
Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC)

 ,
,  , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumorigenic Activity of CDK1 and Anticancer Mechanisms of CDK1 Inhibitors
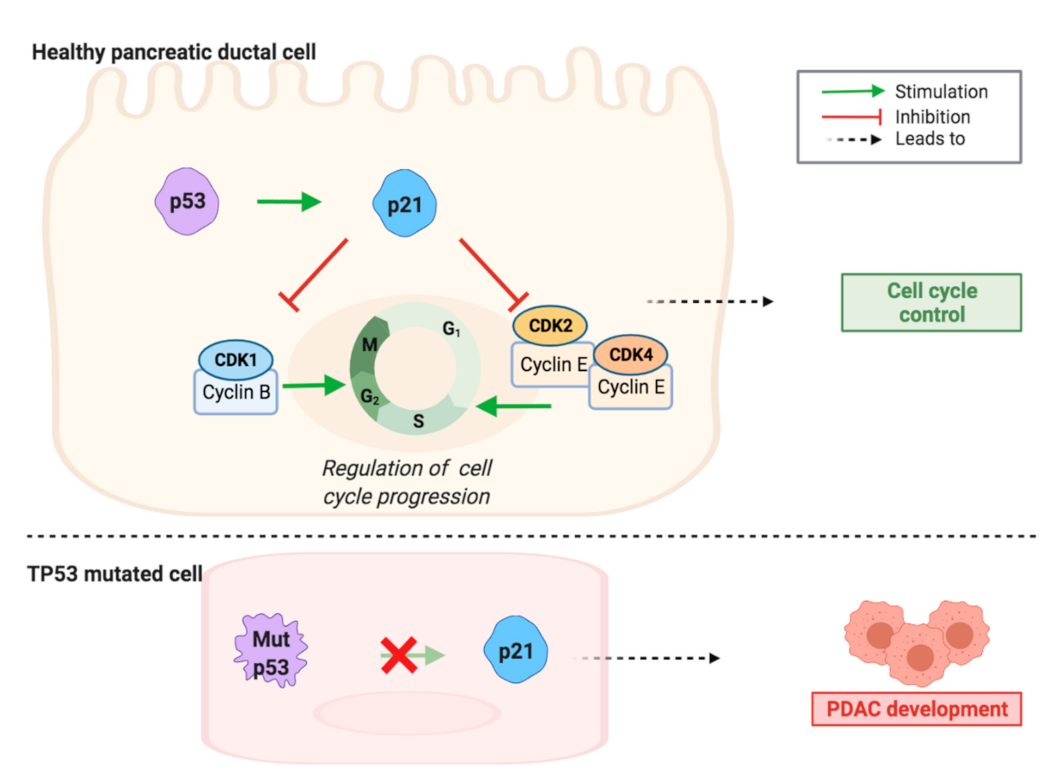
2.1. CDK1 and Cell Cycle
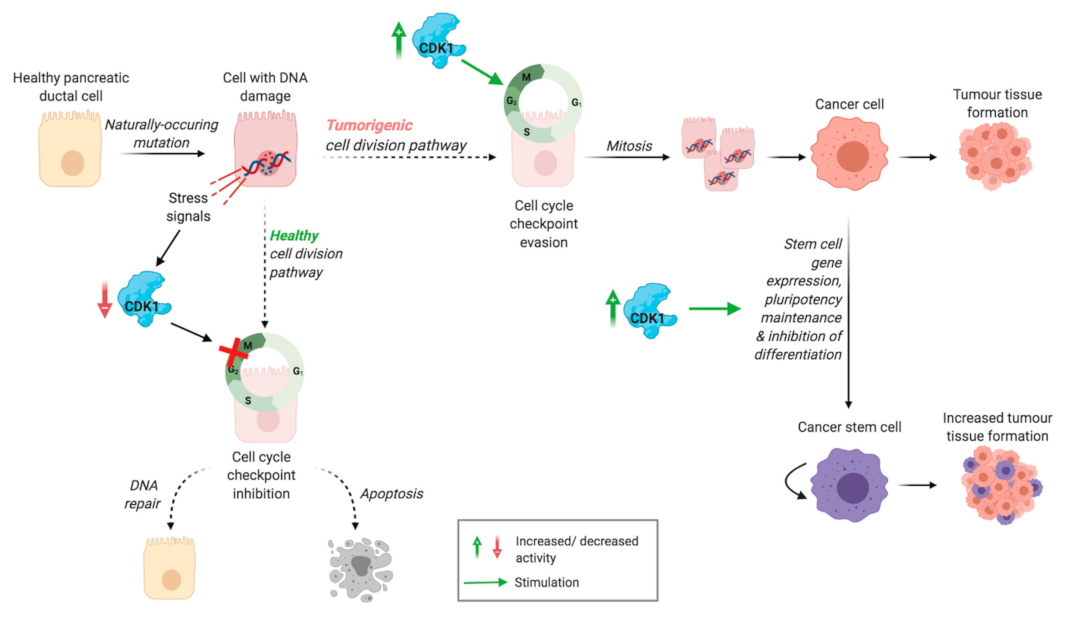
2.2. Bypassing the Cell Cycle Checkpoint
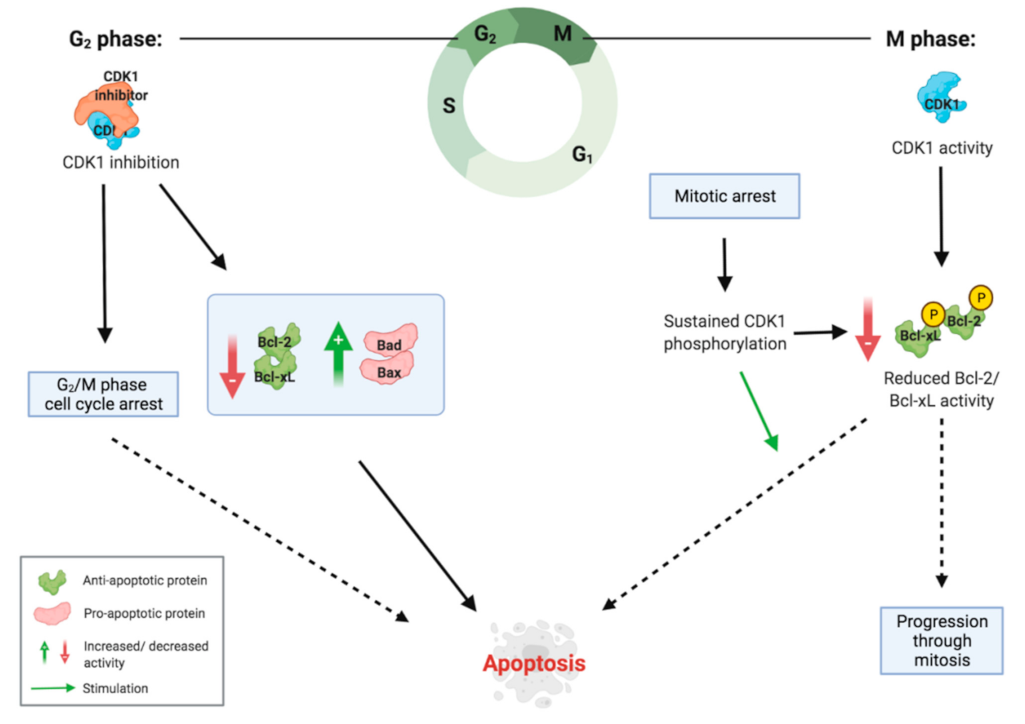
2.3. Inducing G2/M Phase Cell Cycle Arrest
2.4. Inducing Apoptosis
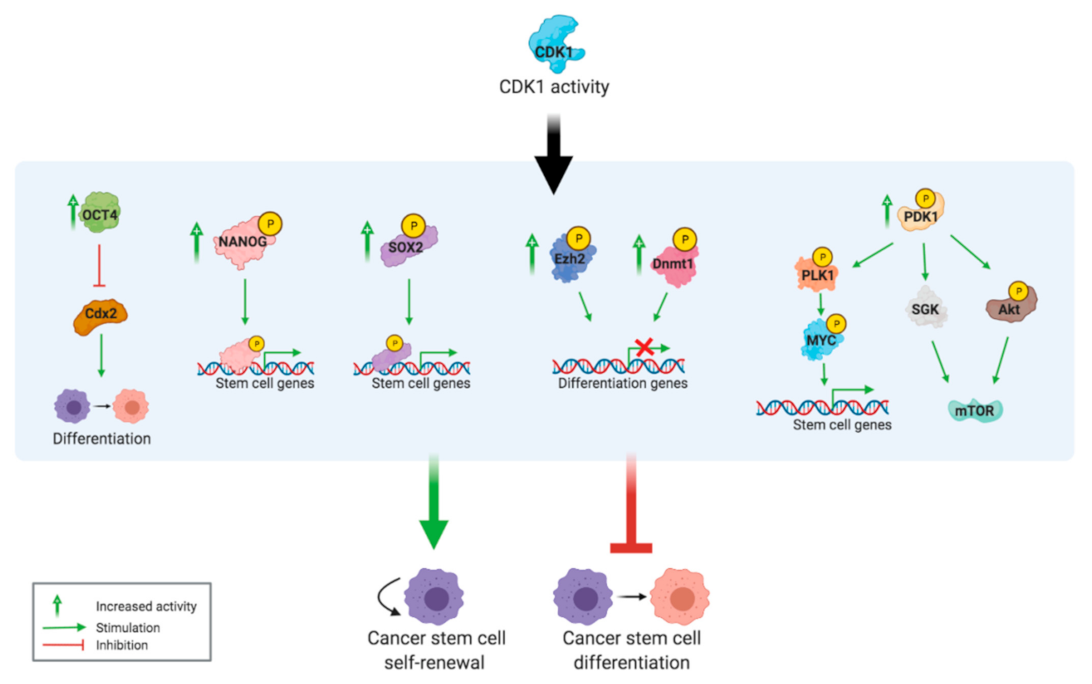
2.5. Inducing and Maintaining Cancer Stem Cell Properties
2.6. Targeting Cancer Stem Cells
3. Therapeutic Potential
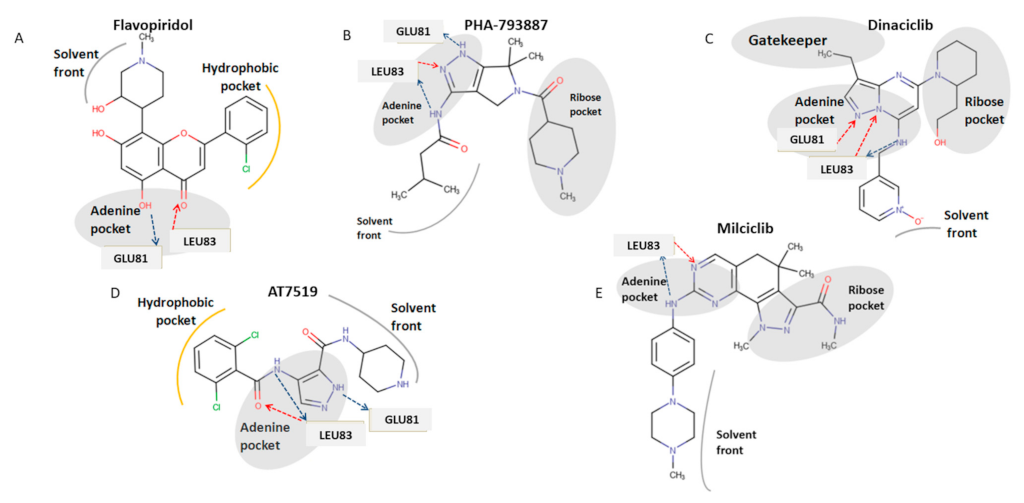
3.1. CDK1 Inhibitors
3.2. Preclinical Studies on CDK1 Inhibition
3.3. Clinical Trials of CDK1 Inhibitors
3.4. Importance of Screening Patients
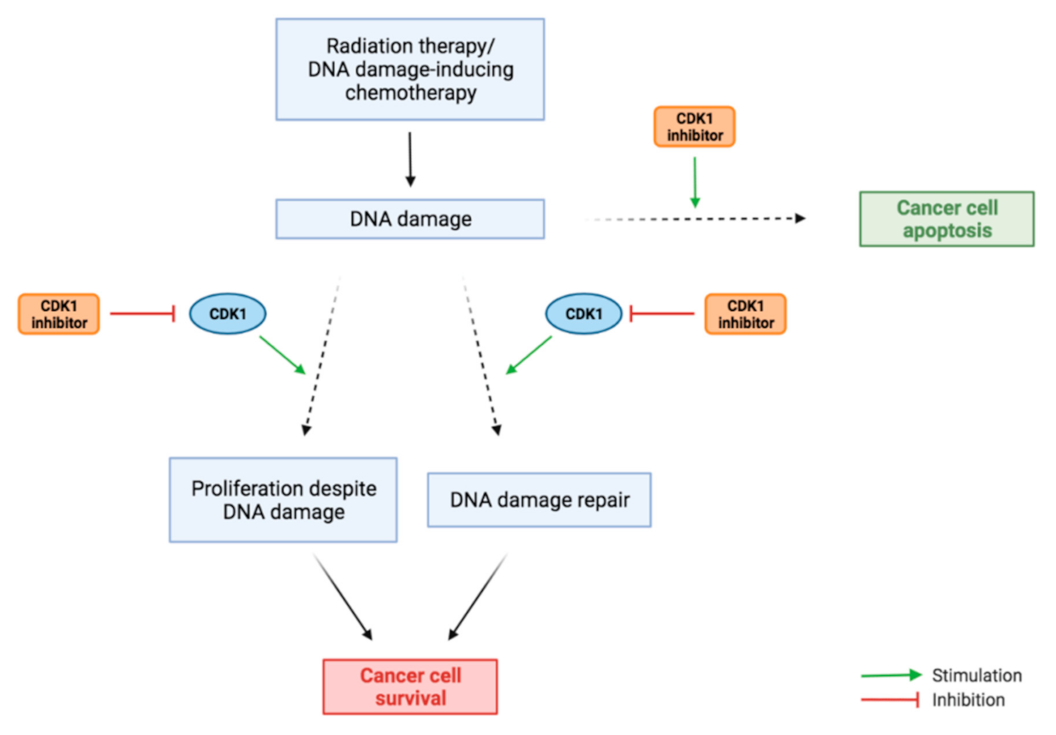
3.5. Combination Therapy
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, A.; Chung, V. Pancreatic Cancer. In Oncology in the Precision Medicine Era; Salgia, R., Ed.; Springer: Cham, Germany, 2020; pp. 97–109. [Google Scholar]
- American Cancer Society. Key Statistics for Pancreatic Cancer. American Cancer Society. Available online: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (accessed on 21 January 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Stoica, A.F.; Chang, C.H.; Pauklin, S. Molecular Therapeutics of Pancreatic Ductal Adenocarcinoma: Targeted Pathways and the Role of Cancer Stem Cells. Trends Pharm. Sci. 2020, 41, 977–993. [Google Scholar] [CrossRef]
- Giovannetti, E.; van der Borden, C.L.; Frampton, A.E.; Ali, A.; Firuzi, O.; Peters, G.J. Never let it go: Stopping key mechanisms underlying metastasis to fight pancreatic cancer. Sem. Cancer Biol. 2017, 44, 43–59. [Google Scholar] [CrossRef]
- Caparello, C.; Meijer, L.L.; Garajova, I.; Falcone, A.; Le Large, T.Y.; Funel, N.; Kazemier, G.; Peters, G.J.; Vasile, E.; Giovannetti, E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J. Gastroenterol. 2016, 22, 6987–7005. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Azim, S.; Zubair, H.; Bhardwaj, A.; Patel, G.K.; Khushman, M.; Singh, S.; Singh, A.P. Molecular Drivers of Pancreatic Cancer Pathogenesis: Looking Inward to Move Forward. Int. J. Mol. Sci. 2017, 18, 779. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, N.M.; Palumbo, A., Jr.; De Martino, M.; Fusco, A.; Pinto, L.F.R.; Nasciutti, L.E. Interplay between HMGA and TP53 in cell cycle control along tumor progression. Cell Mol. Life Sci. 2021, 78, 817–831. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- García-Reyes, B.; Kretz, A.L.; Ruff, J.P.; von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The Emerging Role of Cyclin-Dependent Kinases (CDKs) in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Huang, F.; Zhang, H.; Chen, Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci. Rep. 2019, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Kleeff, J.; Li, J.; Ding, J.; Hammer, J.; Zhao, Y.; Giese, T.; Korc, M.; Büchler, M.W.; Friess, H. Expression and functional significance of CDC25B in human pancreatic ductal adenocarcinoma. Oncogene 2004, 23, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendge, A.K.; Chaudhuri, D.; Mandal, N. The natural flavones, acacetin and apigenin, induce Cdk-Cyclin mediated G2/M phase arrest and trigger ROS-mediated apoptosis in glioblastoma cells. Mol. Biol. Rep. 2021, 48, 539–549. [Google Scholar] [CrossRef]
- Huang, J.; Chen, P.; Liu, K.; Liu, J.; Zhou, B.; Wu, R.; Peng, Q.; Liu, Z.-X.; Li, C.; Kroemer, G.; et al. CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer. Gut 2020, 70, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Seddon, A.M.; Dalgleish, A.G.; Khelwatty, S.; Ioannou, N.; Mudan, S.; Modjtahedi, H. Synergistic activity of agents targeting growth factor receptors, CDKs and downstream signaling molecules in a panel of pancreatic cancer cell lines and the identi cation of antagonistic combinations: Implications for future clinical trials in pancreatic. Ongol. Rep. 2020, 44, 2581–2594. [Google Scholar]
- Prevo, R.; Pirovano, G.; Puliyadi, R.; Herbert, K.J.; Rodriguez-Berriguete, G.; O’Docherty, A.; Greaves, W.; McKenna, W.G.; Higgins, G.S. CDK1 inhibition sensitizes normal cells to DNA damage in a cell cycle dependent manner. Cell Cycle 2018, 17, 1513–1523. [Google Scholar] [CrossRef]
- Mostafa, M.E.; Erbarut-Seven, I.; Pehlivanoglu, B.; Adsay, V. Pathologic classification of “pancreatic cancers”: Current concepts and challenges. Chin. Clin. Onc. 2017, 6, 59. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Novak, B.; Tyson, J.; Gyorffy, B.; Csikasz-Nagy, A. Irreversible cell-cycle transitions are due to systems-level feedback. Nat. Cell Biol. 2007, 9, 724–728. [Google Scholar] [CrossRef]
- Sung, W.W.; Lin, Y.M.; Wu, P.R.; Yen, H.H.; Lai, H.W.; Su, T.C.; Huang, R.H.; Wen, C.K.; Chen, C.Y.; Chen, C.J.; et al. High nuclear/cytoplasmic ratio of Cdk1 expression predicts poor prognosis in colorectal cancer patients. BMC Cancer 2014, 14, 915. [Google Scholar] [CrossRef] [Green Version]
- Neganova, I.; Tilgner, K.; Buskin, A.; Paraskevopoulou, I.; Atkinson, S.P.; Peberdy, D.; Passos, J.F.; Lako, M. CDK1 plays an important role in the maintenance of pluripotency and genomic stability in human pluripotent stem cells. Cell Death Dis. 2014, 5, 1508. [Google Scholar] [CrossRef] [Green Version]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2009, 488, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic Kinases as Regulators of Cell Division and its Checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Bendris, N.; Lemmers, B.; Blanchard, J.M. Cell cycle, cytoskeleton dynamics and beyond: The many functions of cyclins and CDK inhibitors. Cell Cycle 2015, 14, 1786–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Q.; Lo, C.M.; Chen, L.; Ngan, E.S.; Xu, A.; Poon, R.Y. CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ. 2017, 24, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Hsia, S.M.; Yu, C.C.; Shih, Y.H.; Yuanchien Chen, M.; Wang, T.H.; Huang, Y.T.; Shieh, T.M. Isoliquiritigenin as a cause of DNA damage and inhibitor of ataxia-telangiectasia mutated expression leading to G2/M phase arrest and apoptosis in oral squamous cell carcinoma. Head Neck 2016, 38, E360–E371. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Zhu, B.T. Upregulated cyclin B1/CDK1 mediates apoptosis following 2- methoxyestradiol-induced mitotic catastrophe: Role of Bcl-XL phosphorylation. Steroids 2019, 150, 108381. [Google Scholar] [CrossRef]
- Zhang, S.; Bao, Y.; Ju, X.; Li, K.; Shang, H.; Ha, L.; Qian, Y.; Zou, L.; Sun, X.; Li, J.; et al. BA-j as a novel CDK1 inhibitor selectively induces apoptosis in cancer cells by regulating ROS. 1. Sci. Rep. 2015, 5, 13626. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [Green Version]
- Costa-Cabral, S.; Brough, R.; Konde, A.; Aarts, M.; Campbell, J.; Marinari, E.; Riffell, J.; Bardelli, A.; Torrance, C.; Lord, C.J.; et al. CDK1 Is a Synthetic Lethal Target for KRAS Mutant Tumours. PLoS ONE. 2016, 11, e0149099. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Ichimaru, Y.; Kurita, M.; Hayashi, E.; Homma, T.; Saito, H.; Masuda, S.; Nemoto, N.; Hemmi, A.; Suzuki, T.; et al. Induction of cell death in pancreatic ductal adenocarcinoma by indirubin 30-oxime and 5-methoxyindirubin 30-oxime in vitro and in vivo. Cancer Lett. 2017, 397, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Cai, D.; Zhang, B.; Lou, G.; Zou, X. Combination of HDAC inhibitor TSA and silibinin induces cell cycle arrest and apoptosis by targeting survivin and cyclinB1/Cdk1 in pancreatic cancer cells. Biomed Pharm. 2015, 74, 257–264. [Google Scholar] [CrossRef]
- Zhou, L.; Cai, X.; Han, X.; Xu, N.; Chang, D.C. CDK1 switches mitotic arrest to apoptosis by phosphorylating Bcl-2/Bax family proteins during treatment with microtubule interfering agents. Cell Biol. Int. 2014, 38, 737–746. [Google Scholar] [CrossRef]
- Darweesh, O.; Al-Shehri, E.; Falquez, H.; Lauterwasser, J.; Edlich, F.; Patel, R. Identification of a novel Bax-Cdk1 signalling complex that links activation of the Mitotic Checkpoint to Apoptosis. J. Cell Sci. 2021, 134, 244152. [Google Scholar] [CrossRef] [PubMed]
- Terrano, D.T.; Upreti, M.; Chambers, T.C. Cyclin-Dependent Kinase 1-Mediated Bcl-xL/Bcl-2 Phosphorylation Acts as a Functional Link Coupling Mitotic Arrest and Apoptosis. Mol. Cell Biol. 2010, 30, 640–656. [Google Scholar] [CrossRef] [Green Version]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.X.; Wang, X.Q.; Chok, S.H.; Man, K.; Tsang, S.H.Y.; Chan, A.C.Y.; Ma, K.W.; Xia, W.; Cheung, T.T. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics 2018, 8, 3737–3750. [Google Scholar] [CrossRef]
- Brumbaugh, J.; Russell, J.D.; Yu, P.; Westphall, M.S.; Coon, J.J.; Thomson, J.A. NANOG Is Multiply Phosphorylated and Directly Modified by ERK2 and CDK1 In Vitro. Stem Cell Rep. 2014, 2, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.R.; Luo, Y.; Arcaroli, J.J.; Liu, S.; KrishnanKutty, L.N.; Osborne, D.G.; Li, Y.; Samson, J.M.; Bagby, S.; Tan, A.C.; et al. CDK1 Interacts with Sox2 and Promotes Tumor Initiation in Human Melanoma. Cancer Res. 2018, 78, 6561–6574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Dibble, C.C.; Toker, A.A. P13K signaling in cancer: Beyond Akt. Curr. Opin. Cell Bio. 2017, 45, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy 5. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Ruggero, D. New Connections between Old Pathways: PDK1 Signaling Promotes Cellular Transformation through PLK1-Dependent MYC Stabilization. Cancer Discov. 2013, 3, 1099–1102. [Google Scholar] [CrossRef] [Green Version]
- Hessmann, E.; Schneider, G.; Ellenrieder, V.; Siveke, J.T. MYC in pancreatic cancer: Novel mechanistic insights and their translation into therapeutic strategies. Oncogene 2016, 35, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; Petrenko, O.; Hayman, M.J. Analysis of the tumor-initiating and metastatic capacity of PDX1-positive cells from the adult pancreas. Proc. Natl. Acad. Sci. USA 2014, 111, 3466–3471. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Mueller, M.T.; Heeschen, C. Pancreatic cancer stem Cells—Insights and perspectives. Expert Opin. Biol. Ther. 2009, 9, 1744–7682. [Google Scholar] [CrossRef]
- Casari, I.; Domenichini, A.; Sestito, S.; Capone, E.; Sala, G.; Rapposelli, S.; Falasca, M. Dual PDK1/Aurora Kinase A Inhibitors Reduce Pancreatic Cancer Cell Proliferation and Colony Formation. Cancers 2019, 11, 1695. [Google Scholar] [CrossRef] [Green Version]
- Huskey, N.E.; Guo, T.; Evason, K.J.; Momcilovic, O.; Pardo, D.; Creasman, K.J.; Judson, R.L.; Blelloch, R.; Oakes, S.A.; Hebrok, M.; et al. CDK1 Inhibition Targets the p53-NOXA-MCL1 Axis, Selectively Kills Embryonic Stem Cells, and Prevents Teratoma Formation. Stem Cell Rep. 2015, 4, 374–389. [Google Scholar] [CrossRef] [Green Version]
- Li, V.C.; Ballabeni, A.; Kirschner, M.W. Gap 1 phase length and mouse embryonic stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2012, 109, 12550–12555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharm. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Mariaule, G.; Belmont, P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey. Molecules 2014, 19, 14366–14382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelland, L.R. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: Current status. Expert Opin. Investig. Drugs 2000, 9, 2903–2911. [Google Scholar] [CrossRef]
- Brasca, M.G.; Albanese, C.; Alzani, R.; Amici, R.; Avanzi, N.; Ballinari, D.; Bischoff, J.; Borghi, D.; Casale, E.; Croci, V.; et al. Optimization of 6,6-dimethyl pyrrolo[3,4-c]pyrazoles: Identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing. Bioorg. Med. Chem. 2010, 18, 1844–1853. [Google Scholar] [CrossRef]
- Martin, M.P.; Olesen, S.H.; Georg, G.I.; Schönbrunn, E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, P.G.; Woodhead, A.J.; Berdini, V.; Boulstridge, J.A.; Carr, M.G.; Cross, D.M.; Davis, D.J.; Devine, L.A.; Early, T.R.; Feltell, R.E.; et al. Identification of N-(4-Piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a Novel Cyclin Dependent Kinase Inhibitor Using Fragment-Based X-Ray Crystallography and Structure Based Drug Design. J. Med. Chem. 2008, 51, 4986–4999. [Google Scholar] [CrossRef]
- Brasca, M.G.; Amboldi, N.; Ballinari, D.; Cameron, A.; Casale, E.; Cervi, G.; Colombo, M.; Colotta, F.; Croci, V.; D’Alessio, R.; et al. Identification of N,1,4,4-Tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}-4,5-dihydro1H-pyrazolo[4,3-h]quinazoline-3-carboxamide (PHA-848125), a Potent, Orally Available. J. Med. Chem. 2009, 52, 152–163. [Google Scholar] [CrossRef]
- Feldmann, G.; Mishra, A.; Bisht, S.; Karikari, C.; Garrido-Laguna, I.; Rasheed, Z.; Ottenhof, N.A.; Dadon, T.; Alvarez, H.; Fendrich, V.; et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. Ther. 2011, 12, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hu, S.; Shangguan, J.; Eresen, A.; Li, Y.; Ma, Q.; Yaghmai, V.; Benson, A.B., III; Zhang, Z. Dinaciclib prolongs survival in the LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre (KPC) transgenic murine models of pancreatic ductal adenocarcinoma. Am. J. Transl. Res. 2020, 12, 1031–1043. [Google Scholar] [PubMed]
- Gregory, G.P.; Hogg, S.J.; Kats, L.M.; Vidacs, E.; Baker, A.J.; Gilan, O.; Lefebure, M.; Martin, B.P.; Dawson, M.A.; Johnstone, R.W.; et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2015, 29, 1437–1441. [Google Scholar] [CrossRef]
- Moharram, S.A.; Shah, K.; Khanum, F.; Marhäll, A.; Gazi, M.; Kazi, J.U. Efficacy of the CDK inhibitor dinaciclib in vitro and in vivo in T-cell acute lymphoblastic leukemia. Cancer Lett. 2017, 405, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K. Mayo Phase 2 Consortium. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Mita, A.C.; Moseley, J.L.; Poon, J.; Small, K.A.; Jou, Y.M.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Statkevich, P.; et al. Phase 1 safety, pharmacokinetic and pharmacodynamic study of the cyclin-dependent kinase inhibitor dinaciclib administered every three weeks in patients with advanced malignancies. Br. J. Cancer 2017, 117, 1258–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.G.; Zahurak, M.; Shah, M.; Weekes, C.D.; Hansen, A.; Siu, L.L.; Spreafico, A.; LoConte, N.; Anders, N.M.; Miles, T.; et al. ETCTN-9231 Study Team. A Phase I Study of Dinaciclib in Combination With MK-2206 in Patients with Advanced Pancreatic Cancer. Clin. Transl. Sci. 2020, 13, 1178–1188. [Google Scholar] [CrossRef]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 259. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, R.D.; Tse, A.; Shah, M.A.; Lefkowitz, R.A.; Gonen, M.; Gilman-Rosen, L.; Kortmansky, J.; Kelsen, D.P.; Schwartz, G.K.; O’Reilly, E.M. A Phase II Study of Flavopiridol (Alvocidib) in Combination with Docetaxel in Refractory, Metastatic Pancreatic Cancer. Pancreatology 2009, 9, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Boss, D.S.; Schwartz, G.K.; Middleton, M.R.; Amakye, D.D.; Swaisland, H.; Midgley, R.S.; Ranson, M.; Danson, S.; Calvert, H.; Plummer, R.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol. 2010, 21, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Soria, J.C.; Anthoney, D.A.; Proctor, A.; Scaburri, A.; Pacciarini, M.A.; Laffranchi, B.; Pellizzoni, C.; Kroemer, G.; Armand, J.P.; et al. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle 2011, 10, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Aspeslagh, S.; Shailubhai, K.; Bahleda, R.; Gazzah, A.; Varga, A.; Hollebecque, A.; Massard, C.; Spreafico, A.; Reni, M.; Soria, J.C. Phase I dose-escalation study of milciclib in combination with gemcitabine in patients with refractory solid tumors. Cancer Chemother Pharm. 2017, 79, 1257–1265. [Google Scholar] [CrossRef]
- Villa, E.; Piscaglia, F.; Geva, R.; Dalecos, G.; Papatheodoridis, G.; Ciomei, M.; Davite, C.; Crivori, P.; Palejwala, V.; Jacob, J.; et al. Phase IIa safety and efficacy of milciclib, a pan-cyclin dependent kinase inhibitor, in unresectable, sorafenib-refractory or -intolerant hepatocellular carcinoma patients. J. Clin. Oncol. 2020, 38, e16711. [Google Scholar] [CrossRef]
- Weiss, G.J.; Hidalgo, M.; Borad, M.J.; Laheru, D.; Tibes, R.; Ramanathan, R.K.; Blaydorn, L.; Jameson, G.; Jimeno, A.; Isaacs, J.D.; et al. Phase I study of the safety, tolerability and pharmacokinetics of PHA-848125AC, a dual tropomyosin receptor kinase A and cyclin-dependent kinase inhibitor, in patients with advanced solid malignancies. Investig. New Drugs 2012, 30, 2334–2343. [Google Scholar] [CrossRef]
- Linton, A.; Cheng, Y.Y.; Griggs, K.; Schedlich, L.; Kirschner, M.B.; Gattani, S.; Srikaran, S.; Chuan-Hao Kao, S.; McCaughan, B.C.; Klebe, S.; et al. An RNAi-based screen reveals PLK1, CDK1 and NDC80 as potential therapeutic targets in malignant pleural mesothelioma. Br. J. Cancer 2018, 118, e13. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.R.; Chen, X.J.; Chen, W.; Wu, L.F.; Jiang, H.P.; Lin, D.; Wang, L.J.; Wang, W.; Guo, S.Q. Sp1 contributes to radioresistance of cervical cancer through targeting G2/M cell cycle checkpoint CDK1. Cancer Manag. Res. 2019, 11, 5835–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Geng, X.; Xing, M.; Li, W.; Chen, Z.; Shen, H.; Ying, S. CDK1 promotes nascent DNA synthesis and induces resistance of cancer cells to DNA-damaging therapeutic agents. Oncotarget 2017, 8, 90662–90673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayes, P.A.; Dolloff, N.G.; Daniel, C.J.; Liu, J.J.; Hart, L.S.; Kuribayashi, K.; Allen, J.E.; Jee, D.I.; Dorsey, J.F.; Liu, Y.Y.; et al. Overcoming Hypoxia-Induced Apoptotic Resistance through Combinatorial Inhibition of GSK-3β and CDK1. Cancer Res. 2011, 71, 5265–5275. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Kawakami, H.; Liu, W.; Zeng, X.; Strebhardt, K.; Tao, K.; Huang, S.; Sinicrope, F.A. Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer. Mol. Cancer Res. 2018, 16, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, K.; Zhang, J.; Wang, L.; Sheng, L.; Yan, L. Inhibition of CDK1 Reverses the Resistance of 5-Fu in Colorectal Cancer. Cancer Manag. Res. 2020, 12, 11271–11283. [Google Scholar] [CrossRef]
- Chand, S.; O’Hayer, K.; Blanco, F.F.; Winter, J.M.; Brody, J.R. The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms. Int. J. Biol. Sci. 2016, 12, 273–282. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (Targeted CDKs) | Clinical Trial Phase | Disease (Number of Patients) | Efficacy | Tolerability | Observations | Reference |
|---|---|---|---|---|---|---|
| AZD5438 (1, 2, 9) | Phase I | Advanced solid tumours (64, 8 pancreatic) | − | − − | [71] | |
| Dinaciclib (1, 2, 5, 9) | Phase I | Pancreatic cancer (39) | − | + | In combination with MK-2206 (Akt inhibitor) | [68] |
| Advanced malignancies (61, 5 pancreatic) | − + | + | [67] | |||
| Advanced malignancies(48, 4 pancreatic) | − + | − + | [69] | |||
| Phase II | Advanced breast cancer (39) | − + | + | In comparison vs. capecitabine (similar but not superior anticancer activity) | [67] | |
| Refractory multiple myeloma (27) | ++ | + | [66] | |||
| Phase III | Refractory chronic lymphocytic leukemia (42) | + + | + | In comparison vs. ofatumumab (results suggest superior anticancer activity) | [65] | |
| Flavopiridol (1, 2, 4, 6) | Phase II | Pancreatic cancer (10) | + − | − − | In combination with docetaxel | [70] |
| Milciclib (1, 2, 4, 5) | Phase I | Refractory solid tumours (16, 13 pancreatic) | + | + + | In combination with gemcitabine | [73] |
| Phase II | Hepatocellular carcinoma (14) | + | + + | [74] | ||
| PHA-848125AC (1, 2, 4, 5, 7) | Phase I | Advanced solid malignancies (37, 5 pancreatic) | + | − + | [75] | |
| PHA-793887 (1, 2, 4) | Phase I | Solid tumours (19, 5 pancreatic) | − | − − | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. https://doi.org/10.3390/cancers13174389
Wijnen R, Pecoraro C, Carbone D, Fiuji H, Avan A, Peters GJ, Giovannetti E, Diana P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers. 2021; 13(17):4389. https://doi.org/10.3390/cancers13174389
Chicago/Turabian StyleWijnen, Rosa, Camilla Pecoraro, Daniela Carbone, Hamid Fiuji, Amir Avan, Godefridus J. Peters, Elisa Giovannetti, and Patrizia Diana. 2021. "Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC)" Cancers 13, no. 17: 4389. https://doi.org/10.3390/cancers13174389