
ALT Positivity in Human Cancers: Prevalence and Clinical Insights

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Telomere and Telomere Maintenance Mechanisms
1.2. Commonly Used ALT Biomarkers
1.3. Other Potential ALT Identification Strategies
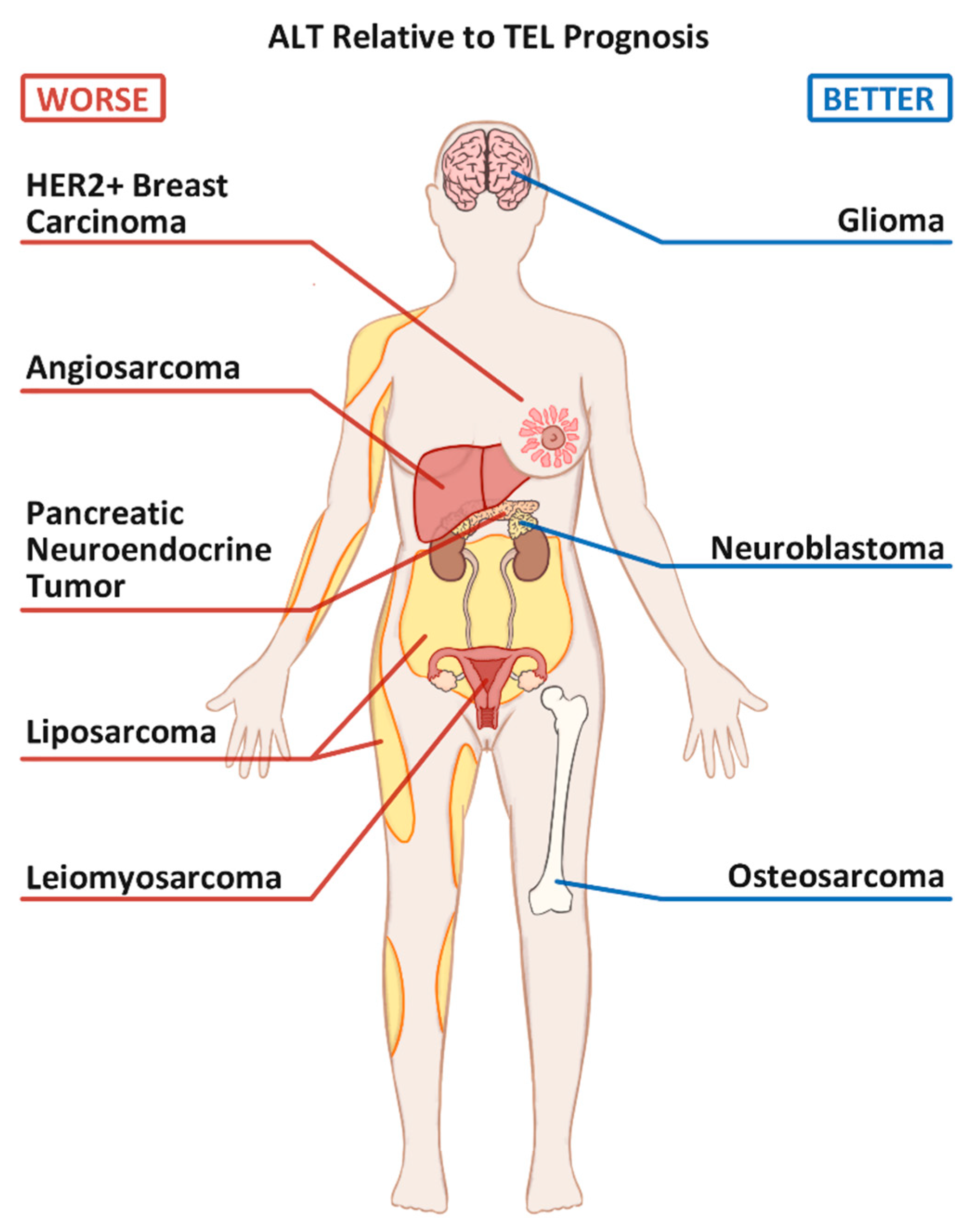
2. ALT Positive Bone Cancers: Osteosarcoma
ALT Positive Osteosarcoma and the Mutation Status of ATRX and DAXX
3. ALT Positive Breast Cancers
4. ALT Positive Central Nervous System (CNS) Tumors
4.1. Gliomas
4.1.1. Genetic Alterations, Cellular Lineage, and Survival in ALT Positive Gliomas
4.1.2. Treatment for ALT Positive Gliomas
4.2. Other CNS Tumors
5. ALT Positive Neuroendocrine Tumors
5.1. Neuroblastoma
5.1.1. ALT Positive Neuroblastoma and the Mutation Status of ATRX/DAXX
5.1.2. MYCN Amplification and ALT Positivity Are Mutually Exclusive and Synthetically Lethal
5.2. Pancreatic Neuroendocrine Tumors (PanNETs)
5.2.1. PanNET and the Mutation Status of MEN1 and ATRX/ DAXX
5.2.2. PanNET Diagnosis
6. ALT Positive Soft Tissue Tumors
6.1. Angiosarcoma
6.2. Leiomyosarcoma
ALT+ Leiomyosarcoma and the Mutation Status of ATRX/DAXX
6.3. Liposarcoma
6.3.1. ALT+ Liposarcoma and the Mutation Status of ATRX/DAXX
6.3.2. Liposarcoma and TMM-Guided Therapies
6.4. Undifferentiated Pleomorphic Sarcoma
7. The Development of Novel Therapies
8. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Maestroni, L.; Matmati, S.; Coulon, S. Solving the Telomere Replication Problem. Genes 2017, 8, 55. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Shay, J.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.J.; Vasu, V.; Griffin, D.K. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- O’Rourke, J.J.; Bythell-Douglas, R.; A Dunn, E.; Deans, A.J. ALT control, delete: FANCM as an anti-cancer target in Alternative Lengthening of Telomeres. Nucl. 2019, 10, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.D.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive Proliferation of Human Cancer Cells with Ever-Shorter Telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef] [Green Version]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.; Sabatier, L.; Marder, B.; Morgan, W. Telomere dynamics in an immortal human cell line. EMBO J. 1994, 13, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- Londoño-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef] [Green Version]
- Tokutake, Y.; Matsumoto, T.; Watanabe, T.; Maeda, S.; Tahara, H.; Sakamoto, S.; Niida, H.; Sugimoto, M.; Ide, T.; Furuichi, Y. Extra-Chromosomal Telomere Repeat DNA in Telomerase-Negative Immortalized Cell Lines. Biochem. Biophys. Res. Commun. 1998, 247, 765–772. [Google Scholar] [CrossRef]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; E Napier, C.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Claude, E.; Decottignies, A. Telomere maintenance mechanisms in cancer: Telomerase, ALT or lack thereof. Curr. Opin. Genet. Dev. 2020, 60, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Reddel, R.R. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Drosopoulos, W.C.; Sethi, L.; Madireddy, A.; Schildkraut, C.L.; Zhang, D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. 2017, 114, E5940–E5949. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110. [Google Scholar] [CrossRef] [Green Version]
- Feng, E.; Batenburg, N.L.; Walker, J.R.; Ho, A.; Mitchell, T.R.H.; Qin, J.; Zhu, X.-D. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020, 133, 133. [Google Scholar] [CrossRef] [PubMed]
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londoño-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731. [Google Scholar] [CrossRef] [Green Version]
- Loe, T.K.; Li, J.S.Z.; Zhang, Y.; Azeroglu, B.; Boddy, M.N.; Denchi, E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020, 34, 650–662. [Google Scholar] [CrossRef]
- Dellaire, G. The Nuclear Protein Database (NPD): Sub-nuclear localisation and functional annotation of the nuclear proteome. Nucleic Acids Res. 2003, 31, 328–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Cerone, M.A.; Autexier, C.; Londoño-Vallejo, J.A.; Bacchetti, S. A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene 2005, 24, 7893–7901. [Google Scholar] [CrossRef] [Green Version]
- Fasching, C.L.; Bower, K.; Reddel, R.R. Telomerase-Independent Telomere Length Maintenance in the Absence of Alternative Lengthening of Telomeres–Associated Promyelocytic Leukemia Bodies. Cancer Res. 2005, 65, 2722–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, R.A.; Cavazos, D.; Montellano, R.; Chen, Q.; Guarente, L.; Johnson, F.B. A Novel Telomere Structure in a Human Alternative Lengthening of Telomeres Cell Line. Cancer Res. 2005, 65, 2730–2737. [Google Scholar] [CrossRef] [Green Version]
- Henson, J.D.; A Hannay, J.; McCarthy, S.W.; A Royds, J.; Yeager, T.R.; A Robinson, R.; Wharton, S.B.; A Jellinek, D.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [PubMed]
- Zhang, J.-M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.-M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Henson, J.D.; Cao, Y.; I Huschtscha, L.; Chang, A.C.; Au, A.Y.M.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Z.; Shengzhao, G.; Li, X.; Liu, H.; Zhao, Y. Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet. 2019, 15, e1007925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzucco, G.; Huda, A.; Galli, M.; Piccini, D.; Giannattasio, M.; Pessina, F.; Doksani, Y. Telomere damage induces internal loops that generate telomeric circles. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Lau, L.M.; Koch, S.; La Rotta, N.M.; Dagg, R.A.; Reddel, R.R. The C-Circle Assay for alternative-lengthening-of-telomeres activity. Methods 2017, 114, 74–84. [Google Scholar] [CrossRef]
- Lee, M.; Teber, E.T.; Holmes, O.; Nones, K.; Patch, A.-M.; A Dagg, R.; Lau, L.M.S.; Lee, J.H.; E Napier, C.; Arthur, J.W.; et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018, 46, 4903–4918. [Google Scholar] [CrossRef] [Green Version]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef] [Green Version]
- Xin, S.; Wei, G. Prognostic factors in osteosarcoma: A study level meta-analysis and systematic review of current practice. J. Bone Oncol. 2020, 21, 100281. [Google Scholar] [CrossRef]
- Misaghi, A.; Goldin, A.; Awad, M.; A Kulidjian, A. Osteosarcoma: A comprehensive review. SICOT-J. 2018, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Broadhead, M.L.; Clark, J.C.M.; Myers, D.E.; Dass, C.R.; Choong, P.F.M. The Molecular Pathogenesis of Osteosarcoma: A Review. Sarcoma 2011, 2011, 959248. [Google Scholar] [CrossRef]
- Scheel, C.; Schaefer, K.-L.; Jauch, A.; Keller, M.; Wai, D.; Brinkschmidt, C.; Van Valen, F.; Boecker, W.; Dockhorn-Dworniczak, B.; Poremba, C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 2001, 20, 3835–3844. [Google Scholar] [CrossRef] [Green Version]
- Ulaner, G.A.; Huang, H.-Y.; Otero, J.; Zhao, Z.; Ben-Porat, L.; Satagopan, J.M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Huvos, A.G.; et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res. 2003, 63, 1759–1763. [Google Scholar] [PubMed]
- Ulaner, G.A.; Hoffman, A.R.; Otero, J.; Huang, H.-Y.; Zhao, Z.; Mazumdar, M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Ladanyi, M. Divergent patterns of telomere maintenance mechanisms among human sarcomas: Sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing’s sarcomas and osteosarcomas. Genes. Chromosom. Cancer 2004, 41, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.P.; Drissi, R.; Billups, C.A.; Daw, N.C.; Valentine, M.B.; Dome, J.S.; Guillou, L.; Benhattar, J.; Bonichon, F.; Gallagher, G.; et al. Telomerase Expression Predicts Unfavorable Outcome in Osteosarcoma. J. Clin. Oncol. 2004, 22, 3790–3797. [Google Scholar] [CrossRef]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human Sarcomas Are Mosaic for Telomerase-Dependent and Telomerase-Independent Telomere Maintenance Mechanisms. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Tajima, K.; Ohno, A.; Washimi, Y.; Ishimura, D.; Yamada, H. Characterization of human multicentric osteosarcoma using newly established cells derived from multicentric osteosarcoma. J. Cancer Res. Clin. Oncol. 2010, 137, 423–433. [Google Scholar] [CrossRef]
- Mason-Osann, E.; Gali, H.; Flynn, R.L. Resolving Roadblocks to Telomere Replication. Methods Mol. Biol. 2019, 1999, 31–57. [Google Scholar] [CrossRef]
- Kim, J.; Sun, C.; Tran, A.D.; Chin, P.-J.; Ruiz, P.D.; Wang, K.; Gibbons, R.J.; Gamble, M.J.; Liu, Y.; Oberdoerffer, P. The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening. Nat. Struct. Mol. Biol. 2019, 26, 213–219. [Google Scholar] [CrossRef]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, J.; Shi, G.; Zhang, L.; Yuanling, J.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, srep32280. [Google Scholar] [CrossRef]
- Heaphy, C.M.; De Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; De Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Liau, J.-Y.; Lee, J.-C.; Tsai, J.-H.; Yang, C.-Y.; Liu, T.-L.; Ke, Z.-L.; Hsu, H.-H.; Jeng, Y.-M. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Mod. Pathol. 2015, 28, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Koelsche, C.; Renner, M.; Johann, P.; Leiss, I.; Sahm, F.; Schimmack, S.; Wardelmann, E.; Renker, E.-K.; Schirmacher, P.; Korshunov, A.; et al. Differential nuclear ATRX expression in sarcomas. Histopathology 2015, 68, 738–745. [Google Scholar] [CrossRef]
- Mason-Osann, E.; Dai, A.; Floro, J.; Lock, Y.J.; Reiss, M.; Gali, H.; Matschulat, A.; Labadorf, A.; Flynn, R.L. Identification of a novel gene fusion in ALT positive osteosarcoma. Oncotarget 2018, 9, 32868–32880. [Google Scholar] [CrossRef] [Green Version]
- Yost, K.E.; Soper, S.F.C.; Walker, R.L.; Pineda, M.A.; Zhu, Y.J.; Ester, C.D.; Showman, S.; Roschke, A.V.; Waterfall, J.J.; Meltzer, P.S. Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci. Rep. 2019, 9, 4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.-J.; Panel Members. Strategies for subtypes—Dealing with the diversity of breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Subhawong, A.P.; Heaphy, C.M.; Argani, P.; Konishi, Y.; Kouprina, N.; Nassar, H.; Vang, R.; Meeker, A.K. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod. Pathol. 2009, 22, 1423–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Peng, M.; Song, Q. The co-expression of telomerase and ALT pathway in human breast cancer tissues. Tumor Biol. 2013, 35, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Robinson, N.J.; Morrison-Smith, C.D.; Gooding, A.J.; Schiemann, B.J.; Jackson, M.W.; Taylor, D.J.; Schiemann, W.P. SLX4IP and telomere dynamics dictate breast cancer metastasis and therapeutic responsiveness. Life Sci. Alliance 2020, 3, e201900427. [Google Scholar] [CrossRef] [Green Version]
- Panier, S.; Maric, M.; Hewitt, G.; Mason-Osann, E.; Gali, H.; Dai, A.; Labadorf, A.; Guervilly, J.-H.; Ruis, P.; Segura-Bayona, S.; et al. SLX4IP Antagonizes Promiscuous BLM Activity during ALT Maintenance. Mol. Cell 2019, 76, 27–43e.11. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Crowe, D.L. Molecular and cellular basis of mammary gland fibrosis and cancer risk. Int. J. Cancer 2019, 144, 2239–2253. [Google Scholar] [CrossRef]
- Patel, A.P.; Fisher, J.L.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Agius, D.; Alahdab, F.; Alam, T.; et al. Global, regional, and national burden of brain and other CNS cancer, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 376–393. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurother. 2017, 14, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Mellai, M.; Annovazzi, L.; Senetta, R.; Dell’Aglio, C.; Mazzucco, M.; Cassoni, P.; Schiffer, D. Diagnostic revision of 206 adult gliomas (including 40 oligoastrocytomas) based on ATRX, IDH1/2 and 1p/19q status. J. Neurooncol. 2016, 131, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Fogli, A.; Demattei, M.-V.; Corset, L.; Vaurs-Barrière, C.; Chautard, E.; Biau, J.; Kémény, J.-L.; Godfraind, C.; Pereira, B.; Khalil, T.; et al. Detection of the alternative lengthening of telomeres pathway in malignant gliomas for improved molecular diagnosis. J. Neurooncol. 2017, 135, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; De Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Inagaki, A.; Silber, J.; Gorovets, D.; Zhang, J.; Kastenhuber, E.R.; Heguy, A.; Petrini, J.H.; Chan, T.A.; Huse, J.T. Whole exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 2012, 3, 1194–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-Y.; Gerges, N.; Korshunov, A.; Sabha, N.; Khuong-Quang, D.-A.; Fontebasso, A.M.; Fleming, A.; Hadjadj, D.; Schwartzentruber, J.; Majewski, J.; et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012, 124, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Vizcaino, M.A.; Blakeley, J.; Heaphy, C.M. Frequent alternative lengthening of telomeres and ATRX loss in adult NF1-associated diffuse and high-grade astrocytomas. Acta Neuropathol. 2016, 132, 761–763. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Graham, M.K.; Brosnan-Cashman, J.A.; Barber, J.R.; Davis, C.; Vizcaino, M.A.; Palsgrove, D.N.; Giannini, C.; Pekmezci, M.; Dahiya, S.; et al. Telomere alterations in neurofibromatosis type 1-associated solid tumors. Acta Neuropathol. Commun. 2019, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mangerel, J.; Price, A.; Castelo-Branco, P.; Brzezinski, J.; Buczkowicz, P.; Rakopoulos, P.; Merino, D.; Baskin, B.; Wasserman, J.; Mistry, M.; et al. Alternative lengthening of telomeres is enriched in, and impacts survival of TP53 mutant pediatric malignant brain tumors. Acta Neuropathol. 2014, 128, 853–862. [Google Scholar] [CrossRef]
- Minasi, S.; Baldi, C.; Gianno, F.; Antonelli, M.; Buccoliero, A.M.; Pietsch, T.; Massimino, M.; Buttarelli, F.R. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Child Nerv. Syst. 2021, 37, 809–818. [Google Scholar] [CrossRef]
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar] [CrossRef]
- Wiestler, B.; Capper, D.; Holland-Letz, T.; Korshunov, A.; Von Deimling, A.; Pfister, S.M.; Platten, M.; Weller, M.; Wick, W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013, 126, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampl, S.; Pramhas, S.; Stern, C.; Preusser, M.; Marosi, C.; Holzmann, K. Expression of Telomeres in Astrocytoma WHO Grade 2 to 4: TERRA Level Correlates with Telomere Length, Telomerase Activity, and Advanced Clinical Grade. Transl. Oncol. 2012, 5, 56-IN4. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatter, T.; Gifford-Garner, J.; Wiles, A.; Tan, X.; Chen, Y.-J.; Macfarlane, M.; Sullivan, M.; Royds, J.; Hung, N. Pilocytic Astrocytomas Have Telomere-Associated Promyelocytic Leukemia Bodies without Alternatively Lengthened Telomeres. Am. J. Pathol. 2010, 177, 2694–2700. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Brosnan-Cashman, J.A.; Allen, S.J.; Vizcaino, M.A.; Giannini, C.; Camelo-Piragua, S.; Webb, M.; Matsushita, M.; Wadhwani, N.; Tabbarah, A.; et al. Alternative lengthening of telomeres, ATRX loss and H3-K27M mutations in histologically defined pilocytic astrocytoma with anaplasia. Brain Pathol. 2019, 29, 126–140. [Google Scholar] [CrossRef]
- McDonald, K.L.; McDonnell, J.; Muntoni, A.; Henson, J.D.; Hegi, M.E.; Von Deimling, A.; Wheeler, H.R.; Cook, R.J.; Biggs, M.T.; Little, N.S.; et al. Presence of Alternative Lengthening of Telomeres Mechanism in Patients With Glioblastoma Identifies a Less Aggressive Tumor Type With Longer Survival. J. Neuropathol. Exp. Neurol. 2010, 69, 729–736. [Google Scholar] [CrossRef]
- Royds, J.A.; Al Nadaf, S.; Wiles, A.K.; Chen, Y.-J.; Ahn, A.; Shaw, A.; Bowie, S.; Lam, F.; Baguley, B.C.; Braithwaite, A.W.; et al. The CDKN2A G500 Allele Is More Frequent in GBM Patients with No Defined Telomere Maintenance Mechanism Tumors and Is Associated with Poorer Survival. PLoS ONE 2011, 6, e26737. [Google Scholar] [CrossRef]
- Farooqi, A.; Yang, J.; Sharin, V.; Ezhilarasan, R.; Danussi, C.; Alvarez, C.; Dharmaiah, S.; Irvin, D.; Huse, J.; Sulman, E.P. Identification of patient-derived glioblastoma stem cell (GSC) lines with the alternative lengthening of telomeres phenotype. Acta Neuropathol. Commun. 2019, 7, 76. [Google Scholar] [CrossRef]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 2087. [Google Scholar] [CrossRef]
- Hakin-Smith, V.; A Jellinek, D.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.R.; McKay, M.J.; Reddel, R.R.; Royds, J.A. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef]
- Hung, N.; A Eiholzer, R.; Kirs, S.; Zhou, J.; Ward-Hartstonge, K.; Wiles, A.K.; Frampton, C.M.; Taha, A.; A Royds, J.; Slatter, T.L. Telomere profiles and tumor-associated macrophages with different immune signatures affect prognosis in glioblastoma. Mod. Pathol. 2016, 29, 212–226. [Google Scholar] [CrossRef]
- Hung, N.; Chen, Y.-J.; Taha, A.; Olivecrona, M.; Boet, R.; Wiles, A.; Warr, T.; Shaw, A.; Eiholzer, R.; Baguley, B.C.; et al. Increased paired box transcription factor 8 has a survival function in Glioma. BMC Cancer 2014, 14, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.N.; Heaphy, C.M.; De Wilde, R.F.; Orr, B.A.; Odia, Y.; Eberhart, C.G.; Meeker, A.K.; Rodriguez, F.J. Molecular and Morphologic Correlates of the Alternative Lengthening of Telomeres Phenotype in High-Grade Astrocytomas. Brain Pathol. 2012, 23, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.S.V.; Sørensen, M.D.; Pusch, S.; Beier, D.; Bouillon, A.-S.; Kristensen, B.W.; Brümmendorf, T.H.; Beier, C.P.; Beier, F. Alternative lengthening of telomeres is the major telomere maintenance mechanism in astrocytoma with isocitrate dehydrogenase 1 mutation. J. Neurooncol. 2020, 147, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschacher, T.; Wolf, B.; Aschacher, O.; Enzmann, F.; Laszlo, V.; Messner, B.; Türkcan, A.; Weis, S.; Spiegl-Kreinecker, S.; Holzmann, K.; et al. Long interspersed element-1 ribonucleoprotein particles protect telomeric ends in alternative lengthening of telomeres dependent cells. Neoplasia 2020, 22, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Calinescu, A.-A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar] [CrossRef] [Green Version]
- Eid, R.; Demattei, M.-V.; Episkopou, H.; Augé-Gouillou, C.; Decottignies, A.; Grandin, N.; Charbonneau, M. Genetic Inactivation ofATRXLeads to a Decrease in the Amount of Telomeric Cohesin and Level of Telomere Transcription in Human Glioma Cells. Mol. Cell. Biol. 2015, 35, 2818–2830. [Google Scholar] [CrossRef] [Green Version]
- Brosnan-Cashman, J.A.; Yuan, M.; Graham, M.K.; Rizzo, A.J.; Myers, K.M.; Davis, C.; Zhang, R.; Esopi, D.M.; Raabe, E.H.; Eberhart, C.G.; et al. ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS ONE 2018, 13, e0204159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-J.; Hakin-Smith, V.; Teo, M.; Xinarianos, G.E.; Jellinek, D.A.; Carroll, T.; McDowell, D.; Macfarlane, M.R.; Boet, R.; Baguley, B.C.; et al. Association of Mutant TP53 with Alternative Lengthening of Telomeres and Favorable Prognosis in Glioma. Cancer Res. 2006, 66, 6473–6476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Yang, P.; Zhang, C.; Zhang, W.; Liu, Y.; Bao, Z.; Liu, X.; Du, W.; Wang, H.; Jiang, T.; et al. ATRXmRNA expression combined withIDH1/2mutational status and Ki-67 expression refines the molecular classification of astrocytic tumors: Evidence from the whole transcriptome sequencing of 169 samples. Oncotarget 2014, 5, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, J.; Johannessen, T.-C.A.; Ohba, S.; Chow, T.T.; E Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- Hewer, E.; Vajtai, I.; Berezowska, S.; Vassella, E.; Dettmer, M.S. Combined ATRX/IDH1 immunohistochemistry predicts genotype of oligoastrocytomas. Histopathol. 2015, 68, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedalthagafi, M.; Phillips, J.J.; Kim, G.E.; Mueller, S.; Haas-Kogen, D.A.; Marshall, R.E.; Croul, S.E.; Santi, M.R.; Cheng, J.; Zhou, S.; et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: A multi-institutional study of 214 astrocytomas. Mod. Pathol. 2013, 26, 1425–1432. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Bartels, U.; Bouffet, E.; Becher, O.; Hawkins, C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: Diagnostic and therapeutic implications. Acta Neuropathol. 2014, 128, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorris, K.; Sobo, M.; Onar-Thomas, A.; Panditharatna, E.; Stevenson, C.B.; Gardner, S.L.; DeWire, M.D.; Pierson, C.R.; Olshefski, R.; Rempel, S.A.; et al. Prognostic significance of telomere maintenance mechanisms in pediatric high-grade gliomas. J. Neurooncol. 2014, 117, 67–76. [Google Scholar] [CrossRef]
- Han, B.; Cai, J.; Gao, W.; Meng, X.; Gao, F.; Wu, P.; Duan, C.; Wang, R.; Dinislam, M.; Lin, L.; et al. Loss of ATRX suppresses ATM dependent DNA damage repair by modulating H3K9me3 to enhance temozolomide sensitivity in glioma. Cancer Lett. 2018, 419, 280–290. [Google Scholar] [CrossRef]
- Palsgrove, D.N.; Brosnan-Cashman, J.A.; Giannini, C.; Raghunathan, A.; Jentoft, M.; Bettegowda, C.; Gokden, M.; Lin, D.; Yuan, M.; Lin, M.-T.; et al. Subependymal giant cell astrocytoma-like astrocytoma: A neoplasm with a distinct phenotype and frequent neurofibromatosis type-1-association. Mod. Pathol. 2018, 31, 1787–1800. [Google Scholar] [CrossRef]
- Kim, S.; Seo, Y.; Chowdhury, T.; Yu, H.J.; Lee, C.E.; Kim, K.-M.; Kang, H.; Kim, H.J.; Park, S.-J.; Kim, K.; et al. Inhibition of MUC1 exerts cell-cycle arrest and telomerase suppression in glioblastoma cells. Sci. Rep. 2020, 10, 18238. [Google Scholar] [CrossRef]
- Grandin, N.; Network, P.; Pereira, B.; Cohen, C.; Billard, P.; Dehais, C.; Carpentier, C.; Idbaih, A.; Bielle, F.; Ducray, F.; et al. The level of activity of the alternative lengthening of telomeres correlates with patient age in IDH-mutant ATRX-loss-of-expression anaplastic astrocytomas. Acta Neuropathol. Commun. 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Altwairgi, A.K.; Raja, S.; Manzoor, M.; Al Dandan, S.; Al Saeed, E.; Balbaid, A.; Alhussain, H.; Orz, Y.; Lary, A.; Alsharm, A.A. Management and treatment recommendations for World Health Organization Grade III and IV gliomas. Int. J. Health Sci. 2017, 11, 54–62. [Google Scholar]
- Minasi, S.; Baldi, C.; Pietsch, T.; Donofrio, V.; Pollo, B.; Antonelli, M.; Massimino, M.; Giangaspero, F.; Buttarelli, F.R. Telomere elongation via alternative lengthening of telomeres (ALT) and telomerase activation in primary metastatic medulloblastoma of childhood. J. Neurooncol. 2019, 142, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Hoehner, J.C.; Gestblom, C.; Hedborg, F.; Sandstedt, B.; Olsen, L.; Påhlman, S. A developmental model of neuroblastoma: Differentiating stroma-poor tumors’ progress along an extra-adrenal chromaffin lineage. Lab. Investig. 1996, 75, 659–675. [Google Scholar] [PubMed]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koneru, B.; Lopez, G.; Farooqi, A.; Conkrite, K.L.; Nguyen, T.H.; Macha, S.J.; Modi, A.; Rokita, J.L.; Urias, E.; Hindle, A.; et al. Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res. 2020, 80, 2663–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2014, 6, 7493–7503. [Google Scholar] [CrossRef]
- Roderwieser, A.; Sand, F.; Walter, E.; Fischer, J.; Gecht, J.; Bartenhagen, C.; Ackermann, S.; Otte, F.; Welte, A.; Kahlert, Y.; et al. Telomerase Is a Prognostic Marker of Poor Outcome and a Therapeutic Target in Neuroblastoma. JCO Precis. Oncol. 2019, 1–20. [Google Scholar] [CrossRef]
- Ohali, A.; Avigad, S.; Ash, S.; Goshen, Y.; Luria, D.; Feinmesser, M.; Zaizov, R.; Yaniv, I. Telomere length is a prognostic factor in neuroblastoma. Cancer 2006, 107, 1391–1399. [Google Scholar] [CrossRef]
- Onitake, Y.; Hiyama, E.; Kamei, N.; Yamaoka, H.; Sueda, T.; Hiyama, K. Telomere biology in neuroblastoma: Telomere binding proteins and alternative strengthening of telomeres. J. Pediatr. Surg. 2009, 44, 2258–2266. [Google Scholar] [CrossRef]
- Cheung, N.-K.V. Association of Age at Diagnosis and Genetic Mutations in Patients With Neuroblastoma. JAMA 2012, 307, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, S.; Hiyama, E.; Onitake, Y.; Yamaoka, E.; Hiyama, K. Clinical features of ATRX or DAXX mutated neuroblastoma. J. Pediatr. Surg. 2014, 49, 1835–1838. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nat. Cell Biol. 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Federico, S.; Chen, X.; Fan, Y.; Xu, B.; Stewart, E.; Zhou, X.; Jeon, J.; Griffiths, L.; Nguyen, R.; et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosse, Y.P.; Deyell, R.J.; Berthold, F.; Nagakawara, A.; Ambros, P.F.; Monclair, T.; Cohn, S.L.; Pearson, A.D.; London, W.B.; Matthay, K.K. Neuroblastoma in older children, adolescents and young adults: A report from the International Neuroblastoma Risk Group project. Pediatr. Blood Cancer 2013, 61, 627–635. [Google Scholar] [CrossRef]
- Lundberg, G.; Sehic, D.; Länsberg, J.-K.; Øra, I.; Frigyesi, A.; Castel, V.; Navarro, S.; Piqueras, M.; Martinsson, T.; Noguera, R.; et al. Alternative lengthening of telomeres-An enhanced chromosomal instability in aggressive non-MYCN amplified and telomere elongated neuroblastomas. Genes. Chromosom. Cancer 2011, 50, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.S.; Dagg, R.A.; Choi, L.M.R.; Shay, J.W.; Reynolds, C.P.; Lau, L.M.S. Alternative lengthening of telomeres in neuroblastoma cell lines is associated with a lack of MYCN genomic amplification and with p53 pathway aberrations. J. Neurooncol. 2014, 119, 17–26. [Google Scholar] [CrossRef]
- Kawashima, M.; Kojima, M.; Ueda, Y.; Kurihara, S.; Hiyama, E. Telomere biology including TERT rearrangements in neuroblastoma: A useful indicator for surgical treatments. J. Pediatr. Surg. 2016, 51, 2080–2085. [Google Scholar] [CrossRef]
- Kelgiorgi, D.; Dervenis, C. Pancreatic neuroendocrine tumors: The basics, the gray zone, and the target. F1000Research 2017, 6, 663. [Google Scholar] [CrossRef] [Green Version]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.; Perren, A. Loss of DAXX and ATRX Are Associated With Chromosome Instability and Reduced Survival of Patients With Pancreatic Neuroendocrine Tumors. Gastroenterol. 2014, 146, 453–460.e5. [Google Scholar] [CrossRef]
- Vandenbussche, C.J.; Allison, D.B.; Graham, M.K.; Charu, V.; Lennon, A.M.; Wolfgang, C.L.; Hruban, R.H.; Heaphy, C.M. Alternative lengthening of telomeres and ATRX/DAXX loss can be reliably detected in FNAs of pancreatic neuroendocrine tumors. Cancer Cytopathol. 2017, 125, 544–551. [Google Scholar] [CrossRef]
- McGovern, J.M.; Singhi, A.D.; Borhani, A.A.; Furlan, A.; McGrath, K.M.; Zeh, H.J.; Bahary, N.; Dasyam, A.K. CT Radiogenomic Characterization of the Alternative Lengthening of Telomeres Phenotype in Pancreatic Neuroendocrine Tumors. Am. J. Roentgenol. 2018, 211, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Liu, T.-C.; Roncaioli, J.L.; Cao, D.; Zeh, H.J.; Zureikat, A.H.; Tsung, A.; Marsh, J.W.; Lee, K.K.; Hogg, M.E.; et al. Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2017, 23, 600–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Brosnan-Cashman, J.A.; An, S.; Kim, S.J.; Song, K.-B.; Kim, M.-S.; Kim, M.-J.; Hwang, D.W.; Meeker, A.K.; Yu, E.; et al. Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin. Cancer Res. 2016, 23, 1598–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pea, A.; Yu, J.; Marchionni, L.; Noe, M.; Luchini, C.; Pulvirenti, A.; de Wilde, R.F.; Brosens, L.A.; Rezaee, N.; Javed, A.; et al. Genetic Analysis of Small Well-differentiated Pancreatic Neuroendocrine Tumors Identifies Subgroups With Differing Risks of Liver Metastases. Ann. Surg. 2020, 271, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, W.M.; Bs, F.H.M.M.; Moons, L.M.G.; Heaphy, C.M.; Offerhaus, G.J.A.; Dreijerink, K.M.A.; Brosens, L.A.A. Assessment of ARX expression, a novel biomarker for metastatic risk in pancreatic neuroendocrine tumors, in endoscopic ultrasound fine-needle aspiration. Diagn. Cytopathol. 2019, 48, 308–315. [Google Scholar] [CrossRef]
- Yuan, F.; Shi, M.; Ji, J.; Shi, H.; Zhou, C.; Yu, Y.; Liu, B.; Zhu, Z.; Zhang, J. KRAS and DAXX/ATRX Gene Mutations Are Correlated with the Clinicopathological Features, Advanced Diseases, and Poor Prognosis in Chinese Patients with Pancreatic Neuroendocrine Tumors. Int. J. Biol. Sci. 2014, 10, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Chou, A.; Itchins, M.; de Reuver, P.R.; Arena, J.; Clarkson, A.; Sheen, A.; Sioson, L.; Cheung, V.; Perren, A.; Nahm, C.; et al. ATRX loss is an independent predictor of poor survival in pancreatic neuroendocrine tumors. Hum. Pathol. 2018, 82, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Paik, W.H.; Lee, K.; Ryu, J.K.; Lee, S.H.; Kim, Y.-T. DAXX/ATRX and MEN1 genes are strong prognostic markers in pancreatic neuroendocrine tumors. Oncotarget 2017, 8, 49796–49806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogeas, E.; Karagkounis, G.; Heaphy, C.M.; Hirose, K.; Pawlik, T.M.; Wolfgang, C.L.; Meeker, A.; Hruban, R.H.; Cameron, J.L.; Choti, M.A. Alternative Lengthening of Telomeres Predicts Site of Origin in Neuroendocrine Tumor Liver Metastases. J. Am. Coll. Surg. 2014, 218, 628–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pea, A.; Hruban, R.H.; Wood, L.D. Genetics of pancreatic neuroendocrine tumors: Implications for the clinic. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1407–1419. [Google Scholar] [CrossRef] [Green Version]
- Cejas, P.; Drier, Y.; Dreijerink, K.M.A.; Brosens, L.A.A.; Deshpande, V.; Epstein, C.B.; Conemans, E.B.; Morsink, F.H.M.; Graham, M.K.; Valk, G.D.; et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat. Med. 2019, 25, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, W.M.; Schelhaas, W.; Morsink, F.H.M.; Heidsma, C.M.; Van Eeden, S.; Valk, G.D.; Vriens, M.R.; Heaphy, C.M.; Van Dijkum, E.J.M.N.; Offerhaus, G.J.A.; et al. Alternative Lengthening of Telomeres and Differential Expression of Endocrine Transcription Factors Distinguish Metastatic and Non-metastatic Insulinomas. Endocr. Pathol. 2020, 31, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wilde, R.F.; Heaphy, C.M.; Maitra, A.; Meeker, A.K.; Edil, B.H.; Wolfgang, C.L.; A Ellison, T.; Schulick, R.D.; Molenaar, I.Q.; Valk, G.D.; et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod. Pathol. 2012, 25, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, W.M.; Brosens, L.A.; Poruk, K.E.; Noë, M.; Hosoda, W.; Poling, J.S.; Rizzo, A.; Campbell-Thompson, M.; Atkinson, M.A.; Konukiewitz, B.; et al. Aberrant Menin expression is an early event in pancreatic neuroendocrine tumorigenesis. Hum. Pathol. 2016, 56, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, E.K.; Roses, R.E.; Gupta, M.; Shah, P.K.; Shah, K.K.; Zaheer, S.; Wachtel, H.; Kelz, R.R.; Karakousis, G.C.; Fraker, U.L. Surgery for metastatic neuroendocrine tumors with occult primaries. J. Surg. Res. 2013, 184, 221–227. [Google Scholar] [CrossRef]
- Young, R.J.; Brown, N.J.; Reed, M.W.; Hughes, D.; Woll, P.J. Angiosarcoma. Lancet Oncol. 2010, 11, 983–991. [Google Scholar] [CrossRef]
- Panse, G.; Chrisinger, J.S.; Leung, C.H.; Ingram, D.R.; Khan, S.; Wani, K.; Lin, H.; Lazar, A.J.; Wang, W.-L. Clinicopathological analysis of ATRX, DAXX and NOTCH receptor expression in angiosarcomas. Histopathology 2017, 72, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali, R.; Chandramohan, R.; Möller, I.; Scholz, S.L.; Berger, M.; Huberman, K.; Viale, A.; Pirun, M.; Socci, N.D.; Bouvier, N.; et al. Targeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathway. Oncotarget 2015, 6, 36041–36052. [Google Scholar] [CrossRef] [Green Version]
- Liau, J.-Y.; Tsai, J.-H.; Yang, C.-Y.; Lee, J.-C.; Liang, C.-W.; Hsu, H.-H.; Jeng, Y.-M. Alternative lengthening of telomeres phenotype in malignant vascular tumors is highly associated with loss of ATRX expression and is frequently observed in hepatic angiosarcomas. Hum. Pathol. 2015, 46, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; George, S. Leiomyosarcoma. Hematol. Clin. N. Am. 2013, 27, 957–974. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Serrano, C.; Hensley, M.L.; Ray-Coquard, I. Soft Tissue and Uterine Leiomyosarcoma. J. Clin. Oncol. 2018, 36, 144–150. [Google Scholar] [CrossRef]
- Ahvenainen, T.V.; Mäkinen, N.M.; Von Nandelstadh, P.; Vahteristo, M.E.A.; Pasanen, A.M.; Bützow, R.C.; Vahteristo, M.P. Loss of ATRX/DAXX expression and alternative lengthening of telomeres in uterine leiomyomas. Cancer 2018, 124, 4650–4656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, J.-Y.; Tsai, J.-H.; Jeng, Y.-M.; Lee, J.-C.; Hsu, H.-H.; Yang, C.-Y. Leiomyosarcoma With Alternative Lengthening of Telomeres Is Associated With Aggressive Histologic Features, Loss of ATRX Expression, and Poor Clinical Outcome. Am. J. Surg. Pathol. 2015, 39, 236–244. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Park, N.-H.; Lee, H. Prognostic Value of Alternative Lengthening of Telomeres–Associated Biomarkers in Uterine Sarcoma and Uterine Carcinosarcoma. Int. J. Gynecol. Cancer 2012, 22, 434–441. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Liau, J.-Y.; Huang, W.-J.; Chang, Y.-T.; Chang, M.-C.; Lee, J.-C.; Tsai, J.-H.; Su, Y.-N.; Hung, C.-C.; Jeng, Y.-M. Targeted next-generation sequencing of cancer genes identified frequent TP53 and ATRX mutations in leiomyosarcoma. Am. J. Transl. Res. 2015, 7, 2072–2081. [Google Scholar]
- Slatter, T.L.; Hsia, H.; Samaranayaka, A.; Sykes, P.; Clow, W.B.; Devenish, C.J.; Sutton, T.; Royds, J.A.; Pc, P.; Cheung, A.N.; et al. Loss of ATRX and DAXX expression identifies poor prognosis for smooth muscle tumours of uncertain malignant potential and early stage uterine leiomyosarcoma. J. Pathol. Clin. Res. 2015, 1, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, N.; Aavikko, M.; Heikkinen, T.; Taipale, M.; Taipale, J.; Koivisto-Korander, R.; Bützow, R.; Vahteristo, P. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet. 2016, 12, e1005850. [Google Scholar] [CrossRef] [PubMed]
- Dall’Asta, A.; Gizzo, S.; Musarò, A.; Quaranta, M.; Noventa, M.; Migliavacca, C.; Sozzi, G.; Monica, M.; Mautone, D.; Berretta, R. Uterine smooth muscle tumors of uncertain malignant potential (STUMP): Pathology, follow-up and recurrence. Int. J. Clin. Exp. Pathol. 2014, 7, 8136–8142. [Google Scholar]
- Gadducci, A.; Zannoni, G.F. Uterine smooth muscle tumors of unknown malignant potential: A challenging question. Gynecol. Oncol. 2019, 154, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Daidone, M.G.; Daprai, L.; Villa, R.; Cantù, S.; Pilotti, S.; Mariani, L.; Gronchi, A.; Henson, J.D.; Reddel, R.R.; et al. Telomere Maintenance Mechanisms in Liposarcomas: Association with Histologic Subtypes and Disease Progression. Cancer Res. 2006, 66, 8918–8924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crago, A.M.; Dickson, M.A. Liposarcoma. Surg. Oncol. Clin. N. Am. 2016, 25, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.T.J.; Thway, K.; Huang, P.H.; Jones, R.L. Clinical and Molecular Spectrum of Liposarcoma. J. Clin. Oncol. 2018, 36, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.N.; Mendez-Bermudez, A.; Zaffaroni, N.; Dubrova, Y.E.; Royle, N.J. Evidence for alternative lengthening of telomeres in liposarcomas in the absence of ALT-associated PML bodies. Int. J. Cancer 2008, 122, 2414–2421. [Google Scholar] [CrossRef]
- Venturini, L.; Motta, R.; Gronchi, A.; Daidone, M.; Zaffaroni, N. Prognostic relevance of ALT-associated markers in liposarcoma: A comparative analysis. BMC Cancer 2010, 10, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cairney, C.J.; Hoare, S.F.; Daidone, M.G.; Zaffaroni, N.; Keith, W.N. High level of telomerase RNA gene expression is associated with chromatin modification, the ALT phenotype and poor prognosis in liposarcoma. Br. J. Cancer 2008, 98, 1467–1474. [Google Scholar] [CrossRef]
- Lee, J.-C.; Jeng, Y.-M.; Liau, J.-Y.; Tsai, J.-H.; Hsu, H.-H.; Yang, C.-Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1064–1073. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.A.; Johnson, J.E.; Pascarelli, K.; Beeharry, N.; Chiourea, M.; Gagos, S.; Lev, D.; von Mehren, M.; Kipling, D.; Broccoli, D. Doxorubicin Resistance in a Novel In vitro Model of Human Pleomorphic Liposarcoma Associated with Alternative Lengthening of Telomeres. Mol. Cancer Ther. 2010, 9, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovatcheva, M.; Liu, D.D.; Dickson, M.A.; Klein, M.E.; O’Connor, R.; Wilder, F.O.; Socci, N.D.; Tap, W.D.; Schwartz, G.K.; Singer, S.; et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget 2015, 6, 8226–8243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laroche-Clary, A.; Chaire, V.; Verbeke, S.; Algéo, M.-P.; Malykh, A.; Le Loarer, F.; Italiano, A. ATR Inhibition Broadly Sensitizes Soft-Tissue Sarcoma Cells to Chemotherapy Independent of Alternative Lengthening Telomere (ALT) Status. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Winchester, D.; Lehman, J.; Tello, T.; Chimato, N.; Hocker, T.; Kim, S.; Chang, J.; Markey, J.; Yom, S.S.; Ryan, W.; et al. Undifferentiated pleomorphic sarcoma: Factors predictive of adverse outcomes. J. Am. Acad. Dermatol. 2018, 79, 853–859. [Google Scholar] [CrossRef]
- De Paiva, A.C.G.; De Abreu, M.A.M.M.; De Souza, M.P. Undifferentiated pleomorphic sarcoma*. An. Bras. de Dermatol. 2018, 93, 154–155. [Google Scholar] [CrossRef] [Green Version]
- Widemann, B.C.; Italiano, A. Biology and Management of Undifferentiated Pleomorphic Sarcoma, Myxofibrosarcoma, and Malignant Peripheral Nerve Sheath Tumors: State of the Art and Perspectives. J. Clin. Oncol. 2018, 36, 160–167. [Google Scholar] [CrossRef]
- Matsuo, T.; Shay, J.W.; Wright, W.E.; Hiyama, E.; Shimose, S.; Kubo, T.; Sugita, T.; Yasunaga, Y.; Ochi, M. Telomere-Maintenance Mechanisms in Soft-Tissue Malignant Fibrous Histiocytomas. J. Bone Jt. Surg. Am. Vol. 2009, 91, 928–937. [Google Scholar] [CrossRef]
- Deeg, K.I.; Chung, I.; Bauer, C.; Rippe, K. Cancer Cells with Alternative Lengthening of Telomeres Do Not Display a General Hypersensitivity to ATR Inhibition. Front. Oncol. 2016, 6, 186. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, T.; Zoumpoulidou, G.; Alvarez-Mendoza, C.; Mancusi, C.; Collopy, L.C.; Strauss, S.J.; Mittnacht, S.; Tomita, K. Selective Elimination of Osteosarcoma Cell Lines with Short Telomeres by Ataxia Telangiectasia and Rad3-Related Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1253–1264. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Soler, M.; Morgado-Palacin, I.; Lafarga, V.; Lecona, E.; Murga, M.; Callen, E.; Azorin, D.; Alonso, J.; Lopez-Contreras, A.J.; Nussenzweig, A.; et al. Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget 2016, 7, 58759–58767. [Google Scholar] [CrossRef] [Green Version]
- Gowan, S.M.; Heald, R.; Stevens, M.F.G.; Kelland, L.R. Potent Inhibition of Telomerase by Small-Molecule Pentacyclic Acridines Capable of Interacting with G-Quadruplexes. Mol. Pharmacol. 2001, 60, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Riou, J.F.; Guittat, L.; Mailliet, P.; Laoui, A.; Renou, E.; Petitgenet, O.; Megnin-Chanet, F.; Helene, C.; Mergny, J.L. Cell senescence and telomere shortening induced by a new series of specific G-quadruplex DNA ligands. Proc. Natl. Acad. Sci. USA 2002, 99, 2672–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennarun, G.; Granotier, C.; Gauthier, L.R.; Gomez, D.; Hoffschir, F.; Mandine, E.; Riou, J.-F.; Mergny, J.-L.; Mailliet, P.; Boussin, F.D.; et al. Apoptosis related to telomere instability and cell cycle alterations in human glioma cells treated by new highly selective G-quadruplex ligands. Oncogene 2005, 24, 2917–2928. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, J.; Matsuo, T.; Shimose, S.; Kubo, T.; Ishikawa, M.; Yasunaga, Y.; Ochi, M. Antitumor effects of telomerase inhibitor TMPyP4 in osteosarcoma cell lines. J. Orthop. Res. 2011, 29, 1707–1711. [Google Scholar] [CrossRef]
- Amato, R.; Valenzuela, M.; Berardinelli, F.; Salvati, E.; Maresca, C.; Leone, S.; Antoccia, A.; Sgura, A. G-quadruplex Stabilization Fuels the ALT Pathway in ALT-positive Osteosarcoma Cells. Genes 2020, 11, 304. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Hsieh, M.; Teng, S. Genistein suppresses the proliferation of telomerase-negative cells. Food Sci. Nutr. 2016, 5, 197–204. [Google Scholar] [CrossRef]
- Sung, J.-Y.; Lim, H.-W.; Joung, J.-G.; Park, W.-Y. Pan-Cancer Analysis of Alternative Lengthening of Telomere Activity. Cancers 2020, 12, 2207. [Google Scholar] [CrossRef]
- Hsieh, M.-H.; Tsai, C.-H.; Lin, C.-C.; Li, T.-K.; Hung, T.-W.; Chang, L.-T.; Hsin, L.-W.; Teng, S.-C. Topoisomerase II inhibition suppresses the proliferation of telomerase-negative cancers. Cell. Mol. Life Sci. 2015, 72, 1825–1837. [Google Scholar] [CrossRef]
- Zencir, S.; Hsieh, M.-H.; Hsu, J.-S.; Ergun, Y.; Chou, G.-L.; Li, T.-K.; Teng, S.-C.; Topcu, Z. Selected ellipticine derivatives, known to target topoisomerase II, suppress the alternative lengthening of telomere (ALT) pathway in telomerase–negative cells. J. Cancer Res.Clin. Oncol. 2020, 146, 1671–1676. [Google Scholar] [CrossRef]
- George, S.L.; Lorenzi, F.; King, D.; Hartlieb, S.; Campbell, J.; Pemberton, H.; Toprak, U.H.; Barker, K.; Tall, J.; da Costa, B.M.; et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 2020, 59, 102971. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Tazawa, H.; Hasei, J.; Kunisada, T.; Yoshida, A.; Hashimoto, Y.; Yano, S.; Yoshida, R.; Uno, F.; Kagawa, S.; et al. Preclinical Evaluation of Telomerase-Specific Oncolytic Virotherapy for Human Bone and Soft Tissue Sarcomas. Clin. Cancer Res. 2011, 17, 1828–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Napier, C.E.; Frölich, S.; Teber, E.; Wong, T.; Noble, J.R.; Choi, E.H.Y.; Everett, R.D.; Cesare, A.J.; Reddel, R.R. Synthetic lethality of cytolytic HSV-1 in cancer cells with ATRX and PML deficiency. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, T.; Kagawa, S.; Kobayashi, N.; Shirakiya, Y.; Umeoka, T.; Teraishi, F.; Taki, M.; Kyo, S.; Tanaka, N.; Fujiwara, T. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res. 2004, 10, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.-A.; Maples, P.B.; Chen, S.; et al. A Phase I Study of Telomerase-specific Replication Competent Oncolytic Adenovirus (Telomelysin) for Various Solid Tumors. Mol. Ther. 2010, 18, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.Y.; Nolan, E.; Lindeman, G.J.; Visvader, J.E. Stem Cells and the Differentiation Hierarchy in Mammary Gland Development. Physiol. Rev. 2020, 100, 489–523. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- Gielen, G.H.; Gessi, M.; Buttarelli, F.R.; Baldi, C.; Hammes, J.; Muehlen, A.Z.; Doerner, E.; Denkhaus, D.; Warmuth-Metz, M.; Giangaspero, F.; et al. Genetic Analysis of Diffuse High-Grade Astrocytomas in Infancy Defines a Novel Molecular Entity. Brain Pathol. 2014, 25, 409–417. [Google Scholar] [CrossRef]
- Yu, E.Y.; Cheung, I.Y.; Feng, Y.; Rabie, M.O.; Roboz, G.J.; Guzman, M.L.; Cheung, N.-K.V.; Lue, N.F. Telomere Trimming and DNA Damage as Signatures of High Risk Neuroblastoma. Neoplasia 2019, 21, 689–701. [Google Scholar] [CrossRef]
- Cives, M.; Ghayouri, M.; Morse, B.; Brelsford, M.; Black, M.; Rizzo, A.; Meeker, A.; Strosberg, J. Analysis of potential response predictors to capecitabine/temozolomide in metastatic pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2016, 23, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.-H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.E.; Varkonyi, R.J.; Schwalm, J.; Cragle, R.; Klein-Szanto, A.; Patchefsky, A.; Cukierman, E.; Von Mehren, M.; Broccoli, D. Multiple Mechanisms of Telomere Maintenance Exist in Liposarcomas. Clin. Cancer Res. 2005, 11, 5347–5355. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Shimose, S.; Kubo, T.; Fujimori, J.; Yasunaga, Y.; Ochi, M. Alternative lengthening of telomeres as a prognostic factor in malignant fibrous histiocytomas of bone. Anticancer. Res. 2010, 30, 4959–4962. [Google Scholar] [PubMed]
- Chami, R.; Marrano, P.; Teerapakpinyo, C.; Arnoldo, A.; Shago, M.; Shuangshoti, S.; Thorner, P.S. Immunohistochemistry for ATRX Can Miss ATRX Mutations. Am. J. Surg. Pathol. 2019, 43, 1203–1211. [Google Scholar] [CrossRef]


{kind=link}
| Biomarkers | Telo-FISH Nuclear Foci | APBs | TIFs | Telomerase Activity | C-circles | tSCE | Telomere Heterogeneity |
|---|---|---|---|---|---|---|---|
| ALT+ | √ | √ | √ | X | √ | √ | √ |
| TEL+ | X | X | X | √ | X | X | X |
| Ever-Shorter Telomeres | X | X | X | X | X | X | X |
| Tumor | % ALT+ | Range * | Total Tumors Tested, n |
|---|---|---|---|
| Bone | |||
| Chondrosarcoma | 48% | N/A | 31 |
| Ewing Sarcoma | 0% | N/A | 62 |
| Osteosarcoma | 63% | 49–86% | 287 |
| Breast | |||
| Overall | 4% | 2–8% | 552 |
| HER2+ Breast Carcinoma | 18% | 14–21% | 50 |
| Central Nervous System | |||
| Overall | 20% | 10–26% | 4386 |
| Glioma | 30% | 13–69% | 912 |
| NF1 loss-associated glioma | 44% | 29–69% | 167 |
| Astrocytoma (Overall) | 23% | 10–78% | 2231 |
| Diffuse astrocytoma (grade II) | 55% | 27–100% | 95 |
| Anaplastic astrocytoma (grade III) | 65% | 21–92% | 118 |
| Adult glioblastoma (grade IV) | 16% | 11–25% | 862 |
| Pediatric glioblastoma (grade IV) | 39% | 12–56% | 89 |
| Oligodendroglioma | 17% | 0–25% | 78 |
| Oligoastrocytoma | 60% | 11–72% | 48 |
| Ependymoma | 0% | N/A | 260 |
| Medulloblastoma | 7% | 2–22% | 237 |
| Choroid plexus carcinomas | 23% | N/A | 31 |
| Neuroendocrine | |||
| Neuroblastoma | 24% | 18–47% | 843 |
| Pancreatic Neuroendocrine Tumor (PanNET) | 32% | 21–61% | 1152 |
| Soft Tissue | |||
| Angiosarcoma | 23% | 11–24% | 79 |
| Leiomyoma | 3% | 0–3% | 217 |
| Leiomyosarcoma | 60% | 52–78% | 331 |
| Liposarcoma | 27% | 25–29% | 566 |
| Undifferentiated Pleomorphic Sarcoma | 59% | 36–77% | 174 |
| Tumor | ATRX Loss Only, n | DAXX Loss Only, n | Both ATRX and DAXX Loss, n | Total Tumors Tested, n | Total Mutant % * | ATRX/DAXX Mutant Cases that are ALT+, % | ALT+ Cases that are ATRX Mutant, % |
|---|---|---|---|---|---|---|---|
| Bone | |||||||
| Chondrosarcoma | 0 ** | N/A *** | N/A | 15 | 0% | N/A | N/A |
| Ewing Sarcoma | 0 | N/A | N/A | 12 | 0% | N/A | N/A |
| Osteosarcoma | 17 | 0 | 0 | 71 | 24% | 100% | 58% |
| Breast | |||||||
| Breast Carcinoma | 0 | 0 | 0 | 96 | 0% | N/A | N/A |
| CNS | |||||||
| Glioma | 403 | 7 | N/A | 1607 | 26% | 74% | 71% |
| Neuroendocrine | |||||||
| Neuroblastoma (NB) | 83 | 1 | 0 | 1052 | 8% | 92% | 67% |
| High Risk NB | 25 | 0 | 0 | 165 | 15% | N/A | 100% |
| PanNET | 218 | 153 | 23 | 1223 | 32% | 96% | 86% |
| Soft Tissue | |||||||
| Angiosarcoma | 16 | 0 | 2 | 77 | 21% | N/A | 88% |
| Leiomyoma | 6 | 1 | 0 | 206 | 3% | 43% | 67% |
| Leiomyosarcoma | 103 | 4 | 0 | 311 | 34% | 83% | 56% |
| Liposarcoma (LPS) | 39 | 1 | N/A | 203 | 20% | 100% | 78% |
| Well differentiated LPS | 0 | N/A | N/A | 6 | 0% | N/A | N/A |
| Dedifferentiated LPS | 28 | 1 | 0 | 52 | 56% | 100% | 93% |
| Myxoid LPS | 0 | N/A | N/A | 55 | 0% | N/A | N/A |
| Pleomorphic LPS | 11 | N/A | N/A | 27 | 41% | 100% | 63% |
| Undifferentiated Pleomorphic Sarcoma | 32 | N/A | N/A | 87 | 37% | 96% | 55% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacKenzie, D., Jr.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers 2021, 13, 2384. https://doi.org/10.3390/cancers13102384
MacKenzie D Jr., Watters AK, To JT, Young MW, Muratori J, Wilkoff MH, Abraham RG, Plummer MM, Zhang D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers. 2021; 13(10):2384. https://doi.org/10.3390/cancers13102384
Chicago/Turabian StyleMacKenzie, Danny, Jr., Andrea K. Watters, Julie T. To, Melody W. Young, Jonathan Muratori, Marni H. Wilkoff, Rita G. Abraham, Maria M. Plummer, and Dong Zhang. 2021. "ALT Positivity in Human Cancers: Prevalence and Clinical Insights" Cancers 13, no. 10: 2384. https://doi.org/10.3390/cancers13102384