
The Stress-Inducible BCL2A1 Is Required for Ovarian Cancer Metastatic Progression in the Peritoneal Microenvironment

 ,
, 
Abstract
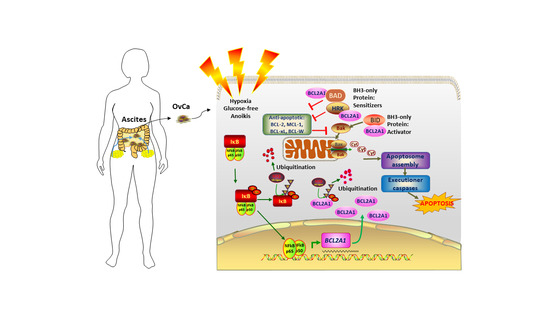
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Clinical Samples
2.2. Plasmids
2.3. RNA Extraction and Real-Time Quantitative PCR (qPCR) Analysis
2.4. XTT Cell Proliferation Assay
2.5. Soft Agar Assay
2.6. Anoikis Assay
2.7. Co-Immunoprecipitation and In Vivo Ubiquitination Assays
2.8. Luciferase Reporter Assays
2.9. In Situ Proximity Ligation Assay
2.10. Western Blot Analysis
2.11. Immunofluorescence Staining
2.12. Immunohistochemical Staining
2.13. Ex Vivo Colonization Assay
2.14. In Vivo Tumor Dissemination Mouse Model
2.15. Statistical Analysis
3. Results
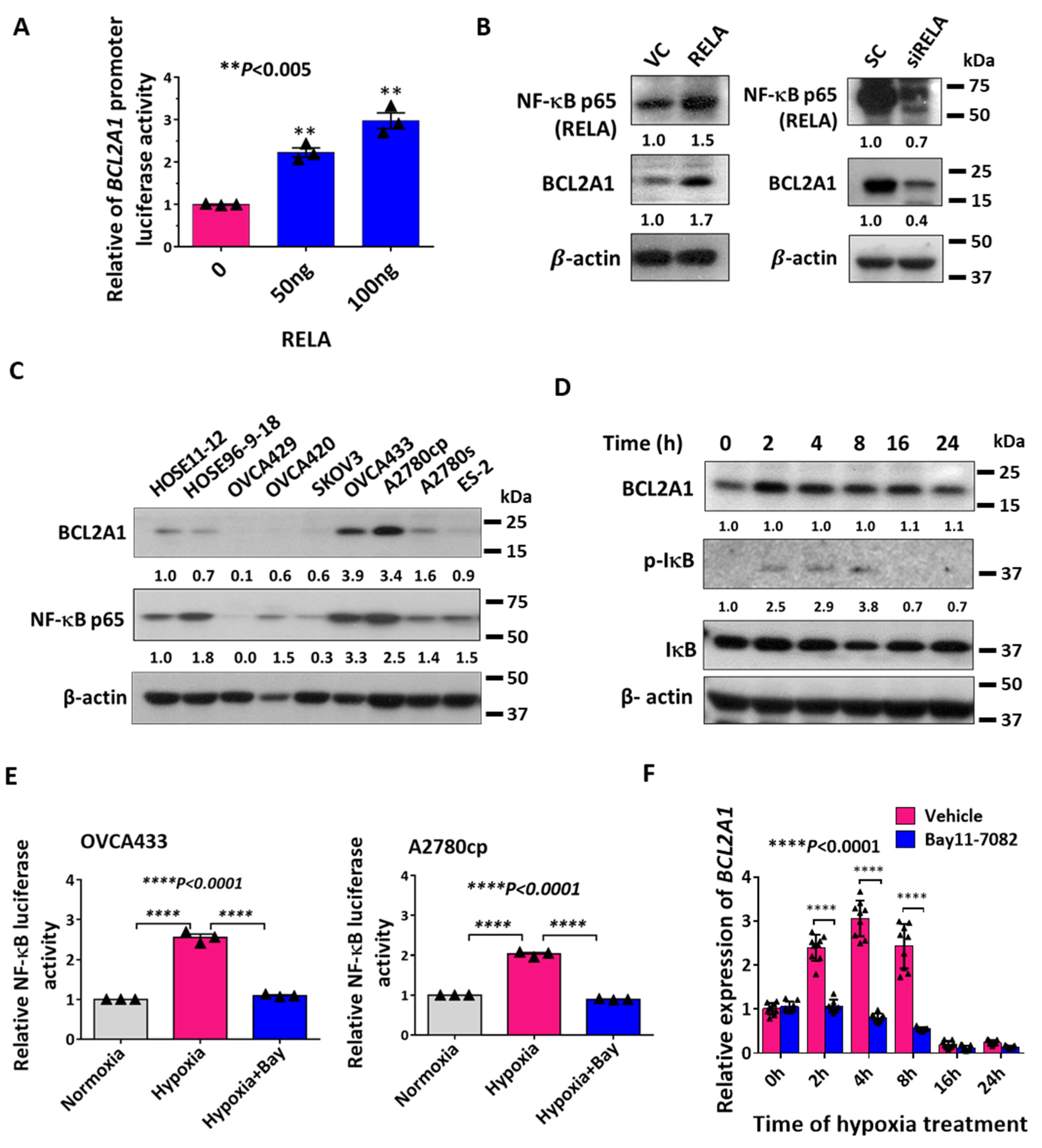
3.1. BCL2A1 Is an Early Response Gene to Hypoxia in Ovarian Cancer Cells
3.2. NF-κB Transcriptionally Regulates Hypoxia-Induced BCL2A1 Upregulation
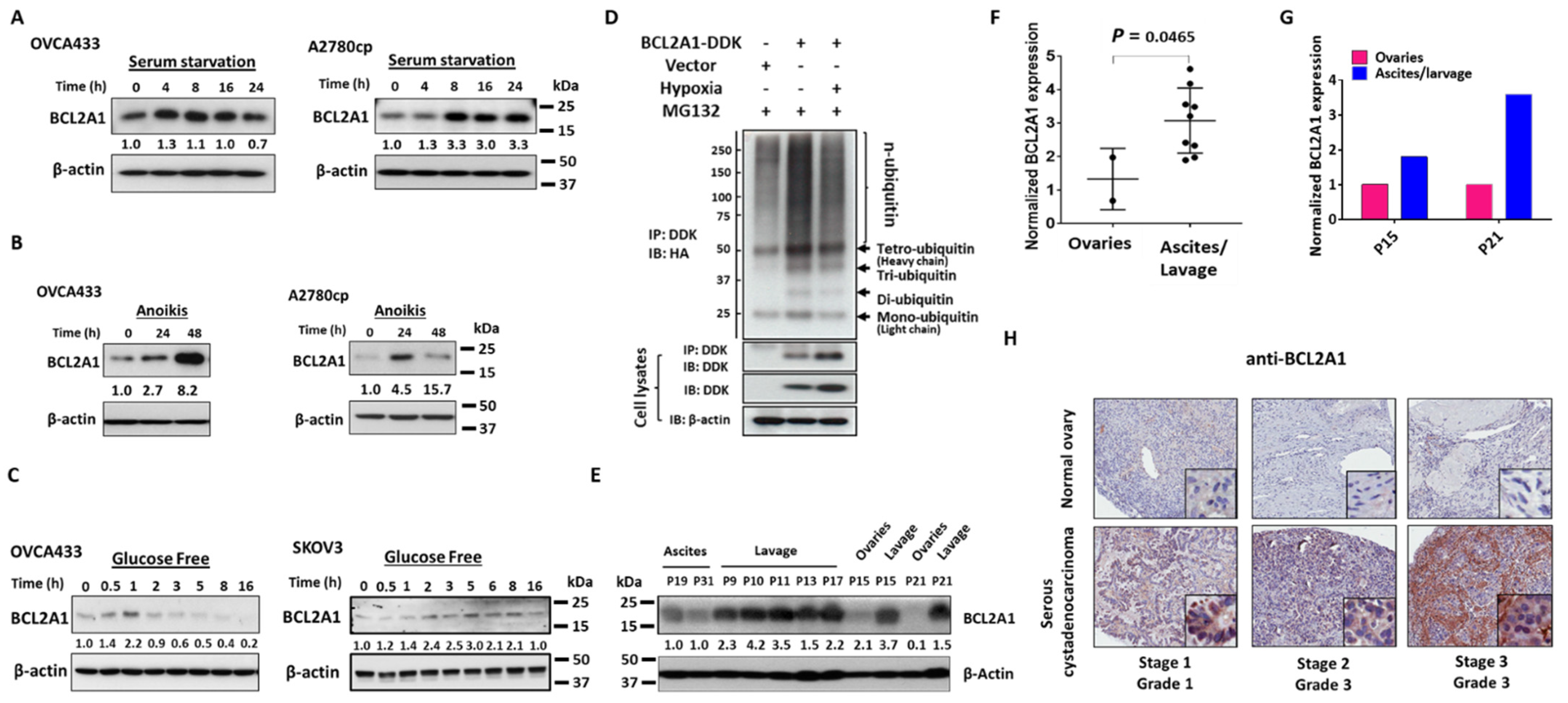
3.3. BCL2A1 Can Be Induced in Ovarian Cancer Cells by Various Physiological Stressors
3.4. BCL2A1 Is Frequently Upregulated in Advanced Metastatic Ovarian Cancers
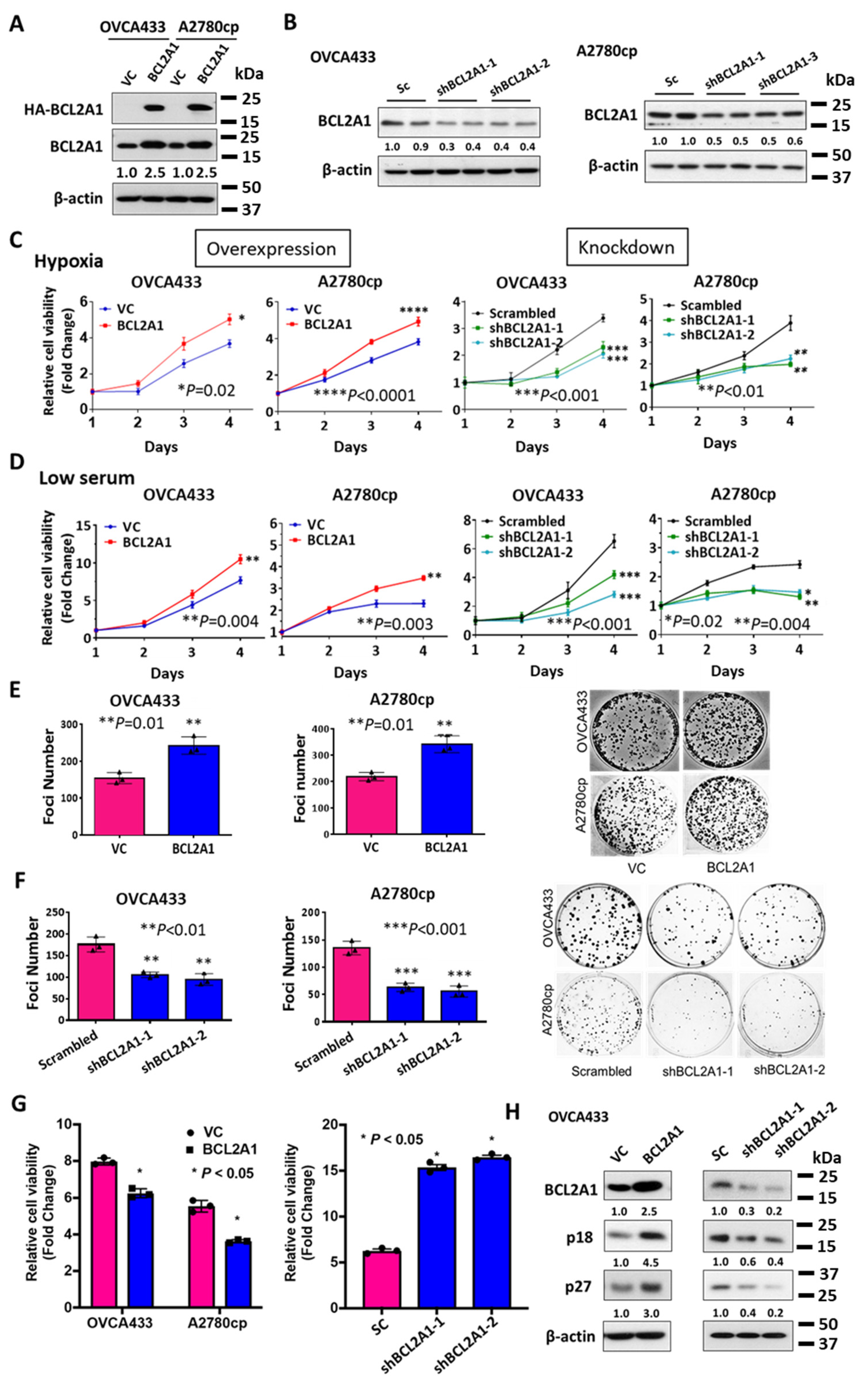
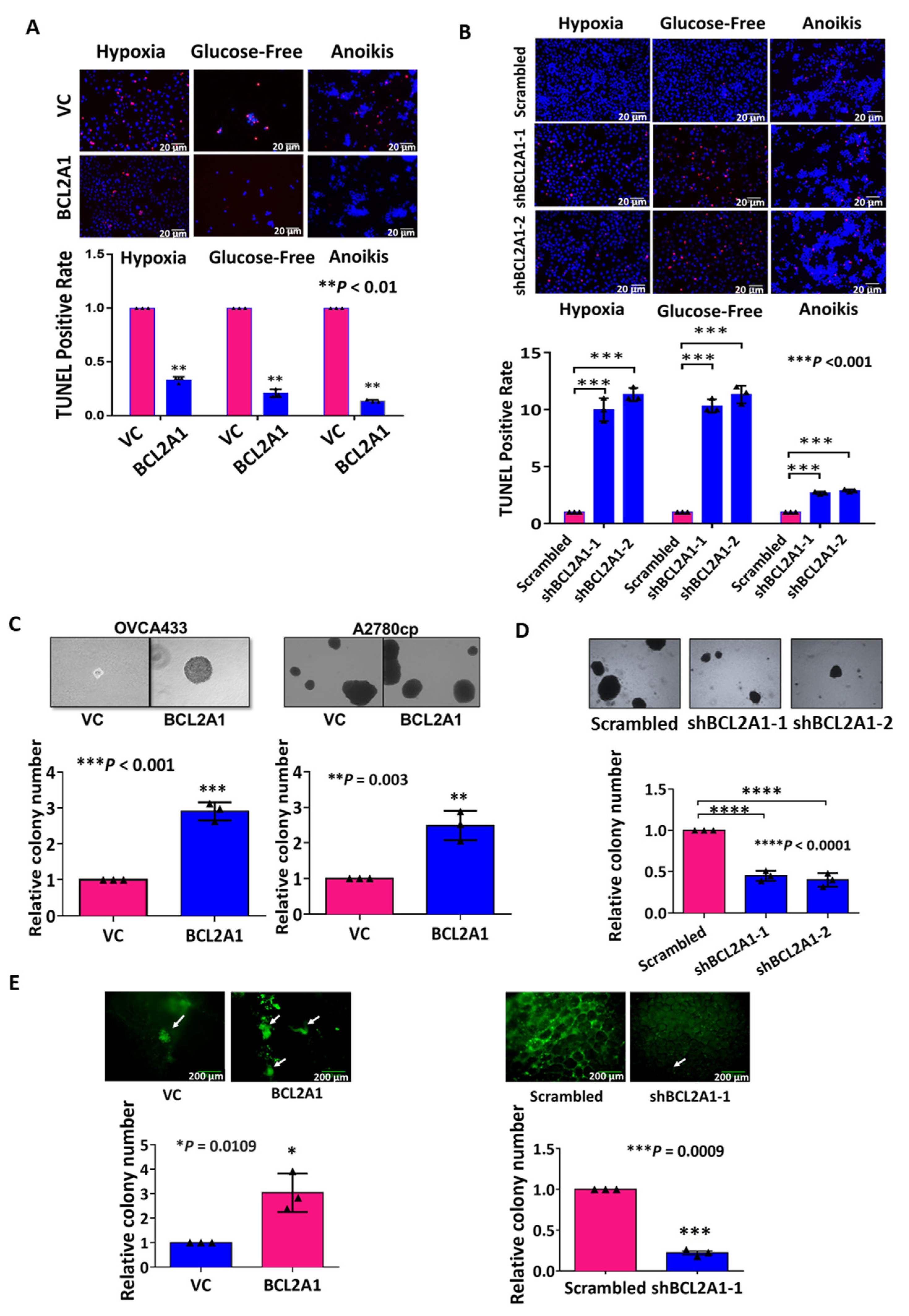
3.5. BCL2A1 Is Required for the Enhanced Cell Viability of Ovarian Cancer Cells under Stress
3.6. BCL2A1 Inhibits Cell Apoptosis and Enhances the Oncogenic Properties of Ovarian Cancer Cells
3.7. BCL2A1 Suppresses Intrinsic Apoptosis by Interacting with HRK/BAD/BID
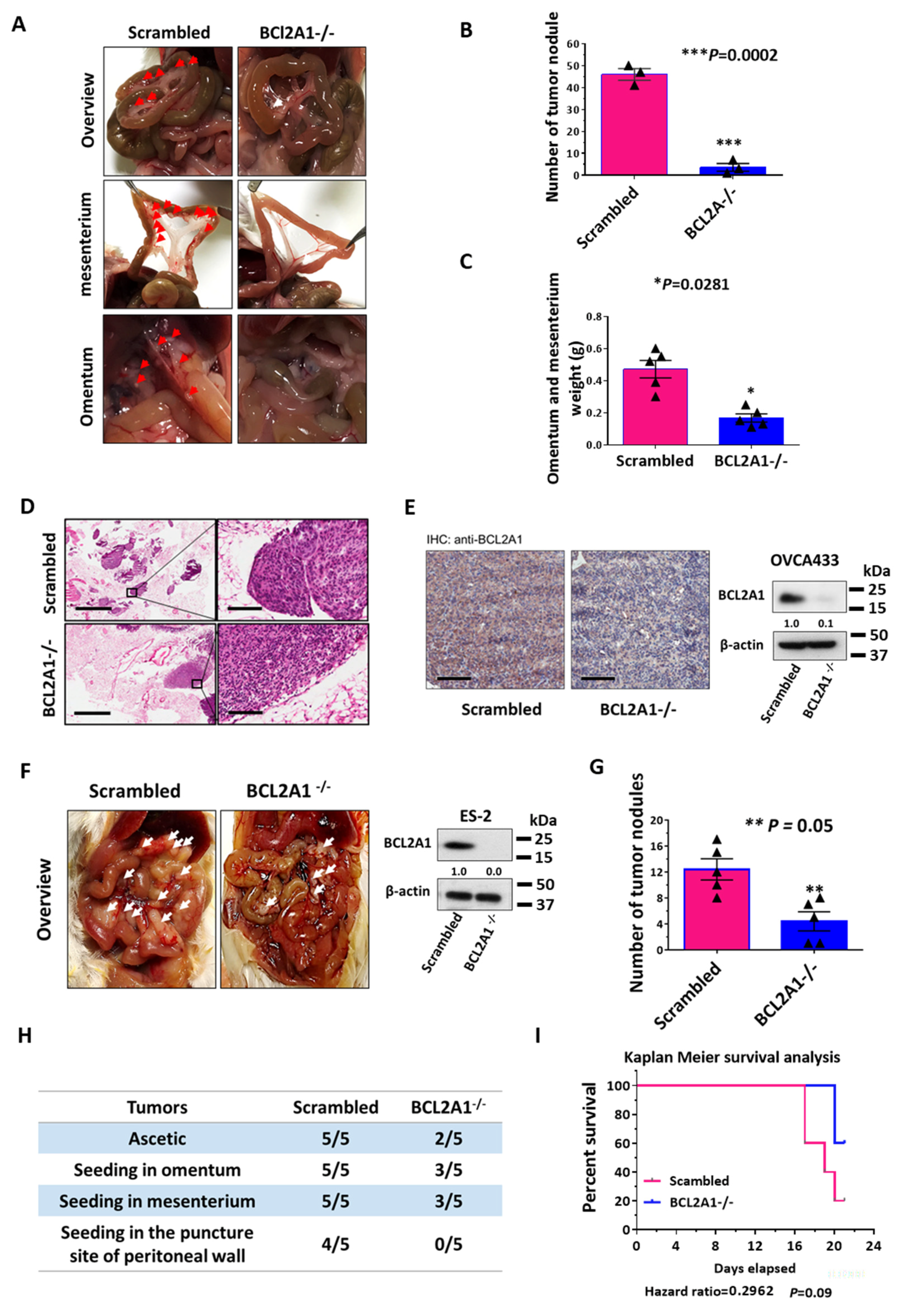
3.8. BCL2A1 Is Required for the In Vivo Tumor Dissemination of Ovarian Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EMT | Epithelial to mesenchymal transition |
| FBS | Fetal bovine serum |
| GLuc | Gaussia luciferase |
| HGSOC | High-grade serous ovarian cancer |
| H2O2 | Hydrogen peroxidase |
| HOSE | Human ovarian surface epithelial |
| i.p. | Intraperitoneally |
| IHC | Immunohistochemical |
| MOMP | Mitochondrial outer membrane permeabilization |
| PLA | in situ Proximity Ligation Assay |
| PVDF | Polyvinylidene difluoride |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| TCGA | The Cancer Genome Atlas |
| TME | Tumor microenvironment |
References
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M. Encyclopedia of Cancer; Springer Science Business Media: Berlin, Germany, 2008. [Google Scholar]
- Ozols, R.F. Ovarian Cancer; PMPH-USA: New Haven, CT, USA, 2003; Volume 1. [Google Scholar]
- Flanagan, M.; Solon, J.; Chang, K.H.; Deady, S.; Moran, B.; Cahill, R.; Shields, C.; Mulsow, J. Peritoneal metastases from extra-abdominal cancer—A population-based study. Eur. J. Surg. Oncol. 2018, 44, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Thibault, B.; Castells, M.; Delord, J.P.; Couderc, B. Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2014, 33, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Biology of Cancer; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Yang, H.; Zou, W.; Chen, B. Overexpression of CD147 in ovarian cancer is initiated by the hypoxic microenvironment. Cell Biol. Int. 2013, 37, 1139–1142. [Google Scholar] [CrossRef]
- Mitra, A.K.; Chiang, C.Y.; Tiwari, P.; Tomar, S.; Watters, K.M.; Peter, M.E.; Lengyel, E. Microenvironment-induced downregulation of miR-193b drives ovarian cancer metastasis. Oncogene 2015, 34, 5923–5932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindl, M.; Schoppmann, S.F.; Samonigg, H.; Hausmaninger, H.; Kwasny, W.; Gnant, M.; Jakesz, R.; Kubista, E.; Birner, P.; Oberhuber, G.; et al. Overexpression of hypoxia-inducible factor 1 alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 2002, 8, 1831–1837. [Google Scholar] [PubMed]
- Birner, P.; Schindl, M.; Obermair, A.; Plank, C.; Breitenecker, G.; Oberhuber, G. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000, 60, 4693–4696. [Google Scholar] [PubMed]
- Sun, L.; Li, H.; Chen, J.; Iwasaki, Y.; Kubota, T.; Matsuoka, M.; Shen, A.; Chen, Q.; Xu, Y. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J. Cell Sci. 2013, 126, 3939–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, S.; Foreman, K.M.; Soriano, M.I.; Rossen, N.S.; Shehade, H.; Fregoso, D.R.; Eggold, J.T.; Krishnan, V.; Dorigo, O.; Krieg, A.J.; et al. Collagen Remodeling in the Hypoxic Tumor-Mesothelial Niche Promotes Ovarian Cancer Metastasis. Cancer Res. 2019, 79, 2271–2284. [Google Scholar] [CrossRef] [Green Version]
- Compton, S.L.E.; Pyne, E.S.; Liu, L.; Guinan, J.; Shea, A.A.; Grieco, J.P.; Frisard, M.I.; Schmelz, E.M. Adaptation of metabolism to multicellular aggregation, hypoxia and obese stromal cell incorporation as potential measure of survival of ovarian metastases. Exp. Cell Res. 2020, 399, 112397. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.C.; D’Orazi, G.; Del Bufalo, D. Targeting hypoxia in tumor: A new promising therapeutic strategy. J. Exp. Clin. Cancer Res. 2020, 39, 8. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Hui, W.W.; Wang, J.J.; Yung, M.M.; Hui, L.M.; Qin, Y.; Liang, R.R.; Leung, T.H.; Xu, D.; Chan, K.K.; et al. DLX1 acts as a crucial target of FOXM1 to promote ovarian cancer aggressiveness by enhancing TGF-beta/SMAD4 signaling. Oncogene 2017, 36, 1404–1416. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Liu, V.W.; Tsao, G.S.; Yao, K.M.; Furukawa, T.; Chan, K.K.; Ngan, H.Y. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 2008, 29, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Mak, C.S.; Yung, M.M.; Hui, L.M.; Leung, L.L.; Liang, R.; Chen, K.; Liu, S.S.; Qin, Y.; Leung, T.H.; Lee, K.F.; et al. MicroRNA-141 enhances anoikis resistance in metastatic progression of ovarian cancer through targeting KLF12/Sp1/survivin axis. Mol. Cancer 2017, 16, 11. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1alpha and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhou, W.; Zhang, Y.; Sun, W.; Yung, M.M.H.; Sun, J.; Li, J.; Chen, C.W.; Li, Z.; Meng, Y.; et al. ERK Regulates HIF1alpha-Mediated Platinum Resistance by Directly Targeting PHD2 in Ovarian Cancer. Clin. Cancer Res. 2019, 26, 5947–5960. [Google Scholar] [CrossRef] [Green Version]
- Piva, R.; Belardo, G.; Santoro, M.G. NF-kappaB: A stress-regulated switch for cell survival. Antioxid. Redox Signal 2006, 8, 478–486. [Google Scholar] [CrossRef]
- Hernandez, L.; Hsu, S.C.; Davidson, B.; Birrer, M.J.; Kohn, E.C.; Annunziata, C.M. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Res. 2010, 70, 4005–4014. [Google Scholar] [CrossRef] [Green Version]
- Simoes, R.V.; Serganova, I.S.; Kruchevsky, N.; Leftin, A.; Shestov, A.A.; Thaler, H.T.; Sukenick, G.; Locasale, J.W.; Blasberg, R.G.; Koutcher, J.A.; et al. Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia 2015, 17, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Ling, Z.Q.; Zhao, T.; Fang, S.H.; Chang, W.C.; Lee, S.C.; Lee, K.R. Genomic-wide analysis of lymphatic metastasis-associated genes in human hepatocellular carcinoma. World J. Gastroenterol. 2009, 15, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wurmbach, E.; Waxman, S.; Jing, Y. Upregulation of Bfl-1/A1 in leukemia cells undergoing differentiation by all-trans retinoic acid treatment attenuates chemotherapeutic agent-induced apoptosis. Leukemia 2006, 20, 1009–1016. [Google Scholar] [CrossRef]
- Ottina, E.; Tischner, D.; Herold, M.J.; Villunger, A. A1/Bfl-1 in leukocyte development and cell death. Exp. Cell Res. 2012, 318, 1291–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, H.A.; Dogan, S.; Zillhardt, M.; Mitra, A.; Yamada, S.D.; Krausz, T.; Lengyel, E. Organotypic models of metastasis: A three-dimensional culture mimicking the human peritoneum and omentum for the study of the early steps of ovarian cancer metastasis. Cancer Treat. Res. 2009, 149, 335–351. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.M.; Funk, H.M.; Thiolloy, S.; Lotan, T.L.; Hickson, J.; Prins, G.S.; Drew, A.F.; Rinker-Schaeffer, C.W. In vitro metastatic colonization of human ovarian cancer cells to the omentum. Clin. Exp. Metastasis 2010, 27, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, N.; Marassi, F.M.; Newmeyer, D.D.; Hanein, D. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 2014, 21, 206–215. [Google Scholar] [CrossRef]
- Kønig, S.M.; Rissler, V.; Terkelsen, T.; Lambrughi, M.; Papaleo, E. Alterations of the interactome of Bcl-2 proteins in breast cancer at the transcriptional, mutational and structural level. PLoS Comput. Biol. 2019, 15, e1007485. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Elangovan, B.; Chinnadurai, G. Functional dissection of the pro-apoptotic protein Bik. Heterodimerization with anti-apoptosis proteins is insufficient for induction of cell death. J. Biol. Chem. 1997, 272, 24494–24498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef]
- Farghaly, S.A. Advances in Diagnosis and Management of Ovarian Cancer; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Koumenis, C.; Hammond, E.; Giaccia, A. Tumor Microenvironment and Cellular Stress; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Witz, I.P. Tumor–microenvironment interactions: Dangerous liaisons. Adv. Cancer Res. 2008, 100, 203–229. [Google Scholar]
- Kim, K.S.; Sengupta, S.; Berk, M.; Kwak, Y.G.; Escobar, P.F.; Belinson, J.; Mok, S.C.; Xu, Y. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006, 66, 7983–7990. [Google Scholar] [CrossRef] [Green Version]
- Cai, P.C.; Shi, L.; Liu, V.W.; Tang, H.W.; Liu, I.J.; Leung, T.H.; Chan, K.K.; Yam, J.W.; Yao, K.M.; Ngan, H.Y.; et al. Elevated TAK1 augments tumor growth and metastatic capacities of ovarian cancer cells through activation of NF-kappaB signaling. Oncotarget 2014, 5, 7549–7562. [Google Scholar] [CrossRef] [Green Version]
- Sen, R.; Baltimore, D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Zong, W.-X.; Edelstein, L.C.; Chen, C.; Bash, J.; Gélinas, C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-κB that blocks TNFα-induced apoptosis. Gene Dev. 1999, 13, 382–387. [Google Scholar] [CrossRef]
- Perkins, N.; Gilmore, T. Good cop, bad cop: The different faces of NF-κB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-κB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Vier, J.; Groth, M.; Sochalska, M.; Kirschnek, S. The anti-apoptotic Bcl-2 family protein A1/Bfl-1 regulates neutrophil survival and homeostasis and is controlled via PI3K and JAK/STAT signaling. Cell Death Dis. 2016, 7, e2103. [Google Scholar] [CrossRef] [PubMed]
- Hind, C.K.; Carter, M.J.; Harris, C.L.; Chan, H.T.; James, S.; Cragg, M.S. Role of the pro-survival molecule Bfl-1 in melanoma. Int. J. Biochem. Cell Biol. 2015, 59, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, M.J.; Fan, G.; Zong, W.X.; Degenhardt, K.; White, E.; Gelinas, C. Bfl-1/A1 functions, similar to Mcl-1, as a selective tBid and Bak antagonist. Oncogene 2008, 27, 1421–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, A.B.; de Vries, E.; Tait, S.W.G.; Bontjer, I.; Borst, J. Bcl-2 family member Bfl-1/A1 sequesters truncated bid to inhibit its collaboration with pro-apoptotic Bak or Bax. J. Biol. Chem. 2002, 277, 22781–22788. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Saez, A. The secrets of the Bcl-2 family. Cell Death Differ. 2012, 19, 1733–1740. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Apoptosis in Cancer. In Mechanisms, Deregulation, and Therapeutic Targeting; Elsevier: Amsterdam, The Netherlands, 2014; pp. 357–390. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Gene Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, W.A.; Ahad, A.; Ahsan, H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update. Arch. Toxicol. 2015, 89, 289–317. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, R.; Yung, M.M.H.; He, F.; Jiao, P.; Chan, K.K.L.; Ngan, H.Y.S.; Chan, D.W. The Stress-Inducible BCL2A1 Is Required for Ovarian Cancer Metastatic Progression in the Peritoneal Microenvironment. Cancers 2021, 13, 4577. https://doi.org/10.3390/cancers13184577
Liang R, Yung MMH, He F, Jiao P, Chan KKL, Ngan HYS, Chan DW. The Stress-Inducible BCL2A1 Is Required for Ovarian Cancer Metastatic Progression in the Peritoneal Microenvironment. Cancers. 2021; 13(18):4577. https://doi.org/10.3390/cancers13184577
Chicago/Turabian StyleLiang, Rui, Mingo M. H. Yung, Fangfang He, Peili Jiao, Karen K. L. Chan, Hextan Y. S. Ngan, and David W. Chan. 2021. "The Stress-Inducible BCL2A1 Is Required for Ovarian Cancer Metastatic Progression in the Peritoneal Microenvironment" Cancers 13, no. 18: 4577. https://doi.org/10.3390/cancers13184577