment of morphology and specific immunohistochemical stains to support the morphological diagnosis. Both benign and malignant smooth muscle tumours express smooth muscle markers, such as smooth muscle actin (SMA), Desmin, and Caldesmon [7]. uLMS tend to have greater expression of Ki−67, mutation type p53 staining, and stronger and more diffuse p16 expression compared with benign smooth muscle tumours [26,27,28]. STUMPs demonstrate positivity for smooth muscle markers similar to leiomyosarcoma and leiomyomas. Immunohistochemistry markers, such as p16, p53, Ki-67, p21, BCL2, ER, and PR, have been used to enhance the diagnosis and prognosis in these tumours without success [29]. Generally, STUMPs should have one of the criteria (diffuse moderate for severe cytological atypia, coagulative tumour necrosis, and increase mitotic activity) used for the diagnosis of leiomyosarcoma, but the histological abnormalities seen in a STUMP fall short of a diagnosis of leiomyosarcoma. Low-grade endometrial stromal sarcomas (LGESS) may show some positivity for smooth muscle markers and will also show diffuse positivity for CD10, ER, and PR, making them very difficult to differentiate from uLMS [30]. HGESS, however, show positivity for Cyclin-D1 and BCOR and will lack expression of CD10, ER, and PR [31]. HGESS also harbour YWHAE::NUTM2A/B gene fusions and BCOR rearrangements, which are diagnostic and not associated with uLMS [32], whilst the gene fusion JAZF1::SUZ12 is most common in LGESS [33]. Beta-catenin may also be used to distinguish ESS from uLMS [34]. PEComas are composed of epithelioid and/or spindle cells and express at least one smooth muscle marker but differ from uLMS in their expression of HMB45 or Melan-A [35]. Detection of TFE3 rearrangement or fusion is helpful for the diagnosis of PEComa as distinct from uLMS [36]. Like uLMS, IMTs frequently express the smooth muscle markers SMA, Desmin, and Caldesmon [37]. ALK expression is also highly sensitive and specific for diagnosis of IMT; however, the degree of positivity varies, with some tumours showing only focal positivity [38]. Undifferentiated uterine sarcoma is a malignant mesenchymal tumour lacking evidence of specific lines of differentiation and therefore is a diagnosis of exclusion [39].
3. Treatment
Surgical removal of uLMS via hysterectomy is typically recommended based on data showing surgical removal with negative surgical margins leads to an increase in survival [6,10]. Importantly, resection of uLMS using power morcellation is associated with reduced survival (FDA warning April 2014; [40]) and thus should be avoided. The addition of bilateral salpingo-oophorectomy (BSO) is considered only if the patient is over the age of 50 years, as BSO is not associated with increased survival in patients with early-stage disease [6,10]. There has been little evidence to support adjuvant therapy following surgical removal of primary disease confined to the uterus. In a recent meta-analysis of 545 patients across nine studies, there was no survival advantage of adjuvant chemotherapy or radiation for patients with early stage, completely resected uLMS compared to observation alone [41]. Another recent analysis performed on 1030 cases collected by the National Cancer Database again showed no increase in survival for patients who received adjuvant chemotherapy (33% of cases), radiation (7.7%), or a combination of the two (6.2%) compared to observation alone following surgical removal of their primary tumour (53.1% of patients, suggesting that selection of patients for adjuvant therapy had taken place) [11]. In the metastatic setting, surgery is still considered beneficial. In a cohort of 96 patients with metastatic uLMS at presentation, treated at Memorial Sloan-Kettering Cancer Centre, the median overall survival (OS) of patients who received surgery with no residual disease was 31.9 months [42]. This is compared to just 5 months in those patients with inoperable disease.
For women with advanced or inoperable uLMS, the principles of management of other soft tissue sarcomas are followed. Chemotherapeutic agents with efficacy include doxorubicin, combination gemcitabine and docetaxel, or trabectedin [43] and to a lesser extent dacarbazine or eribulin [44]. Doxorubicin remains the reference first-line agent following the GeDDiS trial that showed similar efficacy and better tolerance than gemcitabine/docetaxel [45]. The combination of gemcitabine/docetaxel is favoured over single-agent gemcitabine following a study of 122 women who received either combination therapy or gemcitabine alone, with a doubling of progression-free survival (PFS) reported in the combination arm (6.0 vs 3.2 months in the combination arm and single-agent arm, respectively) [46]. Gemcitabine/docetaxel combinations have been trialled in fixed-dose single-arm studies as both a first-line and second-line therapy in advanced uLMS, with PFS reported as 4.4 months and 5.6 months, respectively [47,48]. Ifosfamide, whilst having an important role in the management of other soft tissue sarcomas, has a limited role in LMS due to modest single-agent activity combined with the logistics of administration and its toxicity profile [49,50]. Trabectidin has demonstrated modest efficacy in uLMS, with one trial of 134 unresectable, locally advanced uLMS cases that had received prior chemotherapy, reporting a higher PFS in the trabectedin arm (4.0 months vs 1.5 months for the dacarbazine arm) [51]. A trial of 20 chemotherapy-naïve women with uLMS reported a PFS of 5.8 months, with mean OS of 26.1 months, for trabectedin treatment alone [52]. Similar results were recently observed in a Spanish cohort of patients [53]. For trabectedin in combination with doxorubicin as a first-line therapy in advanced uLMS, 28 out of 47 women achieved partial response, with a further 13 achieving stable disease [54]. Due to the PFS advantage demonstrated in unselected soft tissue sarcomas in the PALETTE trial, pazopanib may also be provided as a second-line therapy although this was not targeted to a specific biomarker [55].
Hormonal therapies have also been explored in uLMS given the high frequency of ER/PR expression in tumours, albeit in small cohorts. In a study involving 16 patients with ER/PR-positive advanced uLMS (12 of which were chemotherapy naïve) treated with an aromatase inhibitor, the authors reported a mean PFS of 14 months [56]. In another small study of newly diagnosed uLMS patients, all four patients receiving letrozole were progression free at both 12 and 24 months, whilst in the observational arm the proportion of individuals progression free at 12 and 24 months had reduced to 80% and 40%, respectively [57]. It was not required that patients be chemotherapy naïve in this study, but the inclusion criteria stipulated a disease-free period of at least five years from any other cancer. In the phase II PARAGON trial of anastrozole in a cohort of 32 patients with ER-positive uLMS, the PFS was 2.8 months, with one patient achieving stable disease for five years [58].
Targeted therapy, in the form of poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi), has shown synthetic lethality in ovarian cancers bearing BRCA1/2 mutations (as will be discussed later). With respect to uLMS, Seligson and colleagues recently found that mutations in BRCA1/2 were enriched within the uterine subtype in a cohort of 170 LMS patients [59]. They and others have subsequently shown that such patients respond very well to PARPi [59,60]. Specifically, Seligson et al. reported durable stable disease in three out of four patients with BRCA2-mutated uLMS treated with olaparib, lasting for 15 months or more, and one partial response [59]. Hensley and colleagues also reported radiographic regression in five patients with BRCA2-mutated uLMS treated with PARPi, with one patient achieving a complete response [60].
Immunotherapies, such as the PD-1 blocking antibodies pembrolizumab and nivolumab, have been explored in relatively small cohorts over the last decade, as they have become increasingly popular in other solid tumours. PD-L1 expression, widely used as a biomarker to indicate immunotherapy response, has been reported as being positive in up to 70% of uLMS cases [61]. Two case studies involving patients with metastatic uLMS reported dramatic reductions in tumour burden and symptomatic disease with immune checkpoint inhibitor therapy [62,63], yet clinical trials on small patient cohorts have as yet failed to show any response to anti PD-1 therapy in uLMS [64,65]. However, in a basket study of doublet immunotherapy involving nivolumab and ipilimumab for four cycles followed by maintenance nivolumab [66], two patients had complete response and a third showed a partial response out of the five uLMS patients enrolled. This observation requires further investigation.
Fifty-six clinical trials are currently listed on the ClinicalTrial.org website testing various combinations of chemotherapy, hormonal therapy, immunotherapy, and targeted agents in uLMS. The few clinical trials involving targeted agents are listed in Table 1 although many of these do not appear to specifically study uLMS. Hence, in keeping with the fact that most targeted therapy trials including uLMS patients are relatively non-specific, meaningful advances in treatment are lacking, and the survival of women with advanced uLMS remains dismal. Whilst there does not appear to be a single driver of this lethal tumour type, there are a number of genomic aberrations frequently observed in uLMS that have been successfully targeted in other more common cancer types. These will be explored in this review.
4. Genomic Landscape
As uLMS is such a rare tumour type, molecular analysis has been limited. With so few samples collected at different sites throughout the world, large-scale analyses have often required input from multiple cohorts as shown in Table 2. To date, 23 and 158 uLMS tumours have undergone whole-genome sequencing (WGS) and whole-exome sequencing (WES), respectively, and been reported in the literature. An additional 434 uLMS samples have received a variation of exon capture, SNP array, or targeted panel sequencing. Collectively, the data show that TP53 is the most frequently mutated gene (frequency of 26–92%), followed by RB1 (27–88%), ATRX (24–34%), PTEN (19–75%), and MED12 (12–21%). Mutations in the homologous recombination (HR) pathway have also been reported in uLMS, with mutations in BRCA2 showing the highest frequency (7–60%).
uLMS tumours typically exhibit a tumour mutational burden (TMB) that is <10 mut/Mb, indicating they may not be strong candidates for immunotherapy given the findings from the recent KEYNOTE-158 study [83]. As with other trials, which defined high TMB as ≥10 mut/Mb [84,85], an objective response rate to pembrolizumab was observed in 29% of patients with a high TMB compared to just 6% of patients in the non-TMB-high group [83]. However, TMB is not indicative of response across all cancer types [86], and “TMB high” cut-offs vary widely [87]. Moreover, as mentioned previously, some success with immunotherapy in uLMS has been observed [62,63,66].
Large structural re-arrangements are common in uLMS. Choi and associates reported 21 cases of uLMS with complex structural rearrangements, 16 of which harboured chromoplexy or chromothripsis [78]. In their cohort of 10 uLMS cases, Chudsama et al. reported four had chromothripsis, one of which was a case that had received no treatment prior to analysis and yet had three affected chromosomes [81]. Structural rearrangements were also detected by Machado-Lopez et al., which resulted in gene fusions in 61.8% of cases, with ATRX the most common fusion partner [82].
The largest study to date on sarcomas, conducted by The Cancer Genome Atlas (TCGA) Program, compared uLMS to other sarcomas, confirming that uLMS is its own distinct tumour type even from its closest relative, soft-tissue LMS (ST-LMS). Compared to ST-LMS, uLMS had different methylation and mRNA expression signatures, with higher DNA-damage response scores and hypomethylation of ESR1 target genes [79]. ESR1 encodes the ER and therefore may explain why some uLMS patients are responsive to anti-endocrine therapies.
Below, we discuss the most frequently mutated genes in uLMS and the emerging therapies targeting these aberrations, which could be potential future treatment options for uLMS patients.
5. TP53
Given their high frequency in all cancers, it is unsurprising that TP53 mutations are the most common genomic aberration observed in uLMS. As with other tumour types [88], mutations in TP53 in uLMS are typically missense mutations occurring in the DNA-binding domain (DBD). In a study assessing the genomic landscape of the largest cohort of uLMS patients (n = 215), 51% of TP53 mutations were missense and found predominantly in the DBD [75]. Of the 40 TP53 mutations reported by Choi and associates in their assessment of the Yale and TCGA uLMS cohorts, 23 (57.5%) were missense, with all except six such mutations located in the DBD [78]. Many of these missense mutations result in changes at critical residues within the DBD reducing proper protein–DNA interactions. Much research focus has therefore been aimed at restoring the conformational changes to the mutant protein enabling activation of key downstream genes. An example of this is APR-246, a methylated analogue of proline-rich membrane anchor 1, or PRIMA-1, which is converted to methylene quinuclidinone (MQ) once in the cell [89]. MQ then binds cysteine residues 124 and 277, which reactivates the stable confirmation of mutant p53 and thus DNA-binding capability [90]. APR-246 has recently shown some promise in a phase Ib/II clinical trial, wherein patients with TP53-mutant myelodysplastic syndromes or acute myeloid leukaemia were treated with a combination of APR-246 (eprenetapopt) and azacytidine [91]. The overall response rate in the 55-patient cohort was 71%, with 44% of patients achieving complete response. Several other clinical trials of APR-246 are underway or have recently been completed: notably two trials combining APR-246 with carboplatin and pegylated doxorubicin in recurrent high grade serous ovarian cancer (HGSOC) (NCT02098343) or pegylated doxorubicin alone in platinum-resistant HGSOC (NCT03268382). Fransson and colleagues have previously demonstrated synergy between APR-246 and carboplatin, cisplatin, and doxorubicin in primary TP53-mutant HGSOC [92], and thus, the results of the aforementioned clinical trials are keenly awaited.
Another exciting avenue of therapeutically targeting mutant p53 is through the inhibition of WEE-1. In TP53-mutant cells the G1/S cell-cycle checkpoint is typically bypassed due to the absence of functional p53 protein; thus, cells become more reliant on the G2/M checkpoint [93]. Transition through this checkpoint and into mitosis is triggered by active cyclin dependent kinase 1 (CDK1). WEE-1 is a tyrosine kinase that inactivates CDK1 via phosphorylation, thereby halting the cell cycle at the G2/M checkpoint [94]. Inhibition of WEE-1 therefore removes the halt on mitosis, and cells already exhibiting a significant amount of DNA damage due to mutation of p53 incur further replication stress, resulting in mitotic catastrophe and cell death. It is thus predicted that just as PARPi show synthetic lethality in tumours bearing a BRCA1/2 mutation, WEE-1 inhibition will be synthetically lethal in p53-mutant cancers. This has certainly been demonstrated both preclinically and clinically. In 2011, Rajeshkumar and associates reported that 25/49 pancreatic xenograft tumours with mutant p53 showed a 50% reduction in initial tumour mass when the WEE-1 inhibitor (WEE-1i) MK-1775 was added to gemcitabine treatment compared to just 7/55 treated with gemcitabine alone [95]. Pancreatic xenograft tumours that were wild type for p53 did not respond to either gemcitabine alone or the combination with MK-1775. Similar findings were observed in pancreatic xenograft tumours treated with a combination of MK-1775 and irinotecan or capecitabine, where effective tumour reductions were only observed in the mutant p53 models [96]. Due to its ability to induce replication stress, MK-1775 has also been shown to increase the cytotoxic capabilities of standard therapeutics in preclinical cancer models of the breast [97,98], cervix [99], oesophagus [100], ovary [101], and even in models of sarcoma [102]. In a clinical trial involving 121 TP53-mutant platinum-sensitive ovarian cancer patients, the addition of adavosertib (MK-1775) to carboplatin and paclitaxel improved progression-free survival by 1.9 months compared to placebo plus the combination chemotherapy. A clinical trial combining adavosertib with carboplatin in TP53-mutated ovarian cancer showed that this WEE-1i had the ability to resensitise tumours that were previously platinum resistant [103]. There are currently 14 active and 13 completed clinical trials involving a WEE-1i on the NIH Clinical Trials website, indicating the significant interest in the potential of this molecule for treating p53-mutant cancers. It should be noted, however, that p53 status cannot always predict response to WEE-1 inhibition. In a panel of eight sarcoma cell lines, Kreahling and associates showed sensitivity to MK-1775 in the nanomolar range irrespective of p53 status [102]. They then went on to show significant synergy when MK-1775 was added to gemcitabine therapy in four sarcoma cell lines, U2O5 (osteosarcoma), MG63 (osteosarcoma), A673 (Ewing sarcoma), and HT-1080 (fibrosarcoma), despite two being p53 wild type (U2O5 and HT-1080) [104]. Similar findings have also been reported in ovarian cancer cell lines [105] and lung and colorectal cancer cell lines [106]. In a clinical trial of 35 patients receiving daily adavosertib, TP53-mutant tumours were observed in both the responder and non-responder groups, noting that some responders were wild type for p53 [107].
6. RB1
RB1, the gene that encodes retinoblastoma protein (Rb), is a tumour suppressor with critical roles in many cellular processes, the most well characterised being a cell cycle regulator. In the resting cell state, Rb is hypophosphorylated and binds to E2F factors, restricting their ability to activate the transcription of genes required for S-phase entry, effectively halting the cell cycle at G1 [108]. In response to mitotic cues, Cyclins C, D, and E and their associated CDKs phosphorylate Rb, releasing E2F and allowing progression through G1 into S phase [108]. Often the second most commonly affected gene after TP53 in uLMS, RB1 is prone to homozygous deletion [75,77,78]. Deletion or inactivating events are classically hard to target therapeutically; however, again, synthetic lethality may be the key. Aurora kinase inhibitors have recently been shown to have selective sensitivity in Rb-null cell lines compared to Rb wild-type cells [109,110]. Aurora kinase A is involved in centrosome formation and mitotic spindle assembly, whilst Aurora kinase B assists with coordinating sister-chromatid cohesion during metaphase [111]. As Rb has also been proposed to contribute to mitotic fidelity through its association with E2F and E2F-independent mechanisms [112], the loss of Rb and the Aurora kinases A and B is thought to be too catastrophic for the cell. Clinical trials of Aurora kinase A inhibitor MLN8237 (Alisertib) have shown promise in ovarian cancer [113] as well as breast cancer, lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma [114]. Following encouraging preclinical evidence of sensitivity to MLN8237 in the vulval LMS cell line SK-LMS-1 both in vitro and in vivo [115], a phase II clinical trial of Alisertib as a monotherapy was conducted in recurrent uLMS patients [116]. Of the 23 patients enrolled there, just one patient was progression-free at six months, with no partial or complete responses recorded. The authors also reported significant toxicity. In another multi-centre phase I/II trial of Alisertib as a single agent, 10 LMS (non-uterine) patients were included [117]. PFS at 12 weeks was 44% with OS reported as 72 weeks for this cohort. The exact molecular profile of these LMS tumours from the aforementioned trials were not disclosed, so patient responses were not stratified according to Rb status. In addition, both trials tested Alisertib as a single-agent. Aurora kinase inhibitors in combination with other agents are yet to be explored clinically in uLMS.
Targeting the proteins that are up-regulated in response to Rb loss may be another mechanism of effective therapeutic control. As discussed, Rb is responsible for binding to and restricting the function of E2Fs. A pan-E2F inhibitor, HLM006474, was developed in 2008 showing efficacy in melanoma and triple-negative breast cancer cell lines [118]. More recently, HLM006474 has been shown to be effective at reducing cell viability in small-cell and non-small-cell lung cancer and synergises with paclitaxel [119]. Additionally, HLM006474 has been shown to inhibit growth of melanoma cells lines and synergises with BRAF-inhibitors [120]. Interestingly, the inhibition of tumour cell growth in this last study was observed only in p53 wild-type cells. This may predict reduced effectiveness of HLM006474 in uLMS, as TP53 and RB1 are frequently co-mutated, and may suggest why no clinical trials for HLM006474 have been initiated.
7. ATRX and Alternative Lengthening of Telomeres (ALT)
Alpha-thalassemia/mental retardation syndrome X-linked (ATRX) is a chromatin-remodelling factor that exists in a complex with death domain-associated protein (DAXX) and is essential for incorporating Histone 3.3 (H3.3) into telomeres [121,122,123,124]. Loss of ATRX or DAXX results in telomere instability and alternative lengthening of telomeres (ALT) [123]. Whilst frequent loss of ATRX has been observed in uLMS (39–52%), mutations in DAXX have been observed but are very rare (0–2%) [80,125]. Many aggressive tumours, such as glioblastoma multiforme [126] and diffuse intrinsic pontine gliomas [127], as well as those of mesenchymal origin [128], such as uLMS, display features of ALT. Indeed, ALT is the predominant telomere maintenance mechanism found in uLMS, with up to 71% of cases being ALT positive (ALT+) [80,125].
In normal cells, telomere maintenance mechanisms are constrained to prevent the formation of cancer. These mechanisms are either disrupted during the process of tumorigenesis or hijacked by cancer cells to enable them to avoid replicative senescence and acquire immortality [129]. Telomere maintenance in most cancer cells occurs due to reactivation of telomerase [130]. However, in 10–15% of cancers, some having a particularly poor prognosis [131,132], telomerase is suppressed, and telomere length is maintained through the ALT mechanism [133]. Cells that possess ALT extend their telomeres by copying other telomeres or extrachromosomal TTAGGG DNA fragments derived from telomeres through HR [134]. ALT telomeres display elevated replicative stress and DNA damage most likely associated with telomeric chromatin changes occurring as a result of mutations in genes that encode H3.3, ATRX, or DAXX [126,135,136]. It has been proposed that when the function of ATRX is lost, DAXX can no longer direct H3.3 to telomeres, leading to the formation of G-quadruplex structures that can give rise to replication stress [137,138]. HR at telomeres then occurs through the MRE11-RAD50-NBS1 complex, leading to ALT [138,139].
Almost all ATRX-mutant tumours are ALT+, but ATRX is not mutated in ~50% of ALT+ tumours, indicating alternative mechanisms driving this phenotype [125]. During DNA recombination, protein kinase ATR is recruited to telomeres by replication protein A, a protein persistently associated with telomeres in ALT+ cells. Therefore, it is hypothesised that ALT+ cancers may display sensitivity to ATR inhibitors. Studies investigating the use of ATR inhibitors (ATRi) to treat ALT+ cancers have produced conflicting results [140,141], so this warrants further assessment but may be a potential avenue to explore for the treatment of uLMS perhaps in combination with other relevant DNA-repair inhibitor therapies. The specialised DNA recombination thought to occur in ALT+ cells may also render them sensitive to inhibitors of DNA synthesis, such as PCNA and BLM inhibitors [142,143]. Furthermore, Liang et al. carried out a CRISPR screen in ATRX-knockout cells and identified components of the cell-cycle checkpoint as being important for cell survival. In particular, inhibition of WEE-1, which as discussed earlier is an important regulator of the G1/S and G2/M cell-cycle checkpoints, was found to be synthetically lethal in ATRX-deficient cells [144,145]. Not only does the presence of TP53 mutations but also the presence of ATRX mutations suggests a subset of patients may benefit from WEE-1i. These have not yet specifically been tested in uLMS although women with uLMS should be encouraged to take part in basket trials of WEE-1i, such as NCT04158336 or NCT0476886.
ATRX is also involved in transcriptional regulation but when recruited to promyelocytic leukaemia nuclear bodies by DAXX, this regulation of gene expression is inhibited [146]. Therefore, ATRX and DAXX function in a wide range of cellular processes both independently as chromatin remodellers as well as in complexes with H3.3 or PML. For example, ATRX associates with H3K9me at repetitive regions (other than telomeres), and reduced H3K9me levels as a result of loss of functional ATRX can give rise to stalled replication forks and genomic instability [139]. This may lead to activation of PARP and ATM, suggesting that ATRX-mutant cells may be responsive to PARPi [147]. ATRX has also been found to regulate the expression of Polycomb responsive genes through interacting with EZH2, indicating that ATRX-mutant cells may be sensitive to EZH2 inhibition [139,148,149].
8. PTEN
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a tumour suppressor whose expression is often lost in tumours [150,151,152]. PTEN is lost or deleted in about 66% of all uterine cancers and in 19% of uLMS specifically [75,153]. As a lipid phosphatase PTEN dephosphorylates phosphatidylinositol-3,4,5-phosphate (PIP3), a critical second messenger in the phosphatidylinositol-3-kinase (PI3K)/AKT signalling pathway [154]. PI3K phosphorylates PIP2 to give rise to PIP3, which is required to recruit AKT to the plasma membrane where it can be phosphorylated and activated by two kinases, PDK1 and mTORC2 [155]. Therefore, PTEN inhibits the PI3K pathway through converting PIP3 back to PIP2, thus regulating cell growth and survival. PTEN can also function as a protein phosphatase, where it has been found to dephosphorylate various proteins involved in cell proliferation and migration, including Cyclin D1, focal adhesion kinase (FAK), and Shc [156,157,158].
Apart from its phosphatase functions, PTEN also has phosphatase-independent functions in the nucleus, where it can regulate chromosome stability (through interacting with various chromosome factors, such as Centromere-Specific Binding Protein and anaphase-promoting complex or cyclosome), DNA repair (through directly interacting with p53, as well as regulating the expression of RAD51), and apoptosis (reviewed in [159,160]). Therefore, PTEN inhibits tumour development through multiple mechanisms and exploiting PTEN loss is the aim of many therapeutic strategies.
PTEN function is not only lost through mutation or genomic deletion of the PTEN gene but also through epigenetic and transcriptional silencing, post-transcriptional and post-translational regulation, and protein–protein interactions [161]. Therefore, the proportion of uLMS harbouring loss-of-function of PTEN could potentially be higher than 19%. Nevertheless, as previously described for loss of p53 and Rb, there are many challenges in therapeutically targeting loss of a protein. The PI3K/AKT/mTOR pathway is one obvious target in PTEN-deficient tumours. Cell lines with PTEN genetic alterations have displayed sensitivity to PI3K inhibitors (AZD6482), AKT inhibitors (MK-2206), and mTORC1 inhibitors (Temsirolimus) [162]. On the other hand, these cells are generally resistant to inhibitors of upstream mediators of PI3K pathway activity, such as receptor tyrosine kinases [162]. Many PI3K/AKT/mTOR inhibitors have been investigated in clinical trials for the treatment of PTEN-deficient tumours with mixed results depending on the molecular background (reviewed in [163,164]). Overall, mTORC1 inhibitors appear to have the greatest efficacy in these trials [163]. For example, mTOR inhibitors have demonstrated some efficacy in clinical trials in STS patients (no uLMS included in these trials) [165,166]. Combination of PI3Ki and the new TORC1/TORC2 inhibitors will likely have greater efficacy given that elevated PI3K/AKT signalling is a known feature of uLMS (due to PTEN loss and up-regulation of other genes in the pathway, such as PIK3CA, AKT1/2/3, and RICTOR) [79].
The interaction between PTEN and FAK may also be exploited therapeutically. Preclinical studies have indicated that PTEN-deficient models of uterine cancer were sensitive to the FAK inhibitor GSK2256098 in vitro and in vivo [167]. The link between PTEN and FAK has been studied in multiple different tumour types, and although many PTEN-deficient tumours display FAK activation, this is not always the case [164]. Whether this correlation is present in uLMS and therefore FAK inhibitors should be considered and needs to be explored. Furthermore, combining FAK and PI3K inhibitors may have greater efficacy, and preclinical studies involving this combination are also worth considering [168]. FAK is also a substrate of Src kinase and along with other Src substrates, such as c-KIT, EGFR, and PDGFR, has been found to be (over)expressed in uLMS [169,170]. Interestingly, the Src inhibitor dasatanib has been found to synergise with gemcitabine and docetaxel independently in uLMS cell lines [171].
Loss of nuclear PTEN function can also be exploited therapeutically. The role of PTEN in the DNA repair process of HR, through regulating RAD51 expression, may render deficient tumours sensitive to PARPi [172]. PARPi have previously demonstrated efficacy in PTEN-deficient endometrial cancers and warrants investigation in uLMS [173,174]. As has been mentioned previously and will become more apparent later in this review, many common aberrations in uLMS may indicate PARPi sensitivity, and this class of drug warrants further investigation in uLMS.
9. MED12
The MED12 gene on Xq13.1 encodes Mediator complex subunit 12 (MED12). The Mediator complex is composed of 25–30 proteins and plays a role in both activating and repressing gene transcription through mediating interactions between RNA Polymerase II (Pol II) at gene promoters and transcription factors at specific enhancers [175,176,177]. MED12 is a subunit of the Kinase module of Mediator, which consists of four proteins (CDK8, Cyclin C, MED12, and MED13) and is required for the kinase activity of the module [178,179]. Up to 86% of uterine leiomyomas have a mutation in MED12 [180]; however, only about 20% of uLMS harbour a mutation in this gene [75,80,181,182], indicating alternative origins for this rare tumour. Most mutations occur in exons 1 and 2, the region required for Cyclin C interaction; however, mutations can also occur in other regions of the protein where they may result in “gain-of-function” of MED12, driving genomic instability [183].
Due to its role in essential developmental and cell fate determination processes, it is not surprising that MED12 deregulation can lead to cancer. Various molecular pathways relevant to tumour development, such as TGF-β signalling [184], Wnt signalling [185], and the p53 network [186], are affected in MED12-mutant cells. Importantly, MED12 inhibition has been found to confer resistance to a number of anti-cancer drugs in the context of specific mutations that are normally targetable [187,188]. Specifically, MEK/ERK activation has been found to remain high following treatment with ALK or EGFR inhibitors when MED12 is lost [189]. A similar effect has been seen in response to BRAF and MEK inhibitors, implicating MED12 in RAS-MEK-ERK signalling [184]. Resistance to the chemotherapeutic agents cisplatin and 5-FU has also been observed [184]. Further investigation found that cytoplasmic MED12 is able to interact with intracellular TGF-βR2, preventing it from being expressed on the cell surface. This leads to increased TGF-β signalling in MED12-mutant cells, resulting in activation of MEK and ERK (even in the presence of specific inhibitors), induction of EMT, and resistance to chemotherapeutic agents [184]. Small-molecule drugs that inhibit TGF-β signalling have been found to overcome drug resistance in MED12-deficient cells [184], indicating TGF-β inhibitors may be viable treatment options for uLMS patients harbouring mutations in MED12. On the other hand, methylation of MED12 has been found to render breast cancer cells sensitive to chemotherapeutic agents, indicating that MED12 has cancer type-specific functions [189]. Whether MED12-mutation confers drug resistance in uLMS that can be overcome by combination treatment strategies involving TGF-β inhibitors requires further investigation.
The action of MED12 in TGF-βR2 regulation is independent of its role in the Kinase module of Mediator. In the Kinase module, MED12 is able to stimulate kinase activity through its interaction with Cyclin C [190]. Recently, it was found that loss of Cyclin C conferred resistance to the ATRi ceralasertib in mouse embryonic cells [191]. Whether this finding will translate to tumour cells is yet to be investigated; however, it suggests that using ATRi to target ALT in uLMS, as proposed earlier, may not be effective in tumours that also harbour MED12 mutations. This is because most of the mutations in MED12 cluster in Exon 1 and 2 [190], the region required for Cyclin C binding [75,80,181,182], and so may result in a similar lack of response to ATRi. Finally, Mediator and the Bromodomain-containing protein 4 (BRD4) have been found to have similar epigenetic functions at super-enhancers in acute myeloid leukaemia (AML) cells [192,193]. Although it is unknown whether BRD4 functionally compensates for loss of MED12, inhibition of BRD4 may also be a treatment strategy for uLMS harbouring a MED12 mutation.
It is intriguing that MED12 mutations are more frequent in uterine leiomyoma (up to 86%) than in uLMS (up to 22%). Furthermore, as MED12 is located on the X-chromosome, the mutation could lie on the inactive X chromosome and thus be irrelevant. Both of these factors suggest MED12 mutations may not function as driver mutations in uLMS, and so, therapeutics that are effective in the context of MED12-deficiency need to be carefully investigated.
10. Homologous Recombination Deficiency
HR and classic non-homologous end joining are the primary repair pathways of DNA double-stranded breaks (reviewed by [194]). Of the two, HR is less error-prone, as it requires the template of a sister chromatid upon which accurate repair can occur. HR deficiency (HRD) is therefore the loss of this high-fidelity system of repair and leads to increases in mutation events that lead to loss of heterozygosity (LOH) and widespread genomic instability. Many proteins are involved in the HR process, including BRCA1, BRCA2, RAD51, and PALB2. Early studies reported modest frequencies of mutations in the HR pathway in uLMS tumours, particularly in BRCA2, but interest in the contribution of HR mutations in this cancer type has recently come under the spotlight due to promising clinical data for drugs targeting these aberrations as mentioned earlier [59,60]. As described above, Seligson and colleagues found that mutations in BRCA1/2 were enriched within the uterine subtype in a cohort of 170 LMS patients [59]. Rosenbaum et al. supported this work in 2020, reporting a frequency of mutations in BRCA2 of 9% in uLMS (n = 121) compared to 2% in ST-LMS (n = 90) [76]. Responsiveness to PARPi has been demonstrated preclinically in uLMS, with Choi et al. showing a favourable response to olaparib in a patient-derived xenograft of HRD uLMS [78]. HRD tumours are also typically sensitive to platinum therapy. Whilst cisplatin was trialled as a therapy for uLMS in the 1980s and 1990s, the findings did not indicate any patient benefit [195,196]. However, these studies were performed on small numbers of women and did not screen uLMS for mutations in the HR pathway; therefore, the full utility of cisplatin in HRD uLMS patients has not yet been thoroughly explored.
Current methods for screening for mutations in the HR pathway in solid tumours involve panel tests, such as the QIAseq Targeted DNA BRCA1 and BRCA2 Panel and FoundationOne Panel, with more challenging options such as WES or WGS typically reserved for more complex or advanced cases in the research setting. Given that BRCA1/2 mutations (predominantly BRCA2) only account for ~10% (but can be as frequent as 60% [81]) of uLMS cases, panel tests appear to be a reasonable alternative. However, hallmarks of genomic instability, such as LOH and telomeric allelic imbalance, are more reliably detected through WGS. The Myriad myChoice CDx test aims to cover these two aspects by calculating a genomic instability score based on the aforementioned measures of genomic instability as well as large-scale state transitions and mutations in BRCA1 and BRCA2; however, at USD 4000, this test may not be much less expensive than WGS but may be more easily available.
11. Other Genomic Characteristics
Epigenomic data on 27 uLMS cases published by TCGA indicated that a unique methylation pattern was observed despite hypomethylation of ESR1 target genes. Overall, uLMS appeared to be hypermethylated compared to ST-LMS [79]. This suggests uLMS may be sensitive to epigenetic modulators, such as DNA Methyltransferase inhibitors (DNMTi). In a preclinical study, the uLMS cell line SK-UT-1 was found to be sensitive to the DNMTi guadecitabine in vitro and in vivo. In vitro efficacy appeared to be correlated with methylation status, as the comparatively hypomethylated cell line SK-LMS-1, which interestingly is a non-uterine LMS cell line, was less responsive [197]. SHARPIN, a protein coding gene involved in TNF signalling, was recently shown to be amplified in uLMS via WES [198]. Investigations of SHARPIN mutations in the TCGA dataset revealed amplification of SHARPIN leads to decreased overall survival. Knockdown of SHARPIN in uLMS cell lines lead to decreased cell proliferation and colony-forming ability [198].
12. Preclinical Models of uLMS
In this review, we detail the frequent genomic aberrations observed in uLMS and the possible targeted agents that are worth investigating further in the context of uLMS (Figure 1, Table 3). In line with this, the field requires well-characterised and validated models of uLMS that represent the heterogeneity observed in patients. The immortalised cell lines SK-UT-1 and SK-UT-1B have been well annotated [81] and are utilized often in uLMS research. In 2008, Press and colleagues reported the generation of a uLMS PDX that reached three generations [199]. Ten uLMS patient-derived xenograft (PDX) models were successfully generated by Cuppens and associates more recently [200], five of which were used in a subsequent study to show the benefit of inhibiting mTOR and PI3K signalling in uLMS (4/5 PDX models showed reduction/stabilisation of tumour growth) [201]. Two uLMS PDX models reported recently with mutations in the HR pathway were treated with single-agent olaparib, pan-PI3K inhibitor, or a BET bromodomain inhibitor, with all three therapeutics reducing tumour growth [78]. The HR status of the other PDX models reported by Cuppens or Press was not included.
Genetically-engineered mouse models (GEMMs) of uLMS have also been attempted. Conditional inactivation of p53 in the mouse reproductive tract through expression of Cre recombinase under the control of the anti-Mullerian hormone type II receptor (Amhr2) gave rise to uLMS in about 50% of mice, with frequency increasing to 82% when BRCA1 was also inactivated [203]. Transgenic mice expressing a growth factor called Cripto-1 (CR-1) under control of the MMTV promoter to activate the Wnt pathway also activated the Src/AKT pathway and gave rise to uLMS in 20% of mice [204]. uLMS GEMMs have also been generated by crossing mice with the T antigens of the SV40 region (SVER) transgene (which encodes the three T antigens of SV40–SV Large T (SVLT), small t, and 17kT) targeted to the mouse Beta-actin locus with mice containing a Cre transgene downstream of the Hsp70-1 heat-shock promoter [205]. Despite almost ubiquitous expression of SVLT, tumour development was restricted to the uterus (with tumours showing uLMS morphology) in female pups and seminal vesicles in male pups. The potential of these GEMMs to drive forward our understanding of uLMS is clear; however, recent advances in this space are lacking, and treatment studies have to date not been reported.
13. Conclusions and Future Perspectives
The high rate of genetic aberrations in uLMS and the increasing advances in targeted therapies provide many possibilities for the future treatment of uLMS. PARPi are one of the most significant advances in the uLMS space, with striking favourable responses akin to what is observed in other HRD gynaecological malignancies. In addition, despite HRD only accounting for a small proportion of cases, PARPi may also be beneficial in HR proficient but PTEN and/or ATRX deficient tumours as discussed above, thus potentially providing benefit to a greater subset of patients. Similarly, PI3K, mTORC1/2, and WEE-1 inhibitors are likely to impact a larger subset of women with uLMS, as they target a number of specific aberrations commonly observed in uLMS and may also provide an opportunity for advantageous combinatorial therapy with PARPi.
As observed for other cancer types, it is unlikely that one therapeutic approach will be of use for all women with uLMS. The large variability in mutation frequencies indicates that a personalised approach may be more beneficial. Whilst this review explores the different mutations and the therapies that can target them in isolation, we have not yet accounted for the complex interactions between concurrent mutations or the ripple effects in downstream signalling pathways that arise as a result of small molecule inhibition. It should also be noted that the molecular profiling of uLMS is still in its infancy compared with other tumour types. With less than 200 reported cases of WGS and WES, researchers have not yet been able to perform the deep sequencing on large data sets that is likely required to understand this clinically elusive, rare tumour type and the molecular subsets within uLMS. In light of this, clinicians should consider WGS or WES in their diagnostic approach to management of women with uLMS not only to discover the potentially actionable mutations described in this review but also to contribute to the global effort in uncovering the multifaceted biology that underpins uLMS.
Author Contributions
Conceptualization and design G.V.D. and H.B.; writing—original draft preparation, G.V.D., A.H., G.R., C.S., H.B.; writing—review and editing, G.V.D., A.H., G.R., C.S., H.B. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are supported by fellowships and grants from the Stafford Fox Medical Research Foundation (G.V.D., C.S. and H.B.); Cancer Council Victoria (Sir Edward Dunlop Fellowship in Cancer Research to C.S. and Ovarian Cancer Research Grant-in-Aid 1186314 to H.B.); the Victorian Cancer Agency (Clinical Fellowships to C.L.S.; CRF10-20, CRF16014, Early Career Fellowship G.V.D.) and Australian and New Zealand Gynaecological Oncology Group Fund For New Research Grant (G.V.D.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
C.L.S. reports membership in advisory boards for AstraZeneca, Clovis Oncology, Roche, Eisai, Sierra Oncology, Takeda, and Merck Sharp & Dohme (no honoraria were received for any advisory boards); grant/research support from Clovis Oncology, Eisai, Sierra Oncology, Roche, BeiGene, AstraZeneca, drug in kind support from Clovis Oncology, Eisai, Sierra Oncology, Roche, and BeiGene; leadership or fiduciary roles with Cancer Trials Australia, the Australia New Zealand Gynaecological Oncology Group, the Cure Cancer Australia Foundation, BioGrid Australia, and OncologyOne; and travel support from AstraZeneca. The other authors made no disclosures.
References
- D’Angelo, E.; Prat, J. Uterine sarcomas: A review. Gynecol. Oncol. 2010, 116, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Skorstad, M.; Kent, A.; Lieng, M. Uterine leiomyosarcoma—Incidence, treatment, and the impact of morcellation. A nationwide cohort study. Acta Obstet. Gynecol. Scand. 2016, 95, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Toro, J.R.; Travis, L.B.; Wu, H.J.; Zhu, K.; Fletcher, C.D.M.; Devesa, S.S. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978–2001: An analysis of 26,758 cases. Int. J. Cancer 2006, 119, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Farid, M.; Ong, W.S.; Tan, M.H.; Foo, L.S.; Lim, Y.K.; Chia, W.K.; Soh, L.T.; Poon, D.; Lee, M.J.; Ho, Z.C.; et al. The influence of primary site on outcomes in leiomyosarcoma: A review of clinicopathologic differences between uterine and extrauterine disease. Am. J. Clin. Oncol. 2013, 36, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Hosh, M.; Antar, S.; Nazzal, A.; Warda, M.; Gibreel, A.; Refky, B. Uterine Sarcoma: Analysis of 13,089 Cases Based on Surveillance, Epidemiology, and End Results Database. Int. J. Gynecol. Cancer 2016, 26, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Kapp, D.S.; Shin, J.Y.; Chan, J.K. Prognostic factors and survival in 1396 patients with uterine leiomyosarcomas: Emphasis on impact of lymphadenectomy and oophorectomy. Cancer 2008, 112, 820–830. [Google Scholar] [CrossRef]
- Ricci, S.; Stone, R.L.; Fader, A.N. Uterine leiomyosarcoma: Epidemiology, contemporary treatment strategies and the impact of uterine morcellation. Gynecol. Oncol. 2017, 145, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Tirumani, S.H.; Deaver, P.; Shinagare, A.B.; Tirumani, H.; Hornick, J.L.; George, S.; Ramaiya, N.H. Metastatic pattern of uterine leiomyosarcoma: Retrospective analysis of the predictors and outcome in 113 patients. J. Gynecol. Oncol. 2014, 25, 306–312. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, E.; Espinosa, I.; Ali, R.; Gilks, C.B.; Rijn, M.v.d.; Lee, C.-H.; Prat, J. Uterine leiomyosarcomas: Tumor size, mitotic index, and biomarkers Ki67, and Bcl-2 identify two groups with different prognosis. Gynecol. Oncol. 2011, 121, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Seagle, B.L.; Sobecki-Rausch, J.; Strohl, A.E.; Shilpi, A.; Grace, A.; Shahabi, S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecol. Oncol. 2017, 145, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Costales, A.B.; Radeva, M.; Ricci, S. Characterizing the efficacy and trends of adjuvant therapy versus observation in women with early stage (uterine confined) leiomyosarcoma: A National Cancer Database study. J. Gynecol. Oncol. 2020, 31, e21. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Kleinerman, R.A.; Seddon, J.M.; Abramson, D.H. Increased risk of secondary uterine leiomyosarcoma in hereditary retinoblastoma. Gynecol. Oncol. 2012, 124, 254–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, E.G.; Tornos, C.S.; Follen-Mitchell, M. Malignant neoplasms of the uterine corpus in patients treated for breast carcinoma: The effects of tamoxifen. Int. J. Gynecol. Pathol. 1994, 13, 248–258. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G.; Varma, M.; Weir, P.; Bharucha, H. Uterine leiomyosarcoma in patient receiving tamoxifen therapy. Acta Obs. Gynecol. Scand. 1996, 75, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.E.; Zhan, M.; Cote, T.; Baquet, C.R. Surveillance, epidemiology, and end results analysis of 2677 cases of uterine sarcoma 1989-1999. Gynecol. Oncol. 2004, 93, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Bodner, K.; Bodner-Adler, B.; Kimberger, O.; Czerwenka, K.; Leodolter, S.; Mayerhofer, K. Estrogen and progesterone receptor expression in patients with uterine leiomyosarcoma and correlation with different clinicopathological parameters. Anticancer. Res. 2003, 23, 729–732. [Google Scholar] [PubMed]
- Zhai, Y.L.; Kobayashi, Y.; Mori, A.; Orii, A.; Nikaido, T.; Konishi, I.; Fujii, S. Expression of steroid receptors, Ki-67, and p53 in uterine leiomyosarcomas. Int. J. Gynecol. Pathol. 1999, 18, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Leitao, M.M., Jr.; Hensley, M.L.; Barakat, R.R.; Aghajanian, C.; Gardner, G.J.; Jewell, E.L.; O’Cearbhaill, R.; Soslow, R.A. Immunohistochemical expression of estrogen and progesterone receptors and outcomes in patients with newly diagnosed uterine leiomyosarcoma. Gynecol. Oncol. 2012, 124, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Leitao, M.M.; Soslow, R.A.; Nonaka, D.; Olshen, A.B.; Aghajanian, C.; Sabbatini, P.; Dupont, J.; Hensley, M.; Sonoda, Y.; Barakat, R.R.; et al. Tissue microarray immunohistochemical expression of estrogen, progesterone, and androgen receptors in uterine leiomyomata and leiomyosarcoma. Cancer 2004, 101, 1455–1462. [Google Scholar] [CrossRef]
- Baek, M.H.; Park, J.Y.; Park, Y.; Kim, K.R.; Kim, D.Y.; Suh, D.S.; Kim, J.H.; Kim, Y.M.; Kim, Y.T.; Nam, J.H. Androgen receptor as a prognostic biomarker and therapeutic target in uterine leiomyosarcoma. J. Gynecol. Oncol. 2018, 29, e30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, T.W.; Borden, E.C.; Goldblum, J.R. Estrogen and progesterone receptor expression in uterine and extrauterine leiomyosarcomas: An immunohistochemical study. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Toledo, G.; Oliva, E. Smooth Muscle Tumors of the Uterus: A Practical Approach. Arch. Pathol. Lab. Med. 2008, 132, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Parra-Herran, C.; Schoolmeester, J.K.; Yuan, L.; Dal Cin, P.; Fletcher, C.D.; Quade, B.J.; Nucci, M.R. Myxoid Leiomyosarcoma of the Uterus: A Clinicopathologic Analysis of 30 Cases and Review of the Literature with Reappraisal of Its Distinction From Other Uterine Myxoid Mesenchymal Neoplasms. Am. J. Surg. Pathol. 2016, 40, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Aynardi, J.T.; Chu, C.S. Uterine leiomyosarcoma: A review of the literature and update on management options. Gynecol. Oncol. 2018, 151, 562–572. [Google Scholar] [CrossRef]
- O’Neill, C.J.; McBride, H.A.; Connolly, L.E.; McCluggage, W.G. Uterine leiomyosarcomas are characterized by high p16, p53 and MIB1 expression in comparison with usual leiomyomas, leiomyoma variants and smooth muscle tumours of uncertain malignant potential. Histopathology 2007, 50, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Oliva, E.; Young, R.H.; Amin, M.B.; Clement, P.B. An immunohistochemical analysis of endometrial stromal and smooth muscle tumors of the uterus: A study of 54 cases emphasizing the importance of using a panel because of overlap in immunoreactivity for individual antibodies. Am. J. Surg. Pathol. 2002, 26, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Lusby, K.; Savannah, K.B.; Demicco, E.G.; Zhang, Y.; Ghadimi, M.P.; Young, E.D.; Colombo, C.; Lam, R.; Dogan, T.E.; Hornick, J.L.; et al. Uterine leiomyosarcoma management, outcome, and associated molecular biomarkers: A single institution’s experience. Ann. Surg. Oncol. 2013, 20, 2364–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, B. Immunohistochemical analysis of p16, p53, and Ki-67 expression in uterine smooth muscle tumors. Int. J. Gynecol. Pathol. 2008, 27, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Matsuo, K.; Duncan, K.; Pakzamir, E.; Pham, H.Q.; Correa, A.; Fedenko, A.; Mhawech-Fauceglia, P. Immunohistochemical panel to differentiate endometrial stromal sarcoma, uterine leiomyosarcoma and leiomyoma: Something old and something new. J. Clin. Pathol. 2015, 68, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Subbaraya, S.; Murthy, S.S.; Devi, G.S. Immunohistochemical and Molecular Characterization of Endometrial Stromal Sarcomas. Clin. Pathol. 2020, 13, 2632010x20916736. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.; Soslow, R.A.; Delair, D.F.; Park, K.J.; Murali, R.; Hollmann, T.J.; Davidson, B.; Micci, F.; Panagopoulos, I.; Hoang, L.N.; et al. ZC3H7B-BCOR high-grade endometrial stromal sarcomas: A report of 17 cases of a newly defined entity. Mod. Pathol. 2018, 31, 674–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrzenjak, A. JAZF1/SUZ12 gene fusion in endometrial stromal sarcomas. Orphanet J. Rare Dis. 2016, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.K.; Jung, J.H.; Lee, A.; Lee, Y.S.; Choi, Y.J.; Yoon, S.K.; Lee, K.Y. Diagnostic use of nuclear beta-catenin expression for the assessment of endometrial stromal tumors. Mod. Pathol. 2008, 21, 756–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Shi, Y.; Zhong, J.; Zhao, M.; Wu, J.; Hai, L.; Xu, X.; Du, H.; Shi, Y. Histopathologic characteristics and immunotypes of perivascular epithelioid cell tumors (PEComa). Int. J. Clin. Exp. Pathol. 2019, 12, 4380–4389. [Google Scholar] [PubMed]
- Bennett, J.A.; Braga, A.C.; Pinto, A.; Van de Vijver, K.; Cornejo, K.; Pesci, A.; Zhang, L.; Morales-Oyarvide, V.; Kiyokawa, T.; Zannoni, G.F.; et al. Uterine PEComas: A Morphologic, Immunohistochemical, and Molecular Analysis of 32 Tumors. Am. J. Surg. Pathol. 2018, 42, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Mandato, V.D.; Valli, R.; Mastrofilippo, V.; Bisagni, A.; Aguzzoli, L.; La Sala, G.B. Uterine inflammatory myofibroblastic tumor: More common than expected: Case report and review. Medicine 2017, 96, e8974. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, N.; Haimes, J.D.; Mishkin, S.; Kudlow, B.A.; Leong, M.Y.; Chew, S.H.; Koay, E.; Whitehouse, A.; Cope, N.; Ali, R.H.; et al. ALK Is a Specific Diagnostic Marker for Inflammatory Myofibroblastic Tumor of the Uterus. Am. J. Surg. Pathol. 2018, 42, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. Female Genital Tumours, 5th ed.; International Agency for Research on Cancer, World Health Organization: Lyon, France, 2020. [Google Scholar]
- Bretthauer, M.; Goderstad, J.M.; Løberg, M.; Emilsson, L.; Ye, W.; Adami, H.O.; Kalager, M. Uterine morcellation and survival in uterine sarcomas. Eur. J. Cancer 2018, 101, 62–68. [Google Scholar] [CrossRef]
- Rizzo, A.; Nannini, M.; Astolfi, A.; Indio, V.; De Iaco, P.; Perrone, A.M.; De Leo, A.; Incorvaia, L.; Di Scioscio, V.; Pantaleo, M.A. Impact of Chemotherapy in the Adjuvant Setting of Early Stage Uterine Leiomyosarcoma: A Systematic Review and Updated Meta-Analysis. Cancers 2020, 12, 1899. [Google Scholar] [CrossRef]
- Leitao, M.M., Jr.; Zivanovic, O.; Chi, D.S.; Hensley, M.L.; O’Cearbhaill, R.; Soslow, R.A.; Barakat, R.R. Surgical cytoreduction in patients with metastatic uterine leiomyosarcoma at the time of initial diagnosis. Gynecol. Oncol. 2012, 125, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Le Cesne, A.; Blay, J.Y.; Cupissol, D.; Italiano, A.; Delcambre, C.; Penel, N.; Isambert, N.; Chevreau, C.; Bompas, E.; Bertucci, F.; et al. A randomized phase III trial comparing trabectedin to best supportive care in patients with pre-treated soft tissue sarcoma: T-SAR, a French Sarcoma Group trial. Ann. Oncol. 2021, 32, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Schöffski, P.; Bauer, S.; Krarup-Hansen, A.; Benson, C.; D’Adamo, D.R.; Jia, Y.; Maki, R.G. Eribulin versus dacarbazine in patients with leiomyosarcoma: Subgroup analysis from a phase 3, open-label, randomised study. Br. J. Cancer 2019, 120, 1026–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, B.; Strauss, S.J.; Whelan, J.; Leahy, M.; Woll, P.J.; Cowie, F.; Rothermundt, C.; Wood, Z.; Benson, C.; Ali, N.; et al. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): A randomised controlled phase 3 trial. Lancet Oncol. 2017, 18, 1397–1410. [Google Scholar] [CrossRef] [Green Version]
- Maki, R.G.; Wathen, J.K.; Patel, S.R.; Priebat, D.A.; Okuno, S.H.; Samuels, B.; Fanucchi, M.; Harmon, D.C.; Schuetze, S.M.; Reinke, D.; et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: Results of sarcoma alliance for research through collaboration study 002 [corrected]. J. Clin. Oncol. 2007, 25, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Hensley, M.L.; Blessing, J.A.; Degeest, K.; Abulafia, O.; Rose, P.G.; Homesley, H.D. Fixed-dose rate gemcitabine plus docetaxel as second-line therapy for metastatic uterine leiomyosarcoma: A Gynecologic Oncology Group phase II study. Gynecol. Oncol. 2008, 109, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, M.L.; Blessing, J.A.; Mannel, R.; Rose, P.G. Fixed-dose rate gemcitabine plus docetaxel as first-line therapy for metastatic uterine leiomyosarcoma: A Gynecologic Oncology Group phase II trial. Gynecol. Oncol. 2008, 109, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Sutton, G.; Blessing, J.A.; Malfetano, J.H. Ifosfamide and doxorubicin in the treatment of advanced leiomyosarcomas of the uterus: A Gynecologic Oncology Group study. Gynecol. Oncol. 1996, 62, 226–229. [Google Scholar] [CrossRef]
- Sutton, G.P.; Blessing, J.A.; Barrett, R.J.; McGehee, R. Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: A Gynecologic Oncology Group study. Am. J. Obs. Gynecol. 1992, 166, 556–559. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef]
- Monk, B.J.; Blessing, J.A.; Street, D.G.; Muller, C.Y.; Burke, J.J.; Hensley, M.L. A phase II evaluation of trabectedin in the treatment of advanced, persistent, or recurrent uterine leiomyosarcoma: A gynecologic oncology group study. Gynecol. Oncol. 2012, 124, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Rubio, M.J.; Lecumberri, M.J.; Varela, S.; Alarcón, J.; Ortega, M.E.; Gaba, L.; Espinós, J.; Calzas, J.; Barretina, P.; Ruiz, I.; et al. Efficacy and safety of trabectedin in metastatic uterine leiomyosarcoma: A retrospective multicenter study of the Spanish ovarian cancer research group (GEICO). Gynecol. Oncol. Rep. 2020, 33, 100594. [Google Scholar] [CrossRef] [PubMed]
- Pautier, P.; Floquet, A.; Chevreau, C.; Penel, N.; Guillemet, C.; Delcambre, C.; Cupissol, D.; Selle, F.; Isambert, N.; Piperno-Neumann, S.; et al. Trabectedin in combination with doxorubicin for first-line treatment of advanced uterine or soft-tissue leiomyosarcoma (LMS-02): A non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2015, 16, 457–464. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.A.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Thanopoulou, E.; Thway, K.; Khabra, K.; Judson, I. Treatment of hormone positive uterine leiomyosarcoma with aromatase inhibitors. Clin. Sarcoma Res. 2014, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Slomovitz, B.M.; Taub, M.C.; Huang, M.; Levenback, C.; Coleman, R.L. A randomized phase II study of letrozole vs. observation in patients with newly diagnosed uterine leiomyosarcoma (uLMS). Gynecol. Oncol. Rep. 2019, 27, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.J.; O’Connell, R.L.; Banerjee, S.; Mileshkin, L.; Sykes, P.; Beale, P.; Fisher, A.; Bonaventura, A.; Millan, D.; Nottley, S.; et al. Phase 2 study of anastrozole in rare cohorts of patients with estrogen receptor/progesterone receptor positive leiomyosarcomas and carcinosarcomas of the uterine corpus: The PARAGON trial (ANZGOG 0903). Gynecol. Oncol. 2021, 163, 524–530. [Google Scholar] [CrossRef]
- Seligson, N.D.; Kautto, E.A.; Passen, E.N.; Stets, C.; Toland, A.E.; Millis, S.Z.; Meyer, C.F.; Hays, J.L.; Chen, J.L. BRCA1/2 Functional Loss Defines a Targetable Subset in Leiomyosarcoma. Oncologist 2019, 24, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Hensley, M.L.; Chavan, S.S.; Solit, D.B.; Murali, R.; Soslow, R.; Chiang, S.; Jungbluth, A.A.; Bandlamudi, C.; Srinivasan, P.; Tap, W.D.; et al. Genomic Landscape of Uterine Sarcomas Defined Through Prospective Clinical Sequencing. Clin. Cancer Res. 2020, 26, 3881–3888. [Google Scholar] [CrossRef] [Green Version]
- Shanes, E.D.; Friedman, L.A.; Mills, A.M. PD-L1 Expression and Tumor-infiltrating Lymphocytes in Uterine Smooth Muscle Tumors: Implications for Immunotherapy. Am. J. Surg. Pathol. 2019, 43, 792–801. [Google Scholar] [CrossRef]
- Wang, Y.J.; Williams, H.R.; Brzezinska, B.N.; Gaidis, A.; Patel, B.; Munroe, J.; White, J.; Rungruang, B. Use of pembrolizumab in MSI-high uterine leiomyosarcoma; a case report and review of the literature. Gynecol. Oncol. Rep. 2021, 35, 100701. [Google Scholar] [CrossRef]
- Miao, D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Amin-Mansour, A.; Carter, S.L.; Wu, C.; Wong, K.-K.; Raut, C.P.; Ott, P.A.; et al. Response and oligoclonal resistance to pembrolizumab in uterine leiomyosarcoma: Genomic, neoantigen, and immunohistochemical evaluation. J. Clin. Oncol. 2016, 34, 11043. [Google Scholar] [CrossRef]
- Tawbi, H.A.-H.; Burgess, M.A.; Crowley, J.; Tine, B.A.V.; Hu, J.; Schuetze, S.; D’Angelo, S.P.; Attia, S.; Priebat, D.A.; Okuno, S.H.; et al. Safety and efficacy of PD-1 blockade using pembrolizumab in patients with advanced soft tissue (STS) and bone sarcomas (BS): Results of SARC028—A multicenter phase II study. J. Clin. Oncol. 2016, 34, 11006. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, O.; Kee, D.; Gao, B.; Markman, B.; da Gama Duarte, J.; Quigley, L.; Jackett, L.; Linklater, R.; Strickland, A.; Scott, C.; et al. Combination immunotherapy with nivolumab and ipilimumab in patients with rare gynecological malignancies: Results of the CA209-538 clinical trial. J. ImmunoTherapy Cancer 2021, 9, e003156. [Google Scholar] [CrossRef] [PubMed]
- Vyse, S.; Thway, K.; Huang, P.H.; Jones, R.L. Next-generation sequencing for the management of sarcomas with no known driver mutations. Curr. Opin. Oncol. 2021, 33, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Hensley, M.L.; Miller, A.; O’Malley, D.M.; Mannel, R.S.; Behbakht, K.; Bakkum-Gamez, J.N.; Michael, H. Randomized phase III trial of gemcitabine plus docetaxel plus bevacizumab or placebo as first-line treatment for metastatic uterine leiomyosarcoma: An NRG Oncology/Gynecologic Oncology Group study. J. Clin. Oncol. 2015, 33, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, S.; Leone Roberti Maggiore, U.; Aiello, N.; Barra, F.; Ditto, A.; Bogani, G.; Raspagliesi, F.; Lorusso, D. Pharmacokinetic drug evaluation of pazopanib for the treatment of uterine leiomyosarcomas. Expert Opin. Drug Metab. Toxicol. 2017, 13, 881–889. [Google Scholar] [CrossRef]
- Pautier, P.; Penel, N.; Ray-Coquard, I.; Italiano, A.; Bompas, E.; Delcambre, C.; Bay, J.O.; Bertucci, F.; Delaye, J.; Chevreau, C.; et al. A phase II of gemcitabine combined with pazopanib followed by pazopanib maintenance, as second-line treatment in patients with advanced leiomyosarcomas: A unicancer French Sarcoma Group study (LMS03 study). Eur. J. Cancer 2020, 125, 31–37. [Google Scholar] [CrossRef]
- George, S.; Merriam, P.; Maki, R.G.; Van den Abbeele, A.D.; Yap, J.T.; Akhurst, T.; Harmon, D.C.; Bhuchar, G.; O’Mara, M.M.; D’Adamo, D.R.; et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J. Clin. Oncol. 2009, 27, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Schoffski, P.; Blay, J.Y.; De Greve, J.; Brain, E.; Machiels, J.P.; Soria, J.C.; Sleijfer, S.; Wolter, P.; Ray-Coquard, I.; Fontaine, C.; et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI). Eur. J. Cancer 2010, 46, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Verweij, J.; van Oosterom, A.; Blay, J.Y.; Judson, I.; Rodenhuis, S.; van der Graaf, W.; Radford, J.; Le Cesne, A.; Hogendoorn, P.C.; di Paola, E.D.; et al. Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur. J. Cancer 2003, 39, 2006–2011. [Google Scholar] [PubMed]
- Knowling, M.; Blackstein, M.; Tozer, R.; Bramwell, V.; Dancey, J.; Dore, N.; Matthews, S.; Eisenhauer, E. A phase II study of perifosine (D-21226) in patients with previously untreated metastatic or locally advanced soft tissue sarcoma: A National Cancer Institute of Canada Clinical Trials Group trial. Investig. New Drugs 2006, 24, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Nannini, M.; Indio, V.; Schipani, A.; Rizzo, A.; Perrone, A.M.; De Iaco, P.; Pirini, M.G.; De Leo, A.; Urbini, M.; et al. Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile. Cancers 2020, 12, 2126. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Jonsson, P.; Seier, K.; Qin, L.-X.; Chi, P.; Dickson, M.; Gounder, M.; Kelly, C.; Keohan, M.L.; Nacev, B.; et al. Clinical Outcome of Leiomyosarcomas With Somatic Alteration in Homologous Recombination Pathway Genes. JCO Precis. Oncol. 2020, 4, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Cuppens, T.; Moisse, M.; Depreeuw, J.; Annibali, D.; Colas, E.; Gil-Moreno, A.; Huvila, J.; Carpen, O.; Zikan, M.; Matias-Guiu, X.; et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int. J. Cancer 2018, 142, 1230–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Manzano, A.; Dong, W.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Yao, X.; Deshpande, A.; Zaidi, S.; Guglielmi, A.; et al. Integrated mutational landscape analysis of uterine leiomyosarcomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2025182118. [Google Scholar] [CrossRef] [PubMed]
- TCGA. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinen, N.; Aavikko, M.; Heikkinen, T.; Taipale, M.; Taipale, J.; Koivisto-Korander, R.; Butzow, R.; Vahteristo, P. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet. 2016, 12, e1005850. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hubschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- Machado-Lopez, A.; Alonso, R.; Lago, V.; Jimenez-Almazan, J.; Garcia, M.; Monleon, J.; Lopez, S.; Barcelo, F.; Torroba, A.; Ortiz, S.; et al. Integrative Genomic and Transcriptomic Profiling Reveals a Differential Molecular Signature in Uterine Leiomyoma versus Leiomyosarcoma. Int. J. Mol. Sci. 2022, 23, 2190. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, D.A.; George, T.J., Jr.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoInt. inhibition. J. Gastrointest Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoInt. blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Stenzinger, A.; Allen, J.D.; Maas, J.; Stewart, M.D.; Merino, D.M.; Wempe, M.M.; Dietel, M. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 2019, 58, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, H.; Norbury, C.J. Therapeutic Exploitation of CheckpoInt. Defects in Cancer Cells Lacking p53 Function. Cell Cycle 2002, 1, 362–368. [Google Scholar] [CrossRef] [PubMed]
- De Witt Hamer, P.C.; Mir, S.E.; Noske, D.; Van Noorden, C.J.F.; Würdinger, T. WEE1 Kinase Targeting Combined with DNA-Damaging Cancer Therapy Catalyzes Mitotic Catastrophe. Clin. Cancer Res. 2011, 17, 4200–4207. [Google Scholar] [CrossRef] [Green Version]
- Rajeshkumar, N.V.; De Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; Demuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, S.J.; Bagby, S.M.; Yacob, B.W.; Simmons, D.M.; MacBeth, M.; Lieu, C.H.; Davis, S.L.; Leal, A.D.; Tentler, J.J.; Diamond, J.R.; et al. WEE1 Inhibition in Combination with Targeted Agents and Standard Chemotherapy in Preclinical Models of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 642328. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.W.; Jin, Z.; Macdonald, D.; Wei, W.; Qian, X.J.; Choi, W.S.; He, R.; Sun, X.; Chan, G. Prolonged mitotic arrest induced by Wee1 inhibition sensitizes breast cancer cells to paclitaxel. Oncotarget 2017, 8, 73705–73722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, T.M.; Simmons, D.M.; Bagby, S.M.; Hartman, S.J.; Yacob, B.W.; Gittleman, B.; Tentler, J.J.; Cittelly, D.; Ormond, D.R.; Messersmith, W.A.; et al. Wee1 Inhibition Enhances the Anti-Tumor Effects of Capecitabine in Preclinical Models of Triple-Negative Breast Cancer. Cancers 2020, 12, 719. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.Y.; Cho, Y.J.; Shin, S.W.; Choi, C.; Ryu, J.Y.; Jeon, H.K.; Choi, J.J.; Hwang, J.R.; Choi, C.H.; Kim, T.J.; et al. Anti-Tumor Effects of Wee1 Kinase Inhibitor with Radiotherapy in Human Cervical Cancer. Sci. Rep. 2019, 9, 15394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shen, C.; Pettit, C.J.; Li, T.; Hu, A.J.; Miller, E.D.; Zhang, J.; Lin, S.H.; Williams, T.M. Wee1 Kinase Inhibitor AZD1775 Effectively Sensitizes Esophageal Cancer to Radiotherapy. Clin. Cancer Res. 2020, 26, 3740–3750. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Gotimer, K.; De Souza, C.; Tepper, C.G.; Karnezis, A.N.; Leiserowitz, G.S.; Chien, J.; Smith, L.H. Short-term organoid culture for drug sensitivity testing of high-grade serous carcinoma. Gynecol. Oncol. 2020, 157, 783–792. [Google Scholar] [CrossRef]
- Kreahling, J.M.; Gemmer, J.Y.; Reed, D.; Letson, D.; Bui, M.; Altiok, S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol. Cancer Ther. 2012, 11, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreahling, J.M.; Foroutan, P.; Reed, D.; Martinez, G.; Razabdouski, T.; Bui, M.M.; Raghavan, M.; Letson, D.; Gillies, R.J.; Altiok, S. Wee1 Inhibition by MK-1775 Leads to Tumor Inhibition and Enhances Efficacy of Gemcitabine in Human Sarcomas. PLoS ONE 2013, 8, e57523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Li, X.; Zhang, Y.; Shi, H.; Zhang, B. WEE1 inhibition by MK1775 as a single-agent therapy inhibits ovarian cancer viability Corrigendum in /10.3892/ol.2018.8757. Oncol. Lett. 2017, 14, 3580–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, A.D.; Li, J.; Liu, Y.; Hurd, M.S.; Schuller, A.G.; Long, B.; Hirsch, H.A.; Feldman, I.; Benita, Y.; Toniatti, C.; et al. Preclinical Evaluation of the WEE1 Inhibitor MK-1775 as Single-Agent Anticancer Therapy. Mol. Cancer Ther. 2013, 12, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lens, S.M.A.; Voest, E.E.; Medema, R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841. [Google Scholar] [CrossRef]
- Manning, A.L.; Dyson, N.J. RB: Mitotic implications of a tumour suppressor. Nat. Rev. Cancer 2012, 12, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sharma, S.; Ghamande, S.; Gordon, M.S.; Del Prete, S.A.; Ray-Coquard, I.; Kutarska, E.; Liu, H.; Fingert, H.; Zhou, X.; et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol. Oncol. 2012, 127, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [CrossRef]
- Brewer Savannah, K.J.; Demicco, E.G.; Lusby, K.; Ghadimi, M.P.; Belousov, R.; Young, E.; Zhang, Y.; Huang, K.-L.; Lazar, A.J.; Hunt, K.K.; et al. Dual targeting of mTOR and aurora-A kinase for the treatment of uterine Leiomyosarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4633–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, D.M.; Sill, M.W.; Lankes, H.A.; Piekarz, R.; Shahin, M.S.; Ridgway, M.R.; Backes, F.; Tenney, M.E.; Mathews, C.A.; Hoffman, J.S.; et al. A phase 2 study of alisertib (MLN8237) in recurrent or persistent uterine leiomyosarcoma: An NRG Oncology/Gynecologic Oncology Group study 0231D. Gynecol. Oncol. 2017, 144, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.A.; Mahoney, M.R.; Tap, W.D.; D’Angelo, S.P.; Keohan, M.L.; Van Tine, B.A.; Agulnik, M.; Horvath, L.E.; Nair, J.S.; Schwartz, G.K. Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1855–1860. [Google Scholar] [CrossRef]
- Ma, Y.; Kurtyka, C.A.; Boyapalle, S.; Sung, S.-S.; Lawrence, H.; Guida, W.; Cress, W.D. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 2008, 68, 6292–6299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtyka, C.A.; Chen, L.; Cress, W.D. E2F Inhibition Synergizes with Paclitaxel in Lung Cancer Cell Lines. PLoS ONE 2014, 9, e96357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouaud, F.; Hamouda-Tekaya, N.; Cerezo, M.; Abbe, P.; Zangari, J.; Hofman, V.; Ohanna, M.; Mograbi, B.; El-Hachem, N.; Benfodda, Z.; et al. E2F1 inhibition mediates cell death of metastatic melanoma. Cell Death Dis. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.D.; George, A.J.; Morgan, K.A.; Mann, J.R.; Choo, K.H. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, J.Y.; Tsai, J.H.; Jeng, Y.M.; Lee, J.C.; Hsu, H.H.; Yang, C.Y. Leiomyosarcoma with alternative lengthening of telomeRes. is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am. J. Surg. Pathol. 2015, 39, 236–244. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeRes. in tumors shows the significance of alternative lengthening of telomeRes. in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [PubMed]
- Shay, J.W.; Wright, W.E. TelomeRes. and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Henson, J.D.; Reddel, R.R. Assaying and investigating Alternative Lengthening of TelomeRes. activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Almouzni, G. Assembly of telomeric chromatin to create ALTernative endings. Trends Cell Biol. 2014, 24, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeRes. telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeRes. pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Taylor, S.; Mitson, M.; Bachrati, C.Z.; Higgs, D.R.; Gibbons, R.J. ATRX dysfunction induces replication defects in primary mouse cells. PLoS ONE 2014, 9, e92915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef]
- Deeg, K.I.; Chung, I.; Bauer, C.; Rippe, K. Cancer Cells with Alternative Lengthening of TelomeRes. Do Not Display a General Hypersensitivity to ATR Inhibition. Front. Oncol. 2016, 6, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeRes. renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Punchihewa, C.; Inoue, A.; Hishiki, A.; Fujikawa, Y.; Connelly, M.; Evison, B.; Shao, Y.; Heath, R.; Kuraoka, I.; Rodrigues, P.; et al. Identification of Small Molecule Proliferating Cell Nuclear Antigen (PCNA) Inhibitor That Disrupts Interactions with PIP-box Proteins and Inhibits DNA Replication. J. Biol. Chem. 2012, 287, 14289–14300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geenen, J.J.J.; Schellens, J.H.M. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 2017, 23, 4540–4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Wu, S.; Liu, H.; Stratt, R.; Barak, O.G.; Shiekhattar, R.; Picketts, D.J.; Yang, X. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 2004, 279, 20369–20377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, M.S.; Ivanochko, D.; Hashem, L.E.; Curtin, M.; Delorme, M.; Goodall, E.; Yan, K.; Picketts, D.J. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016, 7, e2220. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, C.; Timsit, S.; Villard, L.; Khrestchatisky, M.; Fontes, M.; Colleaux, L. Specific interaction between the XNP/ATR-X gene product and the SET domain of the human EZH2 protein. Hum. Mol. Genet. 1998, 7, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, K.; Cifuentes-Rojas, C.; Ergun, A.; Del Rosario, A.; Jeon, Y.; White, F.; Sadreyev, R.; Lee, J.T. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 2014, 159, 869–883. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Li, D.M.; Sun, H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997, 57, 2124–2129. [Google Scholar]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Downes, C.P.; Ross, S.; Maccario, H.; Perera, N.; Davidson, L.; Leslie, N.R. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv. Enzym. Regul. 2007, 47, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 1999, 146, 389–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.P.; Brown, J.L.; Eng, C. PTEN coordinates G(1) arrest by down-regulating cyClin. D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum. Mol. Genet. 2001, 10, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of PTEN. J. Cell Sci. 2008, 121, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfieri, R.; Giovannetti, E.; Bonelli, M.; Cavazzoni, A. New Treatment Opportunities in Phosphatase and Tensin Homolog (PTEN)-Deficient Tumors: Focus on PTEN/Focal Adhesion Kinase Pathway. Front. Oncol. 2017, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Chawla, S.P.; Ray-Coquard, I.; Le Cesne, A.; Staddon, A.P.; Milhem, M.M.; Penel, N.; Riedel, R.F.; Bui-Nguyen, B.; Cranmer, L.D.; et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J. Clin. Oncol. 2013, 31, 2485–2492. [Google Scholar] [CrossRef]
- Trucco, M.M.; Meyer, C.F.; Thornton, K.A.; Shah, P.; Chen, A.R.; Wilky, B.A.; Carrera-Haro, M.A.; Boyer, L.C.; Ferreira, M.F.; Shafique, U.; et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin. Sarcoma Res. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Thanapprapasr, D.; Previs, R.A.; Hu, W.; Ivan, C.; Armaiz-Pena, G.N.; Dorniak, P.L.; Hansen, J.M.; Rupaimoole, R.; Huang, J.; Dalton, H.J.; et al. PTEN Expression as a Predictor of Response to Focal Adhesion Kinase Inhibition in Uterine Cancer. Mol. Cancer 2015, 14, 1466–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, D.; Xin, J.; Volk, A.; Wei, W.; Schmidt, R.; Scurti, G.; Nand, S.; Breuer, E.K.; Kuo, P.C.; Breslin, P.; et al. FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-null T-ALL cells. Cell Rep. 2015, 10, 2055–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, T.M.; Liu, E.T.; Craven, R.J.; Cance, W.G. Expression of growth factor receptors, the focal adhesion kinase, and other tyrosine kinases in human soft tissue tumors. Ann. Surg. Oncol. 1994, 1, 18–27. [Google Scholar] [CrossRef]
- Shank, J.; Frisch, N.; Rhode, J.; Liu, J. Identification of molecular markers of targeted treatment of uterine sarcomas. Gynecol. Oncol. 2013, 131, 282. [Google Scholar] [CrossRef]
- Lopez-Acevedo, M.; Grace, L.; Teoh, D.; Whitaker, R.; Adams, D.J.; Jia, J.; Nixon, A.B.; Secord, A.A. Dasatinib (BMS-35482) potentiates the activity of gemcitabine and docetaxel in uterine leiomyosarcoma cell lines. Gynecol. Oncol. Res. Pract. 2014, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.D.; Dedes, K.J.; Sandhu, S.; Frentzas, S.; Kristeleit, R.; Ashworth, A.; Poole, C.J.; Weigelt, B.; Kaye, S.B.; Molife, L.R. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Robert, F. The Mediator Complex: At the Nexus of RNA Polymerase II Transcription. Trends Cell Biol. 2017, 27, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taatjes, D.J. The human Mediator complex: A versatile, genome-wide regulator of transcription. Trends BioChem. Sci. 2010, 35, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Cantin, G.T.; Stevens, J.L.; Berk, A.J. Characterization of mediator complexes from HeLa cell nuclear extract. Mol. Cell Biol. 2001, 21, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Knuesel, M.T.; Meyer, K.D.; Donner, A.J.; Espinosa, J.M.; Taatjes, D.J. The human CDK8 subcomplex is a histone kinase that requiRes. Med12 for activity and can function independently of mediator. Mol. Cell Biol. 2009, 29, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, H.R.; Pasanen, A.; Heikinheimo, O.; Tanskanen, T.; Palin, K.; Tolvanen, J.; Vahteristo, P.; Sjoberg, J.; Pitkanen, E.; Butzow, R.; et al. Multiple clinical characteristics separate MED12-mutation-positive and -negative uterine leiomyomas. Sci. Rep. 2017, 7, 1015. [Google Scholar] [CrossRef]
- Kampjarvi, K.; Makinen, N.; Kilpivaara, O.; Arola, J.; Heinonen, H.R.; Bohm, J.; Abdel-Wahab, O.; Lehtonen, H.J.; Pelttari, L.M.; Mehine, M.; et al. Somatic MED12 mutations in uterine leiomyosarcoma and colorectal cancer. Br. J. Cancer 2012, 107, 1761–1765. [Google Scholar] [CrossRef] [Green Version]
- Makinen, N.; Kampjarvi, K.; Frizzell, N.; Butzow, R.; Vahteristo, P. Characterization of MED12, HMGA2, and FH alterations reveals molecular variability in uterine smooth muscle tumors. Mol. Cancer 2017, 16, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, P.; Shin, Y.H.; Yatsenko, S.A.; Castro, C.A.; Surti, U.; Rajkovic, A. Med12 gain-of-function mutation causes leiomyomas and genomic instability. J. Clin. Investig. 2015, 125, 3280–3284. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Holzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Xu, X.; Hecht, A.; Boyer, T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, A.J.; Szostek, S.; Hoover, J.M.; Espinosa, J.M. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 2007, 27, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Li, W.; Yao, X.; Lin, Q.J.; Yin, J.W.; Liang, Y.; Heiner, M.; Tian, B.; Hui, J.; Wang, G. Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol. Cell 2012, 45, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R. Mediating resistance in oncogene-driven cancers. N. Engl. J. Med. 2013, 368, 1551–1552. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zeng, H.; Wang, Q.; Zhao, Z.; Boyer, T.G.; Bian, X.; Xu, W. MED12 methylation by CARM1 sensitizes human breast cancer cells to chemotherapy drugs. Sci. Adv. 2015, 1, e1500463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turunen, M.; Spaeth, J.M.; Keskitalo, S.; Park, M.J.; Kivioja, T.; Clark, A.D.; Makinen, N.; Gao, F.; Palin, K.; Nurkkala, H.; et al. Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell Rep. 2014, 7, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, R.L.; Urban, V.; Munoz-Martinez, F.; Ayestaran, I.; Thomas, J.C.; de Renty, C.; O’Connor, M.J.; Forment, J.V.; Galanty, Y.; Jackson, S.P. Loss of CyClin. C or CDK8 provides ATR inhibitor resistance by suppressing transcription-associated replication stress. Nucleic Acids Res. 2021, 49, 8665–8683. [Google Scholar] [CrossRef]
- Bhagwat, A.S.; Roe, J.S.; Mok, B.Y.L.; Hohmann, A.F.; Shi, J.; Vakoc, C.R. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016, 15, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, J.S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell 2015, 58, 1028–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesic, K.; Wakefield, M.; Kondrashova, O.; Scott, C.L.; McNeish, I.A. Targeting DNA repair: The genome as a potential biomarker. J. Pathol. 2018, 244, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, J.T.; Blessing, J.A.; Beecham, J.; Homesley, H.; Yordan, E. Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: A Gynecologic Oncology Group study. J. Clin. Oncol. 1991, 9, 1962–1966. [Google Scholar] [CrossRef]
- Thigpen, J.T.; Blessing, J.A.; Wilbanks, G.D. Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am. J. Clin. Oncol. 1986, 9, 18–20. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho Fischer, C.; Hu, Y.; Morreale, M.; Lin, W.Y.; Wali, A.; Thakar, M.; Karunasena, E.; Sen, R.; Cai, Y.; Murphy, L.; et al. Treatment with epigenetic agents profoundly inhibits tumor growth in leiomyosarcoma. Oncotarget 2018, 9, 19379–19395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, J.; Wu, X.; Zheng, Z. Identification of Somatic Genetic Alterations Using Whole-Exome Sequencing of Uterine Leiomyosarcoma Tumors. Front. Oncol. 2021, 11, 687899. [Google Scholar] [CrossRef] [PubMed]
- Press, J.Z.; Kenyon, J.A.; Xue, H.; Miller, M.A.; De Luca, A.; Miller, D.M.; Huntsman, D.G.; Gilks, C.B.; McAlpine, J.N.; Wang, Y.Z. Xenografts of primary human gynecological tumors grown under the renal capsule of NOD/SCID mice show genetic stability during serial transplantation and respond to cytotoxic chemotherapy. Gynecol. Oncol. 2008, 110, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Cuppens, T.; Depreeuw, J.; Annibali, D.; Thomas, D.; Hermans, E.; Gomme, E.; Trinh, X.B.; Debruyne, D.; Moerman, P.; Lambrechts, D.; et al. Establishment and characterization of uterine sarcoma and carcinosarcoma patient-derived xenograft models. Gynecol. Oncol. 2017, 146, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Cuppens, T.; Annibali, D.; Coosemans, A.; Trovik, J.; Ter Haar, N.; Colas, E.; Garcia-Jimenez, A.; Van de Vijver, K.; Kruitwagen, R.P.; Brinkhuis, M.; et al. Potential Targets’ Analysis Reveals Dual PI3K/mTOR Pathway Inhibition as a Promising Therapeutic Strategy for Uterine Leiomyosarcomas-an ENITEC Group Initiative. Clin. Cancer Res. 2017, 23, 1274–1285. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tu, J.; Zhao, Z.; Chen, L.; Qu, Y.; Li, H.; Yao, H.; Wang, X.; Lee, D.-F.; Shen, J.; et al. Molecular signatuRes. of BRCAness analysis identifies PARP inhibitor Niraparib as a novel targeted therapeutic strategy for soft tissue Sarcomas. Theranostics 2020, 10, 9477–9494. [Google Scholar] [CrossRef]
- Xing, D.; Scangas, G.; Nitta, M.; He, L.; Xu, X.; Ioffe, Y.J.; Aspuria, P.J.; Hedvat, C.Y.; Anderson, M.L.; Oliva, E.; et al. A role for BRCA1 in uterine leiomyosarcoma. Cancer Res. 2009, 69, 8231–8235. [Google Scholar] [CrossRef] [Green Version]
- Strizzi, L.; Bianco, C.; Hirota, M.; Watanabe, K.; Mancino, M.; Hamada, S.; Raafat, A.; Lawson, S.; Ebert, A.D.; D’Antonio, A.; et al. Development of leiomyosarcoma of the uterus in MMTV-CR-1 transgenic mice. J. Pathol. 2007, 211, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Szabolcs, M.; Fisher, P.; Kljuic, A.; Ludwig, T.; Efstratiadis, A. A mouse model of uterine leiomyosarcoma. Am. J. Pathol. 2004, 164, 325–336. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Summary of the common genetic aberrations in uLMS and their potential therapeutic targets.
Figure 1.
Summary of the common genetic aberrations in uLMS and their potential therapeutic targets.


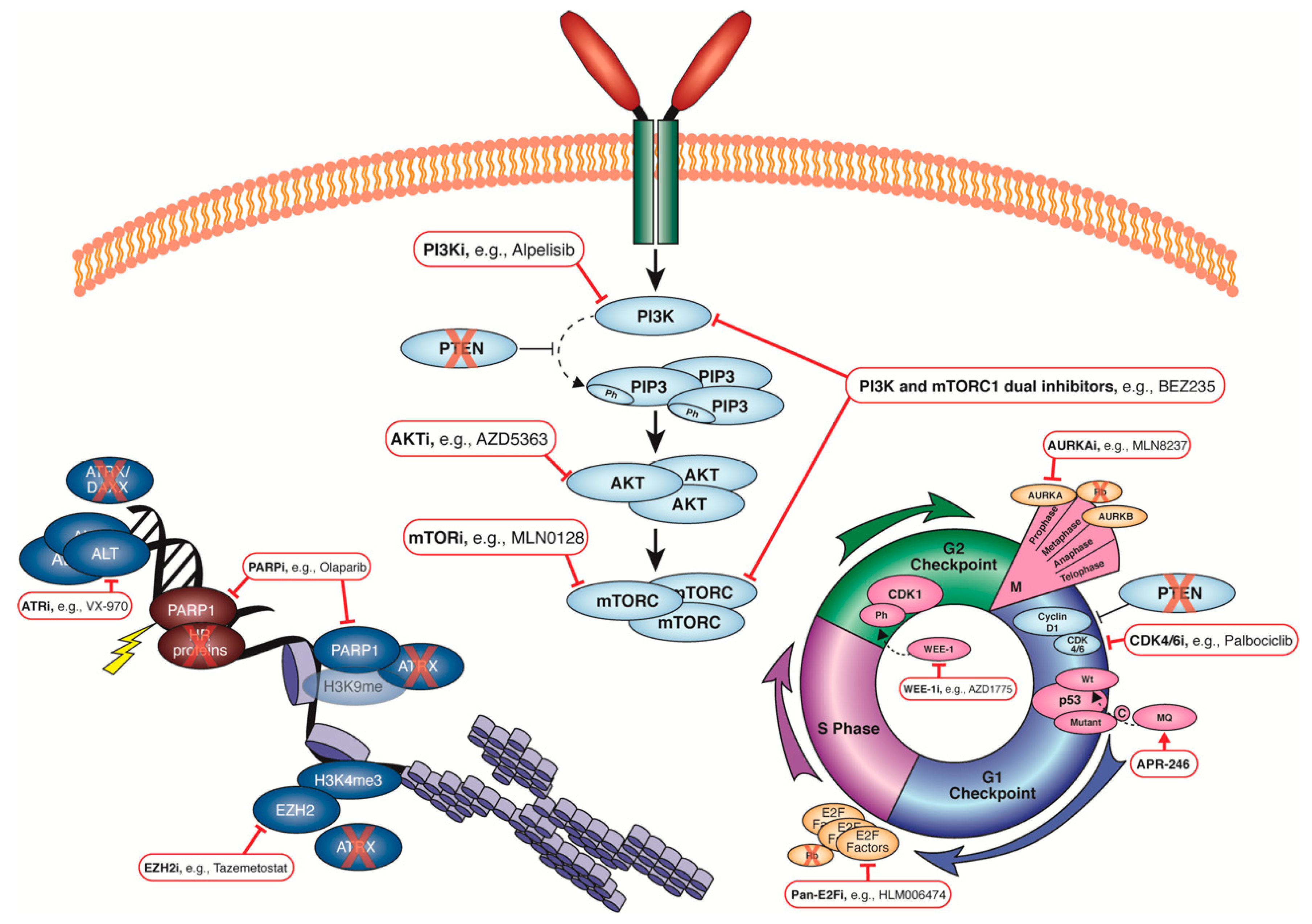{kind=link}
Table 1.
List of clinical trials including uLMS patients.
| Identifier | Target | Interventions | Notes, References |
|---|---|---|---|
| NCT01637961 | AURKA | Alisertib | Phase II; uLMS; completed, no response |
| NCT00378911 | VEGFR1/2/3, PDGFRa/b, c-KIT, RET, GCSFR, FLT-3 | Sunitinib malate | Phase II; uLMS; completed, but no results reported |
| NCT02428192 | CTLA4, PD-1 | Ipilimumab and Nivolumab | Phase II; uLMS; active, no response [67] |
| NCT03880019 | PARP | Olaparib and Temozolomide | Phase II; uLMS; active, no results [67] |
| NCT01012297 | VEGF | Bevacizumab | Phase III; no response [68] |
| NCT02203760 | VEGFR1/2/3, PDGFRa/b, c-KIT | Pazopanib + Gemcitabine | Phase II; metastatic uLMS or UCS; recruiting |
| NCT02601209 | VEGFR1/2/3, PDGFRa/b, c-KIT, mTORC1/mTORC2 | Pazopanib or Sapanisertib | Phase I/II; unresectable LMS and STS; slight benefit with pazopanib [69] |
| NCT04200443 | VEGFR2, c-Met, AXL, RET | Cabozantinib | Phase II; unresectable LMS and STS; recruiting |
| NCT00659360 | Src, Lck, Fyn, Lyn, c-Yes, Blk, Abl EGFRmut | Saracatinib | Phase II; LMS, STS, and uterine sarcoma (US); no response |
| NCT00245102 | VEGFR1/2/3, PDGFRb, RAF-1, BRAF (wt and mut), c-KIT, FLT-3 | Sorafenib tosylate | Phase II; LMS, STS, and US; no response |
| NCT00390234 | VEGF-Trap | Ziv-Aflibercept | Phase II; uLMS, UCS, and US; no response |
| NCT01442662 | VEGFR1/2/3, PDGFRa/b, c-KIT | Pazopanib + Gemcitabine | Phase II; second-line LMS; no response [70] |
| NCT00474994 | VEGFR1/2/3, PDGFRa/b, c-KIT, FLT-3, CSF1R, RET | Sunitinib malate | Phase II; metastatic/recurrent sarcomas; SD best response [71] |
| NCT00526149 | PLK1/2/3, BRD4 | BI-2536 | Phase II; metastatic/recurrent solid tumours; no response [72] |
| NCT00006357 | PDGFR, c-KIT, Abl | Imatinib mesylate | Phase I/II; recurrent/refractor STS; no response [73] |
| NCT00053794 | AKT | Perifosine | Phase II; metastatic STS; no response [74] |
Table 2.
Summary of published uLMS patient cohorts for genomic/genetic analysis.
| Cohort | n | Sequencing (How Many, What Type, Who Analysed/Reported) |
|---|---|---|
| MSKCC | 128 | n = 80 exon capture [60,75] n = 121 exon capture [76] |
| Cuppens | 62 | n = 2 WGS [77] n = 62 SNP array [77] |
| Yale | 55 | n = 11 WGS [78] n = 55 WES, RNA-Seq [78] |
| MSK/Genie | 51 | n = 51 exon-targeted seq [75] |
| Dana-Faber | 39 | n = 39 exon-targeted seq [75] |
| OSU | 34 | n = 35 FoundationOne panel test [59] |
| TCGA | 31 | n = 10 WGS [78,79] n = 27 WES, RNA-Seq [59,78,79] n = 31 exon-targeted seq [75] |
| Helsinki | 19 | n = 19 WES [80] |
| Chudasama | 10 | n = 10 WES [81] |
| U Mich | 8 | n = 8 exon-targeted seq [75] |
| Vanderbilt | 7 | n = 7 exon-targeted seq [75] |
| Spain | 44 | n = 44 WES, RNA-Seq [82] |
WGS, whole-genome sequencing; SNP, single nucleotide polymorphism; WES, whole-exome sequencing.
Table 3.
Summary of the potential therapeutic targets that arise due to genetic aberrations frequently occurring in uLMS.
Table 3.
Summary of the potential therapeutic targets that arise due to genetic aberrations frequently occurring in uLMS.
| Aberration | Frequency | Therapeutic Target | Potential Drugs |
|---|---|---|---|
| TP53 | 26–92% | Mutant p53 [90] | APR-246 [91] |
| WEE-1 [95] | AZD1775/MK-1775 [103,107] | ||
| RB1 | 27–88% | AURKA [109,110] | MLN8237/Alisertib [113,114,117] |
| E2F [108] | HLM006474 [118,119] | ||
| ATRX | 24–34% | ATR | VE-821 [141], VX-970, AZD6738 |
| EZH2 [139,148,149] BLM PCNA PARP WEE-1 | Tazemetostat, GSK-126 ML216 [142] T2AA [143] PJ34 [147] AZD1775 [144,145] | ||
| PTEN | 19–75% | PI3K [163,164] | AZD6482 [162], Buparlisib |
| AKT [163,164] | MK-2206 [162], AZD5363 | ||
| mTORC1 [163,164] SRC FAK | Temsirolimus [162,166], Everolimus, Ridaforolimus [165] Dasatinib [172] VS-4718, VS-6063, PF-573228, PF-562271, GSK2256098 [168] | ||
| PARP | KU0058948 [174], Olaparib [175] | ||
| MED12 | 12–21% | TGF-βR | LY2157299 [185] |
| BET [193] | JQI | ||
| HRD | 7–60% | PARP [78,81,202] | Olaparib [59,60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dall, G.V.; Hamilton, A.; Ratnayake, G.; Scott, C.; Barker, H. Interrogating the Genomic Landscape of Uterine Leiomyosarcoma: A Potential for Patient Benefit. Cancers 2022, 14, 1561. https://doi.org/10.3390/cancers14061561
AMA Style
Dall GV, Hamilton A, Ratnayake G, Scott C, Barker H. Interrogating the Genomic Landscape of Uterine Leiomyosarcoma: A Potential for Patient Benefit. Cancers. 2022; 14(6):1561. https://doi.org/10.3390/cancers14061561
Chicago/Turabian StyleDall, Genevieve V., Anne Hamilton, Gayanie Ratnayake, Clare Scott, and Holly Barker. 2022. "Interrogating the Genomic Landscape of Uterine Leiomyosarcoma: A Potential for Patient Benefit" Cancers 14, no. 6: 1561. https://doi.org/10.3390/cancers14061561
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.