
Understanding the Radiobiology of Vestibular Schwannomas to Overcome Radiation Resistance

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. DNA Damage after Ionizing Radiation
2.1. DNA Oxidation and Oxidative Stress
2.2. DNA Single-Strand Breaks
2.3. DNA Double-Strand Breaks
2.4. Oxidative Clustered DNA Lesions
2.5. Markers of DNA Damage
3. DNA Repair after Ionizing Radiation
3.1. Homologous Recombination
3.2. Nonhomologous End-Joining Recombination
4. Cell Death after Ionizing Radiation
4.1. Apoptosis
4.1.1. Intrinsic Pathway
4.1.2. Extrinsic Pathway
4.2. Caspase-Dependent Cell Death
4.3. Caspase-Independent Cell Death
4.4. Necrosis
4.5. Autophagic Cell Death
4.6. Mitotic Catastrophe
4.7. Cellular Senescence
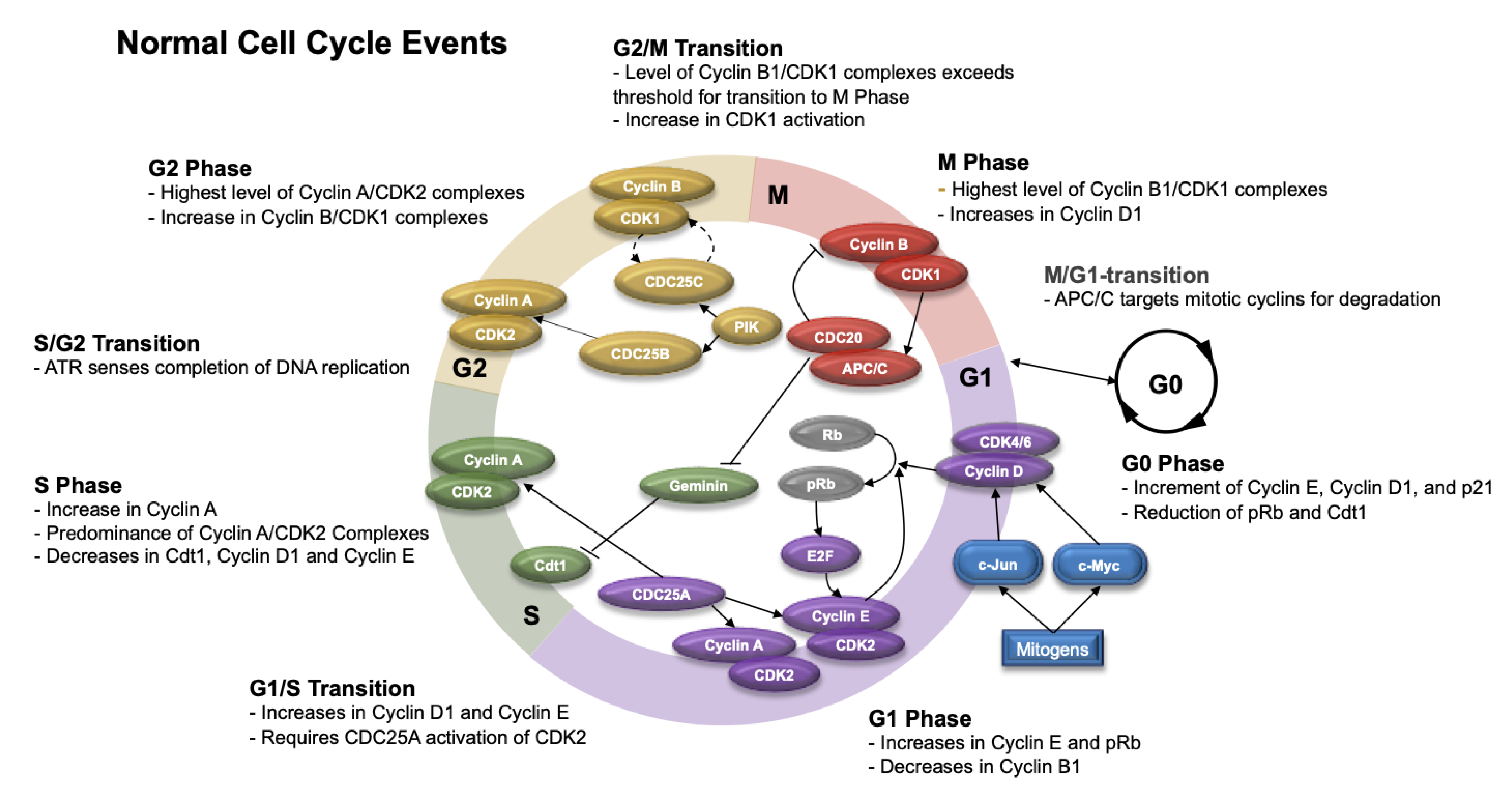
5. Cell Cycle after Ionizing Radiation
5.1. Normal Cell Cycle
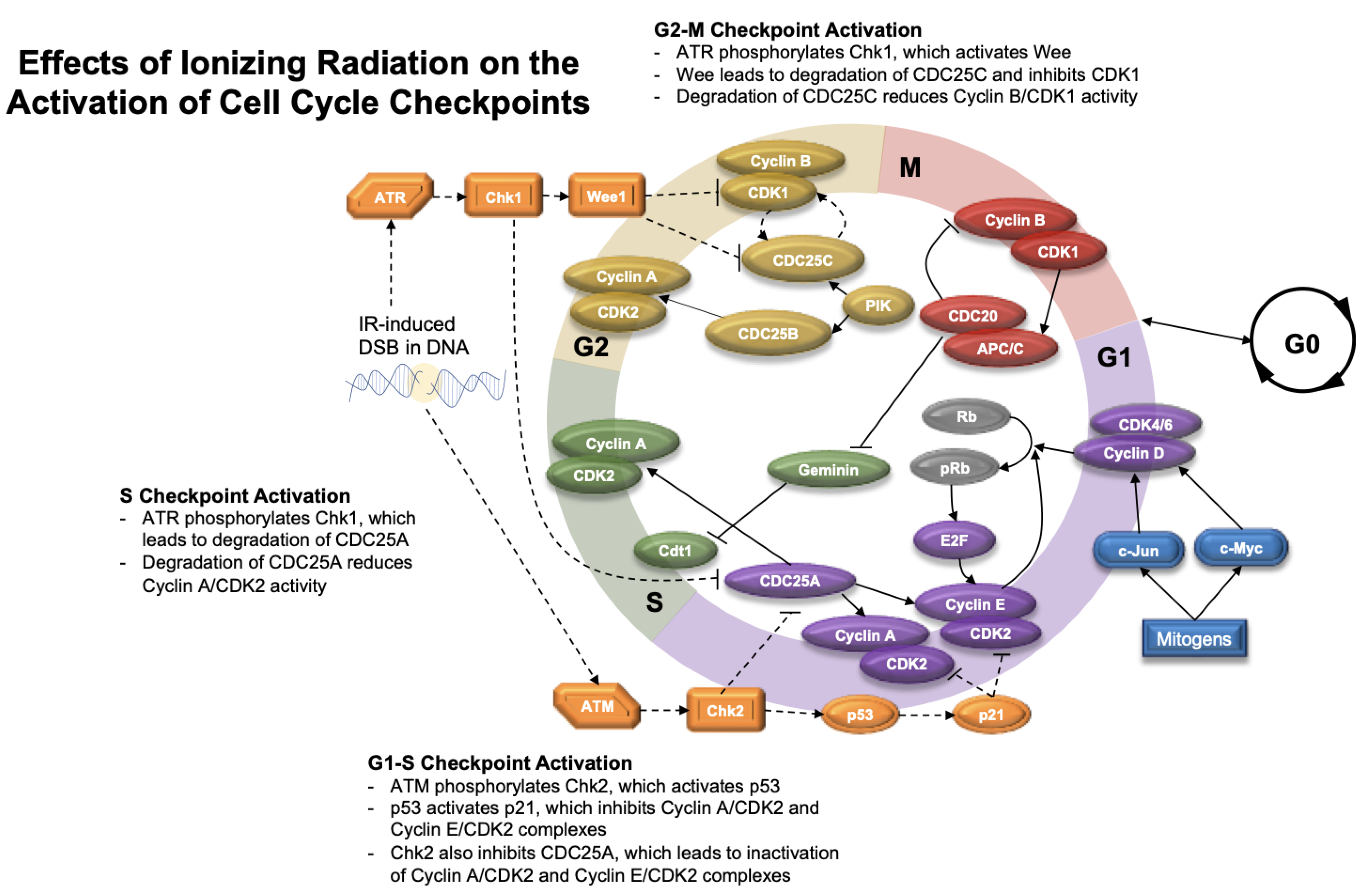
5.2. Cell Cycle Checkpoints after Radiation
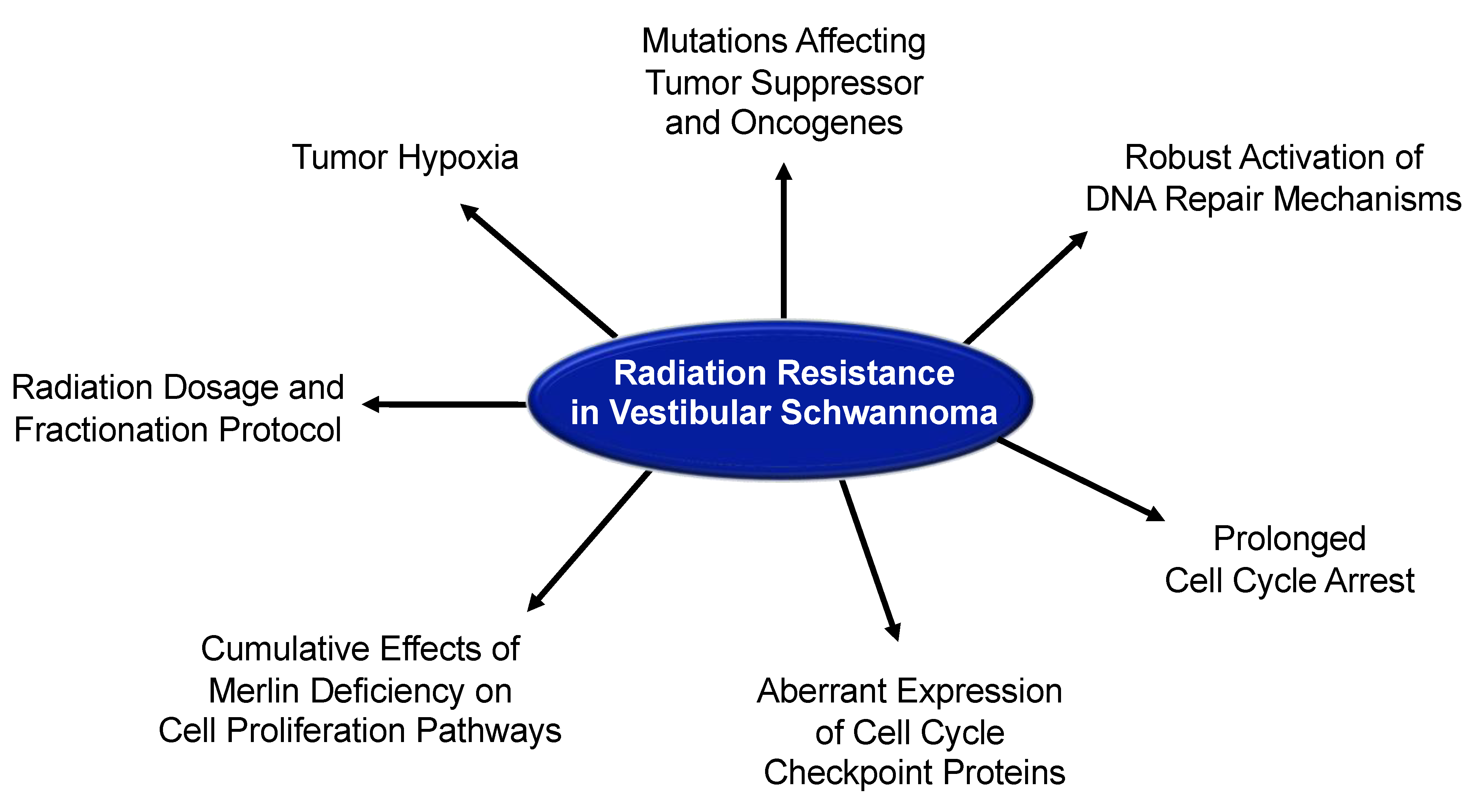
6. General Mechanisms of Radiation Resistance
7. Radiobiology and Radiation Resistance in Vestibular Schwannoma
7.1. Radiation Response in Patients with Vestibular Schwannoma
7.2. Tumor Growth Rate and Radiation Resistance in Vestibular Schwannoma
7.3. DNA Repair and Radiation Resistance in Vestibular Schwannoma
7.4. Tumor Vasculature and Radiation Resistance in Vestibular Schwannoma
7.5. Merlin Deficiency and Radiation Resistance in Vestibular Schwannoma
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stangerup, S.-E.; Caye-Thomasen, P.; Tos, M.; Thomsen, J. The Natural History of Vestibular Schwannoma. Otol. Neurotol. 2006, 27, 547–552. [Google Scholar] [CrossRef]
- Vellin, J.-F.; Grayeli, A.B.; Kalamarides, M.; Fond, C.; Bouccara, D.; Sterkers, O. Intratumoral and Brainstem Hemorrhage in a Patient with Vestibular Schwannoma and Oral Anticoagulant Therapy. Otol. Neurotol. 2006, 27, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Jufas, N. Sudden Death Due to Vestibular Schwannoma: Caution in emergent management. Otol. Neurotol. 2016, 37, 564–567. [Google Scholar] [CrossRef]
- Carlson, M.L.; Rn, N.M.T.; Driscoll, C.L.W.; Van Gompel, J.J.; Lane, J.I.; Raghunathan, A.; Flemming, K.D.; Link, M.J. Clinically significant intratumoral hemorrhage in patients with vestibular schwannoma. Laryngoscope 2016, 127, 1420–1426. [Google Scholar] [CrossRef]
- Jefferis, J.M.; Raoof, N.; Carroll, T.; Salvi, S.M. Optic nerve sheath fenestration in patients with visual failure associated with vestibular schwannoma. Br. J. Neurosurg. 2019, 33, 402–408. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Moran, A.; King, A.; Saeed, S.; Gurusinghe, N.; Ramsden, R. Incidence of Vestibular Schwannoma and Neurofibromatosis 2 in the North West of England over a 10-year Period: Higher Incidence than Previously Thought. Otol. Neurotol. 2005, 26, 93–97. [Google Scholar] [CrossRef]
- Marinelli, J.P.; Grossardt, B.R.; Lohse, C.M.; Carlson, M.L. Prevalence of Sporadic Vestibular Schwannoma: Reconciling Temporal Bone, Radiologic, and Population-based Studies. Otol. Neurotol. 2019, 40, 384–390. [Google Scholar] [CrossRef]
- Arthurs, B.J.; Fairbanks, R.K.; Demakas, J.J.; Lamoreaux, W.T.; Giddings, N.A.; Mackay, A.R.; Cooke, B.S.; Elaimy, A.L.; Lee, C.M. A review of treatment modalities for vestibular schwannoma. Neurosurg. Rev. 2011, 34, 265–279. [Google Scholar] [CrossRef]
- Lu, V.M.; Ravindran, K.; Graffeo, C.S.; Perry, A.; Van Gompel, J.J.; Daniels, D.J.; Link, M.J. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: A systematic review and meta-analysis of treatment outcomes. J. Neuro-Oncol. 2019, 144, 239–248. [Google Scholar] [CrossRef]
- Zou, J.; Hirvonen, T. “Wait and scan” management of patients with vestibular schwannoma and the relevance of non-contrast MRI in the follow-up. J. Otol. 2017, 12, 174–184. [Google Scholar] [CrossRef]
- Leon, J.; Lehrer, E.; Peterson, J.; Vallow, L.; Ruiz-Garcia, H.; Hadley, A.; Herchko, S.; Lundy, L.; Chaichana, K.; Vibhute, P.; et al. Observation or stereotactic radiosurgery for newly diagnosed vestibular schwannomas: A systematic review and meta-analysis. J. Radiosurg. SBRT 2019, 6, 91–100. [Google Scholar]
- Ansari, S.F.; Terry, C.; Cohen-Gadol, A. Surgery for vestibular schwannomas: A systematic review of complications by approach. Neurosurg. Focus 2012, 33, E14. [Google Scholar] [CrossRef] [Green Version]
- Darrouzet, V.; Martel, J.; Enée, V.; Bébéar, J.; Guérin, J. Vestibular Schwannoma Surgery Outcomes: Our Multidisciplinary Experience in 400 Cases Over 17 Years. Laryngoscope 2004, 114, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.K.; Ung, N.; Sheppard, J.P.; Nguyen, T.; Lagman, C.; Choy, W.; Tenn, S.; Pouratian, N.; Lee, P.; Kaprealian, T.; et al. Impact of Cochlear Dose on Hearing Preservation following Stereotactic Radiosurgery and Fractionated Stereotactic Radiotherapy for the Treatment of Vestibular Schwannoma. J. Neurol. Surg. Part B Skull Base 2017, 79, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Apicella, G.; Paolini, M.; Deantonio, L.; Masini, L.; Krengli, M. Radiotherapy for vestibular schwannoma: Review of recent literature results. Rep. Pract. Oncol. Radiother. 2016, 21, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, M.N.; Sahgal, A.; Xu, W.; De Salles, A.; Hayashi, M.; Levivier, M.; Ma, L.; Martinez, R.; Régis, J.; Ryu, S.; et al. Stereotactic radiosurgery for vestibular schwannoma: International Stereotactic Radiosurgery Society (ISRS) Practice Guideline. J. Radiosurg. SBRT 2017, 5, 5–24. [Google Scholar]
- Soltys, S.G.; Milano, M.T.; Xue, J.; Tomé, W.A.; Yorke, E.; Sheehan, J.; Ding, G.X.; Kirkpatrick, J.P.; Ma, L.; Sahgal, A.; et al. Stereotactic Radiosurgery for Vestibular Schwannomas: Tumor Control Probability Analyses and Recommended Reporting Standards. Int. J. Radiat. Oncol. 2021, 110, 100–111. [Google Scholar] [CrossRef]
- Dupic, G.; Urcissin, M.; Mom, T.; Verrelle, P.; Dedieu, V.; Molnar, I.; El-Ouadih, Y.; Chassin, V.; Lapeyre, M.; Lemaire, J.-J.; et al. Stereotactic Radiosurgery for Vestibular Schwannomas: Reducing Toxicity with 11 Gy as the Marginal Prescribed Dose. Front. Oncol. 2020, 10, 598841. [Google Scholar] [CrossRef]
- Watanabe, S.; Yamamoto, M.; Kawabe, T.; Koiso, T.; Yamamoto, T.; Matsumura, A.; Kasuya, H. Stereotactic radiosurgery for vestibular schwannomas: Average 10-year follow-up results focusing on long-term hearing preservation. J. Neurosurg. 2016, 125 (Suppl. 1), 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischer, J.; Gruber, E.; Schöffmann, V.; Ertl, A.; Höftberger, R.; Mallouhi, A.; Wolfsberger, S.; Arnoldner, C.; Eisner, W.; Knosp, E.; et al. Long-term outcome after Gamma Knife radiosurgery for acoustic neuroma of all Koos grades: A single-center study. J. Neurosurg. 2019, 130, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.S.; Barnett, G.H.; Vogelbaum, M.A.; Neyman, G.; Stevens, G.H.J.; Cohen, B.H.; Elson, P.; Vassil, A.D.; Suh, J.H. Long-term outcomes of Gamma Knife radiosurgery in patients with vestibular schwannomas. J. Neurosurg. 2011, 114, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M.L.; Jacob, J.T.; Pollock, B.E.; Neff, B.A.; Tombers, N.M.; Driscoll, C.L.W.; Link, M.J. Long-term hearing outcomes following stereotactic radiosurgery for vestibular schwannoma: Patterns of hearing loss and variables influencing audiometric decline. J. Neurosurg. 2013, 118, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-W.; Tu, H.-T.; Chang, C.-S.; Chou, H.-H.; Lee, M.-T. Gamma Knife radiosurgery for large vestibular schwannomas greater than 3 cm in diameter. J. Neurosurg. 2018, 128, 1380–1387. [Google Scholar] [CrossRef] [Green Version]
- Breshears, J.D.; Chang, J.; Molinaro, A.M.; Sneed, P.K.; McDermott, M.W.; Tward, A.; Theodosopoulos, P.V. Temporal Dynamics of Pseudoprogression After Gamma Knife Radiosurgery for Vestibular Schwannomas—A Retrospective Volumetric Study. Neurosurgery 2018, 84, 123–131. [Google Scholar] [CrossRef]
- Hayhurst, C.; Zadeh, G. Tumor pseudoprogression following radiosurgery for vestibular schwannoma. Neuro-Oncol. 2012, 14, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djalilian, H.R.; Benson, A.G.; Ziai, K.; Safai, Y.; Thakkar, K.H.; Mafee, M.F. Radiation necrosis of the brain after radiosurgery for vestibular schwannoma. Am. J. Otolaryngol. 2007, 28, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, W.R.; Carlson, M.L.; Giannini, C.; Driscoll, C.L.; Link, M.J. Radiation-Induced Sarcoma in a Large Vestibular Schwannoma Following Stereotactic Radiosurgery: Case Report. Neurosurgery 2011, 68, E840–E846, discussion E846. [Google Scholar] [CrossRef]
- Boucher, A.B.; Mendoza, P.; Neill, S.G.; Eaton, B.; Olson, J.J. High-Grade Sarcoma Arising within a Previously Irradiated Vestibular Schwannoma: A Case Report and Literature Review. World Neurosurg. 2020, 144, 99–105. [Google Scholar] [CrossRef]
- Demetriades, A.K.; Saunders, N.; Rose, P.; Fisher, C.; Rowe, J.; Tranter, R.; Hardwidge, C. Malignant Transformation of Acoustic Neuroma/Vestibular Schwannoma 10 Years after Gamma Knife Stereotactic Radiosurgery. Semin. Neurol. 2010, 20, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Langenhuizen, P.P.J.H.; Zinger, S.; Hanssens, P.E.J.; Kunst, H.P.M.; Mulder, J.J.S.; Leenstra, S.; De With, P.H.N.; Verheul, J.B. Influence of pretreatment growth rate on Gamma Knife treatment response for vestibular schwannoma: A volumetric analysis. J. Neurosurg. 2019, 131, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Marston, A.P.; Jacob, J.T.; Carlson, M.L.; Pollock, B.E.; Driscoll, C.L.W.; Link, M.J. Pretreatment growth rate as a predictor of tumor control following Gamma Knife radiosurgery for sporadic vestibular schwannoma. J. Neurosurg. 2017, 127, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, N.N.; Niemierko, A.; Larvie, M.; Curtin, H.; Loeffler, J.S.; McKenna, M.J.; Shih, H.A. Pretreatment Growth Rate Predicts Radiation Response in Vestibular Schwannomas. Int. J. Radiat. Oncol. 2014, 89, 113–119. [Google Scholar] [CrossRef]
- Klijn, S.; Verheul, J.B.; Beute, G.N.; Leenstra, S.; Mulder, J.J.S.; Kunst, H.P.M.; Hanssens, P.E.J. Gamma Knife radiosurgery for vestibular schwannomas: Evaluation of tumor control and its predictors in a large patient cohort in The Netherlands. J. Neurosurg. 2016, 124, 1619–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Liu, A. Long-term follow-up studies of Gamma Knife surgery for patients with neurofibromatosis Type 2. J. Neurosurg. 2014, 121, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Kruyt, I.J.; Verheul, J.B.; Hanssens, P.E.J.; Kunst, H.P.M. Gamma Knife radiosurgery for treatment of growing vestibular schwannomas in patients with neurofibromatosis Type 2: A matched cohort study with sporadic vestibular schwannomas. J. Neurosurg. 2018, 128, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.S.; Singh, R.; Kale, S.S.; Agrawal, D.; Sharma, B.S.; Mahapatra, A.K. Tumor control and hearing preservation after Gamma Knife radiosurgery for vestibular schwannomas in neurofibromatosis type 2. J. Neuro-Oncol. 2010, 98, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.; Zhang, M.; Li, A.; Thompson, P.A.; Tayag, A.T.; Wallach, J.; Gibbs, I.C.; Soltys, S.G.; Hancock, S.L.; Chang, S.D. The Outcome of Hypofractionated Stereotactic Radiosurgery for Large Vestibular Schwannomas. World Neurosurg. 2016, 93, 398–409. [Google Scholar] [CrossRef]
- Lee, D.J.; Westra, W.H.; Staecker, H.; Long, D.; Niparko, J.K. Clinical and Histopathologic Features of Recurrent Vestibular Schwannoma (Acoustic Neuroma) after Stereotactic Radiosurgery. Otol. Neurotol. 2003, 24, 650–660, discussion 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, Y.; Fukushima, T.; Watanabe, K.; Friedman, A.H.; Iii, C.D.C.; Zomorodi, A.R. Surgical management of vestibular schwannomas after failed radiation treatment. Neurosurg. Rev. 2016, 39, 303–312, discussion 312. [Google Scholar] [CrossRef]
- Wise, S.C.; Carlson, M.L.; Tveiten, Ø.V.; Driscoll, C.L.; Myrseth, E.; Lund-Johansen, M.; Link, M.J. Surgical salvage of recurrent vestibular schwannoma following prior stereotactic radiosurgery. Laryngoscope 2016, 126, 2580–2586. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.-L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edumont, E.; Monari, A. Understanding DNA under oxidative stress and sensitization: The role of molecular modeling. Front. Chem. 2015, 3, 43. [Google Scholar]
- Chepelev, N.L.; Kennedy, D.A.; Gagne, R.; White, T.; Long, A.S.; Yauk, C.; White, P.A. HPLC Measurement of the DNA Oxidation Biomarker, 8-oxo-7,8-dihydro-2′-deoxyguanosine, in Cultured Cells and Animal Tissues. J. Vis. Exp. 2015, 2015, e52697. [Google Scholar]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of Ionizing Radiation on Biological Molecules—Mechanisms of Damage and Emerging Methods of Detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; Yang, E.S. The Impact of DNA Repair Pathways in Cancer Biology and Therapy. Cancers 2017, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- El-Khamisy, S.F.; Hartsuiker, E.; Caldecott, K.W. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair 2007, 6, 1485–1495. [Google Scholar] [CrossRef]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy—A double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Starcher, C.L.; Pay, S.L.; Singh, N.; Yeh, I.-J.; Bhandare, S.B.; Su, X.; Huang, X.; Bey, E.A.; Motea, E.A.; Boothman, D.A. Targeting Base Excision Repair in Cancer: NQO1-Bioactivatable Drugs Improve Tumor Selectivity and Reduce Treatment Toxicity Through Radiosensitization of Human Cancer. Front. Oncol. 2020, 10, 1575. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Heeres, J.T.; Hergenrother, P.J. Poly(ADP-ribose) makes a date with death. Curr. Opin. Chem. Biol. 2007, 11, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.-J.; Elosta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltiel, I.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hayashi, N.; Takata, M.; Yamamoto, K.-I. NBS1 directly activates ATR independently of MRE11 and TOPBP1. Genes Cells 2013, 18, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX Is Phosphorylated in an ATR-dependent Manner in Response to Replicational Stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Soutoglou, E.; Dorn, J.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007, 9, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, T.C.; El-Osta, A. Epigenetic changes activate widespread signals in response to doublestrand breaks. Cancer Biol. Ther. 2004, 3, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free. Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Gollapalle, E.; Wang, R.; Adetolu, R.; Tsao, D.; Francisco, D.; Sigounas, G.; Georgakilas, A.G. Detection of Oxidative Clustered DNA Lesions in X-Irradiated Mouse Skin Tissues and Human MCF-7 Breast Cancer Cells. Radiat. Res. 2007, 167, 207–216. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Pilch, D.R.; Redon, C.; Bonner, W.M.; Martin, O.A. Involvement of H2AX in the DNA Damage and Repair Response. Cancer Biol. Ther. 2003, 2, 233–235. [Google Scholar] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. -H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Howley, P.M.; Israel, M.A.; Gray, J.W.; Thompson, C. The Molecular Basis of Cancer, 4th ed.; Saunders/Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Ahmed, E.A.; Rosemann, M.; Scherthan, H. NHEJ Contributes to the Fast Repair of Radiation-induced DNA Double-strand Breaks at Late Prophase I Telomeres. Health Phys. 2018, 115, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genom. Integr. 2012, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Reilly, N.M.; Yard, B.D.; Pittman, D.L. Homologous Recombination-Mediated DNA Repair and Implications for Clinical Treatment of Repair Defective Cancers. Breast Cancer 2019, 1999, 3–29. [Google Scholar]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Deriano, L.; Roth, D.B. Modernizing the Nonhomologous End-Joining Repertoire: Alternative and Classical NHEJ Share the Stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Ha, K.; Kim, M.-S.; Noh, Y.-W.; Lin, H.; Tang, L.; Zhu, Q.; Zhang, D.; Chen, H.; Han, S.; et al. The anaphase promoting complex promotes NHEJ repair through stabilizing Ku80 at DNA damage sites. Cell Cycle 2018, 17, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Ochi, T.; Wu, Q.; Blundell, T.L. The spatial organization of non-homologous end joining: From bridging to end joining. DNA Repair 2014, 17, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Käshammer, L.; Saathoff, J.-H.; Lammens, K.; Gut, F.; Bartho, J.; Alt, A.; Kessler, B.; Hopfner, K.-P. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell 2019, 76, 382–394.e6. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, H.; Kodama, S.; Matsuda, N.; Suzuki, K.; Watanabe, M. Involvement of Reactive Oxygen Species (ROS) in the Induction of Genetic Instability by Radiation. J. Radiat. Res. 2004, 45, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Bhatt, D.; Oltvai, Z.N.; Greenberger, J.S.; Bahar, I. Significance of p53 dynamics in regulating apoptosis in response to ionizing radiation and polypharmacological strategies. Sci. Rep. 2014, 4, srep06245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-L.; Blum, J.M.; Kirsch, D.G. Role of p53 in regulating tissue response to radiation by mechanisms independent of apoptosis. Transl. Cancer Res. 2013, 2, 412–421. [Google Scholar]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.-L. Modulating Both Tumor Cell Death and Innate Immunity Is Essential for Improving Radiation Therapy Effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef]
- Dinh, C.T.; Goncalves, S.; Bas, E.; Van De Water, T.R.; Zine, A. Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma. Front. Cell. Neurosci. 2015, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, L. No PUMA, no death: Implications for p53-dependent apoptosis. Cancer Cell 2003, 4, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Caspase-dependent and -independent cell death pathways after DNA damage (Review). Oncol. Rep. 2005, 14, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [Green Version]
- Dickens, L.S.; Powley, I.R.; Hughes, M.A.; MacFarlane, M. The ‘complexities’ of life and death: Death receptor signalling platforms. Exp. Cell Res. 2012, 318, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.; Cain, K.; MacFarlane, M. A Death Effector Domain Chain DISC Model Reveals a Crucial Role for Caspase-8 Chain Assembly in Mediating Apoptotic Cell Death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Goldschneider, D.; Mehlen, P. Dependence receptors: A new paradigm in cell signaling and cancer therapy. Oncogene 2010, 29, 1865–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratton, S.B.; Walker, G.; Srinivasula, S.M.; Sun, X.; Butterworth, M.; Alnemri, E.S.; Cohen, G.M. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001, 20, 998–1009. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nat. Cell Biol. 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Sahara, S.; Aoto, M.; Eguchi, Y.; Imamoto, N.; Yoneda, Y.; Tsujimoto, Y. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nat. Cell Biol. 1999, 401, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics, M.; Martinon, F.; Micheau, O.; Bodmer, J.-L.; Hofmann, K.; Tschopp, J. Overexpression of Helicard, a CARD-Containing Helicase Cleaved during Apoptosis, Accelerates DNA Degradation. Curr. Biol. 2002, 12, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Denault, J.-B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. XIAP inhibits caspase-3 and -7 using two binding sites: Evolutionarily conserved mechanism of IAPs. EMBO J. 2005, 24, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V. Mitochondrial and Nuclear Cross Talk in Cell Death: Parthanatos. Ann. New York Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prévost, M.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Takahashi-Niki, K.; Akagi, T.; Hashikawa, T.; Takahashi, R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2003, 11, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef] [Green Version]
- Borges, H.; Linden, R.; Wang, J.Y. DNA damage-induced cell death: Lessons from the central nervous system. Cell Res. 2007, 18, 17–26. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [Green Version]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.; Ii, G.W.D.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nat. Cell Biol. 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Lieberman, B.; Kirshenbaum, L.A. Cyclophilin D phosphorylation is critical for mitochondrial calcium uniporter regulated permeability transition pore sensitivity. Cardiovasc. Res. 2018, 115, 261–263. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- McComb, S.; Cheung, H.H.; Korneluk, R.G.; Wang, S.; Krishnan, L.; Sad, S. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 2012, 19, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silke, J.; Strasser, A. The FLIP Side of Life. Sci. Signal. 2013, 6, pe2. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Romero, H.; Ros, U.; Garcia-Saez, A.J. Pore formation in regulated cell death. EMBO J. 2020, 39, e105753. [Google Scholar] [CrossRef]
- Shimizu, S. Autophagic cell death and cancer chemotherapeutics. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 219–226. [Google Scholar]
- Eliopoulos, A.; Havaki, S.; Gorgoulis, V. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.-L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.-L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef]
- Mansilla, S.; Priebe, W.; Portugal, J. Mitotic Catastrophe Results in Cell Death by Caspase-Dependentand Caspase-Independent Mechanisms. Cell Cycle 2005, 5, 53–60. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; You, L.; Xue, J.; Lu, Y. Ionizing Radiation-Induced Cellular Senescence in Normal, Non-transformed Cells and the Involved DNA Damage Response: A Mini Review. Front. Pharmacol. 2018, 9, 522. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.-I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.-P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological Adjustment of Senescent Cells by Modulating Caveolin-1 Status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Golias, C.; Charalabopoulos, A. Cell proliferation and cell cycle control: A mini review. Int. J. Clin. Pract. 2004, 58, 1134–1141. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Dean, D.C. Rb function in cell-cycle regulation and apoptosis. Nat. Cell Biol. 2000, 2, E65–E67. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef]
- Topacio, B.R.; Zatulovskiy, E.; Cristea, S.; Xie, S.; Tambo, C.S.; Rubin, S.M.; Sage, J.; Kõivomägi, M.; Skotheim, J.M. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol. Cell 2019, 74, 758–770.e754. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [Green Version]
- Boudolf, V.; Lammens, T.; Boruc, J.; Van Leene, J.; Daele, H.V.D.; Maes, S.; Van Isterdael, G.; Russinova, E.; Kondorosi, E.; Witters, E.; et al. CDKB1;1 Forms a Functional Complex with CYCA2;3 to Suppress Endocycle Onset. Plant Physiol. 2009, 150, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, E.V. The role of c-myc in cellular growth control. Oncogene 1999, 18, 2988–2996. [Google Scholar] [CrossRef] [Green Version]
- Shen, T. The Role of Cdc25A in the Regulation of Cell Proliferation and Apoptosis. Anti-Cancer Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef]
- Gookin, S.; Min, M.; Phadke, H.; Chung, M.; Moser, J.; Miller, I.; Carter, D.; Spencer, S.L. A map of protein dynamics during cell-cycle progression and cell-cycle exit. PLoS Biol. 2017, 15, e2003268. [Google Scholar] [CrossRef] [Green Version]
- Buck, S.B.; Bradford, J.; Gee, K.R.; Agnew, B.J.; Clarke, S.T.; Salic, A. Detection of S-phase cell cycle progression using 5-ethynyl-2′-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2′-deoxyuridine antibodies. Biotechniques 2008, 44, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, 2045. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Stacey, D.W.; Hitomi, M. Post-transcriptional regulation of cyclin D1 expression during G2 phase. Oncogene 2002, 21, 7545–7556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhang, P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Qiao, R.; Weissmann, F.; Yamaguchi, M.; Brown, N.; VanderLinden, R.; Imre, R.; Jarvis, M.A.; Brunner, M.R.; Davidson, I.F.; Litos, G.; et al. Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2016, 113, E2570–E2578. [Google Scholar] [CrossRef] [Green Version]
- Clijsters, L.; Ogink, J.; Wolthuis, R.M. The spindle checkpoint, APC/CCdc20, and APC/CCdh1 play distinct roles in connecting mitosis to S phase. J. Cell Biol. 2013, 201, 1013–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarry, T.J.; Kirschner, M.W. Geminin, an Inhibitor of DNA Replication, Is Degraded during Mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.; Bagella, L.; Tutton, S.; Romano, G.; Giordano, A. From G0 to S phase: A view of the roles played by the retinoblastoma (Rb) family members in the Rb-E2F pathway. J. Cell. Biochem. 2007, 102, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to Cell Cycle Regulation by the Chk2 Protein Kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nat. Cell Biol. 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef]
- Gamper, A.M.; Rofougaran, R.; Watkins, S.C.; Greenberger, J.S.; Beumer, J.H.; Bakkenist, C.J. ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 2013, 41, 10334–10344. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Block, W.D.; Lees-Miller, S.P. The role of ATM and ATR in DNA damage-induced cell cycle control. Prog. Cell Cycle Res. 2003, 5, 393–411. [Google Scholar] [PubMed]
- Pandita, T.K.; Lieberman, H.B.; Lim, D.-S.; Dhar, S.; Zheng, W.; Taya, Y.; Kastan, M.B. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 2000, 19, 1386–1391. [Google Scholar] [CrossRef] [Green Version]
- Bertino, J.R. Encyclopedia of Cancer, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Shimono, H.; Kaida, A.; Homma, H.; Nojima, H.; Onozato, Y.; Harada, H.; Miura, M. Fluctuation in radioresponse of HeLa cells during the cell cycle evaluated based on micronucleus frequency. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Liu, C.; Nie, J.; Wang, R.; Mao, W. The Cell Cycle G2/M Block Is an Indicator of Cellular Radiosensitivity. Dose-Response 2019, 17, 1559325819891008. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 1–27. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Y.; Mei, J.; Li, S.; Xu, H.; Xiong, H.; Wang, X.; He, X. Targeting RAD50 increases sensitivity to radiotherapy in colorectal cancer cells. Neoplasma 2018, 65, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Abbott, D.; Thompson, M.E.; Robinson-Benion, C.; Tomlinson, G.; Jensen, R.A.; Holt, J.T. BRCA1 Expression Restores Radiation Resistance in BRCA1-defective Cancer Cells through Enhancement of Transcription-coupled DNA Repair. J. Biol. Chem. 1999, 274, 18808–18812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negroni, A.; Stronati, L.; Grollino, M.G.; Barattini, P.; Gumiero, D.; Danesi, D.T. Radioresistance in a tumour cell line correlates with radiation inducible Ku 70/80 end-binding activity. Int. J. Radiat. Biol. 2008, 84, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Hu, Y.; Zhang, Y.; Peng, T.; Ji, W. Effect of Ku70 expression on radiosensitivity in renal carcinoma 786-O cells. Cancer Cell Int. 2014, 14, 44. [Google Scholar] [CrossRef] [Green Version]
- Shintani, S.; Mihara, M.; Li, C.; Nakahara, Y.; Hino, S.; Nakashiro, K.-I.; Hamakawa, H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003, 94, 894–900. [Google Scholar] [CrossRef]
- Velegzhaninov, I.O.; Belykh, E.S.; Rasova, E.E.; Pylina, Y.I.; Shadrin, D.M.; Klokov, D.Y. Radioresistance, DNA Damage and DNA Repair in Cells With Moderate Overexpression of RPA1. Front. Genet. 2020, 11, 855. [Google Scholar] [CrossRef]
- Jun, S.; Jung, Y.-S.; Na Suh, H.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 10994. [Google Scholar] [CrossRef] [Green Version]
- Aebersold, D.M.; Burri, P.; Beer, K.T.; Laissue, J.; Djonov, V.; Greiner, R.H.; Semenza, G.L. Expression of hypoxia-inducible factor-1alpha: A novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001, 61, 2911–2916. [Google Scholar] [PubMed]
- Menezes, A.; dos Reis, G.H.; Oliveira-Nunes, M.C.; Mariath, F.; Cabanel, M.; Pontes, B.; Castro, N.G.; de Brito, J.M.; Carneiro, K. Live Cell Imaging Supports a Key Role for Histone Deacetylase as a Molecular Target during Glioblastoma Malignancy Downgrade through Tumor Competence Modulation. J. Oncol. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PosthumaDeBoer, J.; Würdinger, T.; Graat, H.C.; van Beusechem, V.W.; Helder, M.N.; van Royen, B.J.; Kaspers, G.J. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer 2011, 11, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Fan, M.; Candas, D.; Qin, L.; Zhang, X.; Eldridge, A.; Zou, J.X.; Zhang, T.; Juma, S.; Jin, C.; et al. CDK1-Mediated SIRT3 Activation Enhances Mitochondrial Function and Tumor Radioresistance. Mol. Cancer Ther. 2015, 14, 2090–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-J.; Wu, S.-P.; Liu, J.-B.; Shi, Y.-S.; Huang, X.; Zhang, Q.-B.; Yao, K.-T. MYC Regulation of CHK1 and CHK2 Promotes Radioresistance in a Stem Cell-like Population of Nasopharyngeal Carcinoma Cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Alsubhi, N.; Middleton, F.; Abdel-Fatah, T.M.; Stephens, P.; Doherty, R.; Arora, A.; Moseley, P.; Chan, S.; Aleskandarany, M.; Green, A.; et al. Chk1 phosphorylated at serine345 is a predictor of early local recurrence and radio-resistance in breast cancer. Mol. Oncol. 2015, 10, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.; Pena, S.; Mei, C.; Bracho, O.; Marples, B.; Elsayyad, N.; Goncalves, S.; Ivan, M.; Monje, P.V.; Liu, X.-Z.; et al. Merlin-Deficient Schwann Cells Are More Susceptible to Radiation Injury than Normal Schwann Cells In Vitro. J. Neurol. Surg. Part B Skull Base 2021. [Google Scholar] [CrossRef]
- Deckbar, D.; Jeggo, P.A.; Löbrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.C.; Komata, T.; Kondo, Y.; Kanzawa, T.; Kondo, S.; Germano, I.M. Molecular response of human glioblastoma multiforme cells to ionizing radiation: Cell cycle arrest, modulation of the expression of cyclin-dependent kinase inhibitors, and autophagy. J. Neurosurg. 2003, 98, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.H.; Sughrue, M.E.; Kane, A.J.; Tihan, T.; Cheung, S.W.; Parsa, A.T. Radiobiology of vestibular schwannomas: Mechanisms of radioresistance and potential targets for therapeutic sensitization. Neurosurg. Focus 2009, 27, E2. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Bakanauskas, V.J.; Cerniglia, G.J.; Cheng, Y.; Bernhard, E.J.; Muschel, R.J.; McKenna, W.G. The Ras radiation resistance pathway. Cancer Res. 2001, 61, 4278–4282. [Google Scholar] [PubMed]
- Fernandez, L.A.; Squatrito, M.; Northcott, P.; Awan, A.; Holland, E.C.; Taylor, M.; Nahlé, Z.; Kenney, A.M. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene 2011, 31, 1923–1937. [Google Scholar] [CrossRef] [Green Version]
- Kasid, U.; Pfeifer, A.; Weichselbaum, R.; Dritschilo, A.; Mark, G. The raf oncogene is associated with a radiation-resistant human laryngeal cancer. Science 1987, 237, 1039–1041. [Google Scholar] [CrossRef]
- Hein, A.L.; Ouellette, M.M.; Yan, Y. Radiation-induced signaling pathways that promote cancer cell survival (Review). Int. J. Oncol. 2014, 45, 1813–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanità, P.; Monache, S.D.; Musio, D.; De Felice, F.; di Cesare, E.; et al. c-Myc Sustains Transformed Phenotype and Promotes Radioresistance of Embryonal Rhabdomyosarcoma. Cell Lines Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef]
- Lee, J.M.; Bernstein, A. p53 mutations increase resistance to ionizing radiation. Proc. Natl. Acad. Sci. USA 1993, 90, 5742–5746. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.R.; Loke, W.K. Molecular mechanisms of low dose ionizing radiation-induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int. J. Radiat. Biol. 2014, 91, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Weichselbaum, R.R.; Beckett, M.A.; Vokes, E.E.; Brachman, D.G.; Haraf, D.; Hallahan, D.; Kufe, D. Cellular and molecular mechanisms of radioresistance. Cancer Treat. Res. 1995, 74, 131–140. [Google Scholar]
- Okaichi, K.; Nose, K.; Kotake, T.; Izumi, N.; Kudo, T. Phosphorylation of p53 modifies sensitivity to ionizing radiation. Anticancer. Res. 2011, 31, 2255–2258. [Google Scholar]
- Chiang, T.-M.; Sawyers, C.L.; Bride, M. Oncogene Expression and Cellular Radiation Resistance: A Modulatory Role for c-myc. Mol. Diagn. 1998, 3, 21–27. [Google Scholar] [CrossRef]
- Christensen, M.; Najy, A.J.; Snyder, M.; Movilla, L.S.; Kim, H.-R.C. A Critical Role of the PTEN/PDGF Signaling Network for the Regulation of Radiosensitivity in Adenocarcinoma of the Prostate. Int. J. Radiat. Oncol. 2014, 88, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Lawrence, T.S.; Xu, L. Overcoming cancer therapy resistance by targeting inhibitors of apoptosis proteins and nuclear factor-kappa B. Am. J. Transl. Res. 2009, 1, 1–15. [Google Scholar] [PubMed]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Chiao, P.J.; Andreeff, M.; Meyn, R.E. Inhibition of constitutively activated nuclear factor-kappaB radiosensitizes human melanoma cells. Mol. Cancer Ther. 2004, 3, 985–992. [Google Scholar] [PubMed]
- Essmann, F.; Engels, I.H.; Totzke, G.; Schulze-Osthoff, K.; Jänicke, R.U. Apoptosis Resistance of MCF-7 Breast Carcinoma Cells to Ionizing Radiation Is Independent of p53 and Cell Cycle Control but Caused by the Lack of Caspase-3 and a Caffeine-Inhibitable Event. Cancer Res. 2004, 64, 7065–7072. [Google Scholar] [CrossRef] [Green Version]
- Winter, R.N.; Rhee, J.G.; Kyprianou, N. Caspase-1 enhances the apoptotic response of prostate cancer cells to ionizing radiation. Anticancer. Res. 2004, 24, 1377–1386. [Google Scholar] [PubMed]
- Osato, K.; Sato, Y.; Ochiishi, T.; Osato, A.; Zhu, C.; Sato, M.; Swanpalmer, J.; Modjtahedi, N.; Kroemer, G.; Kuhn, H.-G.; et al. Apoptosis-inducing factor deficiency decreases the proliferation rate and protects the subventricular zone against ionizing radiation. Cell Death Dis. 2010, 1, e84. [Google Scholar] [CrossRef]
- Leon, J.; Trifiletti, D.M.; Waddle, M.R.; Vallow, L.; Ko, S.; May, B.; Tzou, K.; Garcia, H.R.; Lundy, L.; Chaichana, K.; et al. Trends in the initial management of vestibular schwannoma in the United States. J. Clin. Neurosci. 2019, 68, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Kano, H.; Faramand, A.; Pease, M.; Nakamura, A.; Hassib, M.; Spencer, D.; Sisterson, N.; Faraji, A.H.; Arai, Y.; et al. Long term results of primary radiosurgery for vestibular schwannomas. J. Neuro-Oncol. 2019, 145, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Saadatmand, H.J.; Wu, C.-C.; Black, P.J.; Wuu, Y.-R.; Lesser, J.; Horan, M.; Isaacson, S.R.; Wang, T.J.C.; Sisti, M.B. Treatment Outcomes and Dose Rate Effects Following Gamma Knife Stereotactic Radiosurgery for Vestibular Schwannomas. Neurosurgery 2019, 85, E1084–E1094. [Google Scholar] [CrossRef] [Green Version]
- Kaylie, D.M.; Horgan, M.J.; Delashaw, J.B.; McMenomey, S.O. A Meta-analysis Comparing Outcomes of Microsurgery and Gamma Knife Radiosurgery. Laryngoscope 2000, 110, 1850–1856. [Google Scholar] [CrossRef]
- Watanabe, S.; Yamamoto, M.; Kawabe, T.; Koiso, T.; Aiyama, H.; Kasuya, H.; Barfod, B.E. Long-term follow-up results of stereotactic radiosurgery for vestibular schwannomas larger than 8 cc. Acta Neurochir. 2019, 161, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Foote, R.L.; Coffey, R.J.; Swanson, J.W.; Harner, S.G.; Beatty, C.W.; Kline, R.W.; Stevens, L.N.; Hu, T.C. Stereotactic radiosurgery using the gamma knife for acoustic neuromas. Int. J. Radiat. Oncol. 1995, 32, 1153–1160. [Google Scholar] [CrossRef]
- Kondziolka, D.; Lunsford, L.D.; McLaughlin, M.R.; Flickinger, J. Long-Term Outcomes after Radiosurgery for Acoustic Neuromas. N. Engl. J. Med. 1998, 339, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.R.; Clark, J.J.; Gantz, B.J.; Goswami, P.C. Effects of ErbB2 Signaling on the Response of Vestibular Schwannoma Cells to Gamma-Irradiation. Laryngoscope 2008, 118, 1023–1030. [Google Scholar] [CrossRef]
- Kirkpatrick, J.P.; Soltys, S.G.; Lo, S.S.; Beal, K.; Shrieve, D.C.; Brown, P.D. The radiosurgery fractionation quandary: Single fraction or hypofractionation? Neuro-Oncology 2017, 19, ii38–ii49. [Google Scholar] [CrossRef] [Green Version]
- Thielhelm, T.P.; Goncalves, S.; Welford, S.; Mellon, E.A.; Bracho, O.; Estivill, M.; Brown, C.; Morcos, J.; Ivan, M.E.; Telischi, F.; et al. Primary Vestibular Schwannoma Cells Activate p21 and RAD51-Associated DNA Repair Following Radiation-Induced DNA Damage. Otol. Neurotol. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Yaes, R.J. Tumor heterogeneity, tumor size, and radioresistance. Int. J. Radiat. Oncol. 1989, 17, 993–1005. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayé-Thomasen, P.; Werther, K.; Nalla, A.; Bøg-Hansen, T.C.; Nielsen, H.J.; Stangerup, S.-E.; Thomsen, J. VEGF and VEGF Receptor-1 Concentration in Vestibular Schwannoma Homogenates Correlates to Tumor Growth Rate. Otol. Neurotol. 2005, 26, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Cayé-Thomasen, P.; Baandrup, L.; Jacobsen, G.K.; Thomsen, J.; Stangerup, S.-E. Immunohistochemical Demonstration of Vascular Endothelial Growth Factor in Vestibular Schwannomas Correlates to Tumor Growth Rate. Laryngoscope 2003, 113, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, Y.; Stemmer-Rachamimov, A.O.; Liu, H.; Huang, P.; Chin, S.; Selig, M.K.; Plotkin, S.R.; Jain, R.K.; Xu, L. Anti-VEGF treatment improves neurological function and augments radiation response in NF2 schwannoma model. Proc. Natl. Acad. Sci. USA 2015, 112, 14676–14681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, A.; Brat, D.J. Practical Surgical Neuropathology: A Diagnostic Approach: A Volume in the Pattern Recognition Series. Churchill Livingstone: Philadelphia, PA, USA, 2010. [Google Scholar]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, M.C.; Shibata, S.B.; Hansen, M.R. The biological underpinnings of radiation therapy for vestibular schwannomas: Review of the literature. Laryngosc. Investig. Otolaryngol. 2021, 6, 458–468. [Google Scholar] [CrossRef]
- Dinh, C.T.; Nisenbaum, E.; Chyou, D.; Misztal, C.; Yan, D.; Mittal, R.; Young, J.; Tekin, M.; Telischi, F.; Fernandez-Valle, C.; et al. Genomics, Epigenetics, and Hearing Loss in Neurofibromatosis Type 2. Otol. Neurotol. 2020, 41, e529–e537. [Google Scholar] [CrossRef]
- Gugel, I.; Ebner, F.H.; Grimm, F.; Czemmel, S.; Paulsen, F.; Hagel, C.; Tatagiba, M.; Nahnsen, S.; Tabatabai, G. Contribution of mTOR and PTEN to Radioresistance in Sporadic and NF2-Associated Vestibular Schwannomas: A Microarray and Pathway Analysis. Cancers 2020, 12, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radu, A.; Neubauer, V.; Akagi, T.; Hanafusa, H.; Georgescu, M.-M. PTEN Induces Cell Cycle Arrest by Decreasing the Level and Nuclear Localization of Cyclin D1. Mol. Cell. Biol. 2003, 23, 6139–6149. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.J.; Albanese, C.; Fu, M.; D’Amico, M.; Lin, B.; Watanabe, G.; Haines, G.K.; Siegel, P.M.; Hung, M.-C.; Yarden, Y.; et al. Cyclin D1 Is Required for Transformation by Activated Neu and Is Induced through an E2F-Dependent Signaling Pathway. Mol. Cell. Biol. 2000, 20, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Hitomi, M.; Stacey, D.W. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006, 1, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.Y.; Clark, J.J.; Fernando, A.; Domann, F.; Hansen, M.R. Contribution of persistent C-Jun N-terminal kinase activity to the survival of human vestibular schwannoma cells by suppression of accumulation of mitochondrial superoxides. Neuro-Oncology 2011, 13, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, G.; Tsuji, T.; Cross, J.V.; Davis, R.J.; Templeton, D.J.; Jiang, W.; Ronai, Z.A. JNK-mediated Phosphorylation of Cdc25C Regulates Cell Cycle Entry and G2/M DNA Damage Checkpoint. J. Biol. Chem. 2010, 285, 14217–14228. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.Y.; Clark, J.J.; Telisak, M.; Hansen, M.R. Inhibition of c-Jun N-Terminal Kinase Activity Enhances Vestibular Schwannoma Cell Sensitivity to Gamma Irradiation. Neurosurgery 2013, 73, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Lasak, J.M.; Welling, D.B.; Akhmametyeva, E.M.; Salloum, M.; Chang, L.-S. Retinoblastoma-Cyclin-Dependent Kinase Pathway Deregulation in Vestibular Schwannomas. Laryngoscope 2002, 112, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Cress, W.D.; Engel, B.E.; Santiago-Cardona, P.G. The retinoblastoma protein: A master tumor suppressor acts as a link between cell cycle and cell adhesion. Cell Health Cytoskelet. 2014, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- May, M.; Schelle, I.; Brakebusch, C.; Rottner, K.; Genth, H. Rac1-dependent recruitment of PAK2 to G2 phase centrosomes and their roles in the regulation of mitotic entry. Cell Cycle 2014, 13, 2210–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thielhelm, T.P.; Goncalves, S.; Welford, S.M.; Mellon, E.A.; Cohen, E.R.; Nourbakhsh, A.; Fernandez-Valle, C.; Telischi, F.; Ivan, M.E.; Dinh, C.T. Understanding the Radiobiology of Vestibular Schwannomas to Overcome Radiation Resistance. Cancers 2021, 13, 4575. https://doi.org/10.3390/cancers13184575
Thielhelm TP, Goncalves S, Welford SM, Mellon EA, Cohen ER, Nourbakhsh A, Fernandez-Valle C, Telischi F, Ivan ME, Dinh CT. Understanding the Radiobiology of Vestibular Schwannomas to Overcome Radiation Resistance. Cancers. 2021; 13(18):4575. https://doi.org/10.3390/cancers13184575
Chicago/Turabian StyleThielhelm, Torin P., Stefania Goncalves, Scott M. Welford, Eric A. Mellon, Erin R. Cohen, Aida Nourbakhsh, Cristina Fernandez-Valle, Fred Telischi, Michael E. Ivan, and Christine T. Dinh. 2021. "Understanding the Radiobiology of Vestibular Schwannomas to Overcome Radiation Resistance" Cancers 13, no. 18: 4575. https://doi.org/10.3390/cancers13184575