
Efficacy of Targeted Radionuclide Therapy Using [131I]ICF01012 in 3D Pigmented BRAF- and NRAS-Mutant Melanoma Models and In Vivo NRAS-Mutant Melanoma

 , , , , , , , and
, , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Conditions and Spheroid Collection
2.2. [131I]ICF01012 TRT and MEK Inhibitor Treatment of Spheroids
2.3. Colony Formation Assay
2.4. Apoptosis Assay
2.5. Cell Cycle Analysis
2.6. Western Blotting
2.7. Murine Models
2.8. Biodistribution Study
2.9. Dosimetry
2.10. [131I]ICF01012 Treatment
2.11. SPECT-CT Imaging
2.12. Melanin Determination
2.13. RT-qPCR Analyses
2.14. Transcriptomic Analysis
2.15. Statistical Analyses
3. Results
3.1. MEKi Radio-Sensitizes BRAF- and NRAS-Mutant Melanoma Spheroids for [131I]ICF01012 by Increasing Apoptosis
3.2. MEKi Inhibits [131I]ICF01012-Induced ERK1/2 Phosphorylation in BRAF- and NRAS-Mutant Melanoma Spheroid Models
3.3. MEKi Enhances the Radiosensitivity of BRAF- and NRAS-Mutant Melanoma Spheroids, Leading to Increased DNA Double-Strand Breaks
3.4. [131I]ICF01012 Accumulates in NRAS 1007 Tumors, with a Favorable Dosimetry for TRT
3.5. [131I]ICF01012 Induces Significant Tumor Regression and Extended Survival for Mice Bearing NRAS 1007 Tumors
3.6. [131I]ICF01012 Reduces Lymph-Node Metastases in the NRAS 1007 Model
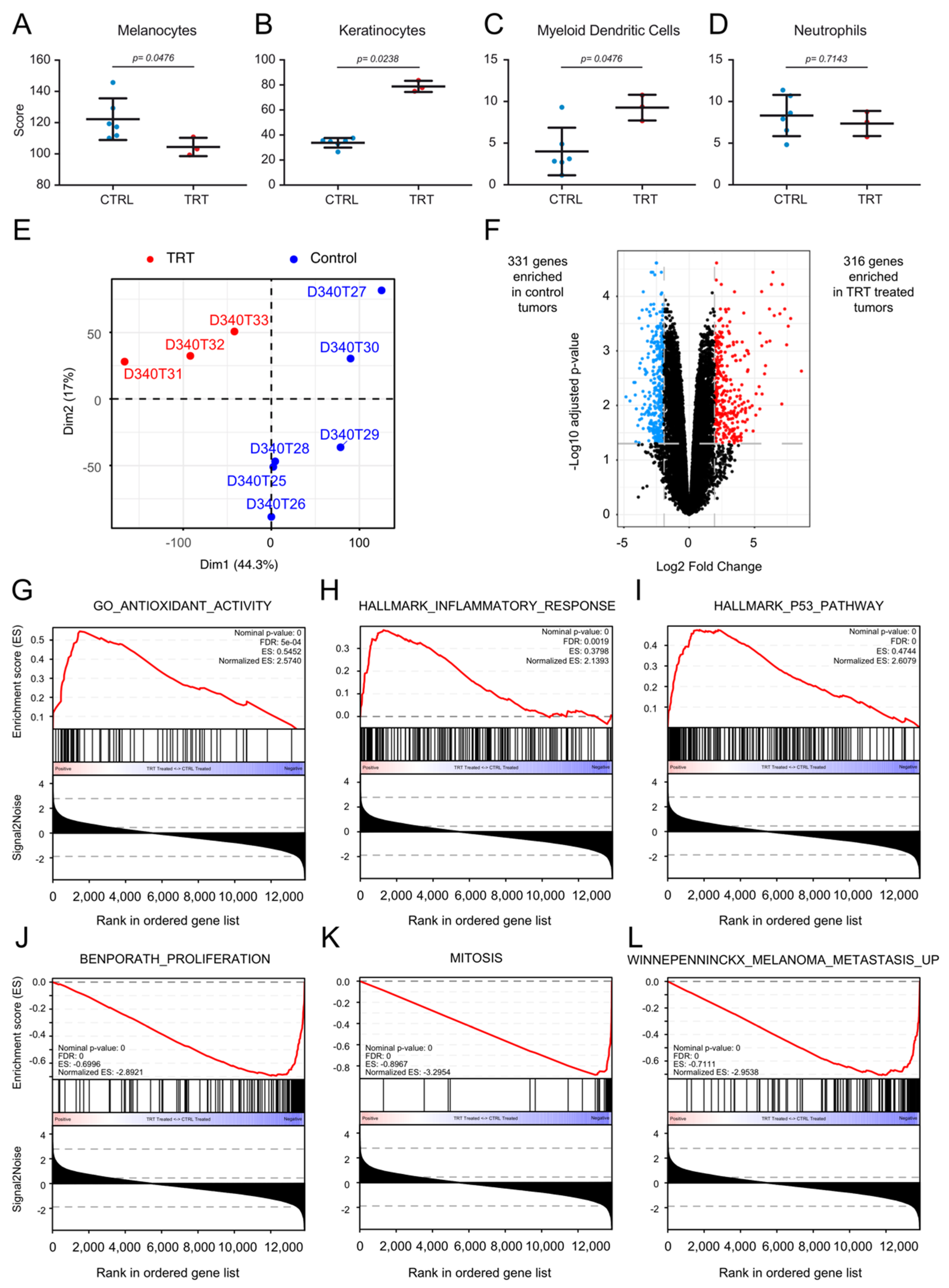
3.7. [131I]ICF01012 Modifies Oxidative Stress, Inflammatory, and P53 Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK signaling pathway in melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesbert, F.; Larue, L. Cutaneous melanoma: From rationalized models to patients care. Med. Sci. 2018, 34, 407–416. [Google Scholar] [CrossRef] [Green Version]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Prasad, C.P.; Mohapatra, P.; Andersson, T. Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer? Cancers 2015, 7, 1900–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, M.; Holzel, M. The impact of melanoma genetics on treatment response and resistance in clinical and experimental studies. Cancer Metastasis Rev. 2017, 36, 53–75. [Google Scholar] [CrossRef]
- Mason, R.; Au, L.; Ingles Garces, A.; Larkin, J. Current and emerging systemic therapies for cutaneous metastatic melanoma. Expert Opin. Pharm. 2019, 20, 1135–1152. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-year outcomes with dabrafenib plus trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Salama, A.K.; Niedzwiecki, D.; Johnson, J.; Linette, G.; Bucher, C.; Blaskovich, M.A.; Sebti, S.M.; Haluska, F. Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104). J. Transl. Med. 2012, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Smalley, K.S.; Eisen, T.G. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int. J. Cancer 2003, 105, 165–175. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Norain, A.; Dadachova, E. Targeted radionuclide therapy of melanoma. Semin. Nucl. Med. 2016, 46, 250–259. [Google Scholar] [CrossRef]
- Degoul, F.; Borel, M.; Jacquemot, N.; Besse, S.; Communal, Y.; Mishellany, F.; Papon, J.; Penault-Llorca, F.; Donnarieix, D.; Doly, M.; et al. In vivo efficacy of melanoma internal radionuclide therapy with a 131I-labelled melanin-targeting heteroarylcarboxamide molecule. Int. J. Cancer 2013, 133, 1042–1053. [Google Scholar] [CrossRef]
- Viallard, C.; Perrot, Y.; Boudhraa, Z.; Jouberton, E.; Miot-Noirault, E.; Bonnet, M.; Besse, S.; Mishellany, F.; Cayre, A.; Maigne, L.; et al. [123I]ICF01012 melanoma imaging and [131I]ICF01012 dosimetry allow adapted internal targeted radiotherapy in preclinical melanoma models. Eur. J. Dermatol. 2015, 25, 29–35. [Google Scholar] [CrossRef]
- Bonnet, M.; Mishellany, F.; Papon, J.; Cayre, A.; Penault-Llorca, F.; Madelmont, J.C.; Miot-Noirault, E.; Chezal, J.M.; Moins, N. Anti-melanoma efficacy of internal radionuclide therapy in relation to melanin target distribution. Pigment Cell Melanoma Res. 2010, 23, e1–e11. [Google Scholar] [CrossRef]
- Bonnet-Duquennoy, M.; Papon, J.; Mishellany, F.; Labarre, P.; Guerquin-Kern, J.L.; Wu, T.D.; Gardette, M.; Maublant, J.; Penault-Llorca, F.; Miot-Noirault, E.; et al. Targeted radionuclide therapy of melanoma: Anti-tumoural efficacy studies of a new 131I labelled potential agent. Int. J. Cancer 2009, 125, 708–716. [Google Scholar] [CrossRef]
- Viallard, C.; Chezal, J.M.; Mishellany, F.; Ranchon-Cole, I.; Pereira, B.; Herbette, A.; Besse, S.; Boudhraa, Z.; Jacquemot, N.; Cayre, A.; et al. Targeting DNA repair by coDbait enhances melanoma targeted radionuclide therapy. Oncotarget 2016, 7, 12927–12936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chezal, J.M.; Papon, J.; Labarre, P.; Lartigue, C.; Galmier, M.J.; Decombat, C.; Chavignon, O.; Maublant, J.; Teulade, J.C.; Madelmont, J.C.; et al. Evaluation of radiolabeled (hetero)aromatic analogues of N-(2-diethylaminoethyl)-4-iodobenzamide for imaging and targeted radionuclide therapy of melanoma. J. Med. Chem. 2008, 51, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Akil, H.; Rouanet, J.; Viallard, C.; Besse, S.; Auzeloux, P.; Chezal, J.M.; Miot-Noirault, E.; Quintana, M.; Degoul, F. Targeted radionuclide therapy decreases melanoma lung invasion by modifying epithelial-mesenchymal transition-like mechanisms. Transl. Oncol. 2019, 12, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Schick, U.; Kyula, J.; Barker, H.; Patel, R.; Zaidi, S.; Gregory, C.; Hafsi, H.; Roulstone, V.; Deutsch, E.; McLaughlin, M.; et al. Trametinib radiosensitises RAS- and BRAF-mutated melanoma by perturbing cell cycle and inducing senescence. Radiother. Oncol. 2015, 117, 364–375. [Google Scholar] [CrossRef]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Biological Rationale for Targeting MEK/ERK Pathways in anti-cancer therapy and to potentiate tumour responses to radiation. Int. J. Mol. Sci. 2019, 20, 2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, M.; Zimmer, L.; Loquai, C.; Weishaupt, C.; Gutzmer, R.; Schuster, B.; Gleisner, S.; Schulze, B.; Goldinger, S.M.; Berking, C.; et al. Radiosensitization by BRAF inhibitor therapy-mechanism and frequency of toxicity in melanoma patients. Ann. Oncol. 2015, 26, 1238–1244. [Google Scholar] [CrossRef]
- Petit, V.; Raymond, J.; Alberti, C.; Pouteaux, M.; Gallagher, S.J.; Nguyen, M.Q.; Aplin, A.E.; Delmas, V.; Larue, L. C57BL/6 congenic mouse NRAS(Q61K) melanoma cell lines are highly sensitive to the combination of Mek and Akt inhibitors in vitro and in vivo. Pigment Cell Melanoma Res. 2019. [Google Scholar] [CrossRef]
- Buch, K.; Peters, T.; Nawroth, T.; Sanger, M.; Schmidberger, H.; Langguth, P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay--a comparative study. Radiat. Oncol. 2012, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Akil, H.; Abbaci, A.; Lalloue, F.; Bessette, B.; Costes, L.M.; Domballe, L.; Charreau, S.; Guilloteau, K.; Karayan-Tapon, L.; Bernard, F.X.; et al. IL22/IL-22R pathway induces cell survival in human glioblastoma cells. PLoS ONE 2015, 10, e0119872. [Google Scholar] [CrossRef] [Green Version]
- Perrot, Y.; Degoul, F.; Auzeloux, P.; Bonnet, M.; Cachin, F.; Chezal, J.M.; Donnarieix, D.; Labarre, P.; Moins, N.; Papon, J.; et al. Internal dosimetry through GATE simulations of preclinical radiotherapy using a melanin-targeting ligand. Phys. Med. Biol. 2014, 59, 2183–2198. [Google Scholar] [CrossRef] [Green Version]
- Stabin, M.G. Developments in the internal dosimetry of radiopharmaceuticals. Radiat. Prot. Dosim. 2003, 105, 575–580. [Google Scholar] [CrossRef]
- Rioux, B.; Rouanet, J.; Akil, H.; Besse, S.; Debiton, E.; Bouchon, B.; Degoul, F.; Quintana, M. Determination of eumelanin and pheomelanin in melanomas using solid-phase extraction and high performance liquid chromatography-diode array detection (HPLC-DAD) analysis. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2019, 1113, 60–68. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Sievert, C. Interactive Web-based Data Visualization with R, Plotly, and Shiny; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2020. [Google Scholar]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward minimal residual disease-directed therapy in melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.M.; Zhang, G.; Bastian, B.C.; Arcasoy, M.O.; Karande, P.; Pushparajan, A.; Acs, G.; Xu, X. Erythropoietin receptor contributes to melanoma cell survival in vivo. Oncogene 2012, 31, 1649–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, M.R.; Pedersen, S.R.; Lindkvist, A.; Christensen, J.P.; Thomsen, A.R. Quantification of B16 melanoma cells in lungs using triplex Q-PCR--a new approach to evaluate melanoma cell metastasis and tumor control. PLoS ONE 2014, 9, e87831. [Google Scholar] [CrossRef]
- Munshi, A.; Ramesh, R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Pomp, J.; Ouwerkerk, I.J.; Hermans, J.; Wondergem, J.; Cornelisse, C.J.; Leer, J.W.; Schrier, P.I. The influence of the oncogenes NRAS and MYC on the radiation sensitivity of cells of a human melanoma cell line. Radiat Res. 1996, 146, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Rutter, C.E.; Johung, K.L.; Yao, X.; Lu, A.Y.; Jilaveanu, L.B.; Yu, J.B.; Contessa, J.N.; Kluger, H.M.; Chiang, V.L.S.; Bindra, R.S. Demonstration of differential radiosensitivity based upon mutation profile in metastatic melanoma treated with stereotactic radiosurgery. J. Radiosurg. SBRT 2016, 4, 97–106. [Google Scholar] [PubMed]
- Conde-Perez, A.; Larue, L. Human relevance of NRAS/BRAF mouse melanoma models. Eur. J. Cell Biol. 2014, 93, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Jouberton, E.; Perrot, Y.; Dirat, B.; Billoux, T.; Auzeloux, P.; Cachin, F.; Chezal, J.M.; Filaire, M.; Labarre, P.; Miot-Noirault, E.; et al. Radiation dosimetry of [131I]ICF01012 in rabbits: Application to targeted radionuclide therapy for human melanoma treatment. Med. Phys. 2018, 45, 5251–5262. [Google Scholar] [CrossRef]
- Mier, W.; Kratochwil, C.; Hassel, J.C.; Giesel, F.L.; Beijer, B.; Babich, J.W.; Friebe, M.; Eisenhut, M.; Enk, A.; Haberkorn, U. Radiopharmaceutical therapy of patients with metastasized melanoma with the melanin-binding benzamide 131I-BA52. J. Nucl. Med. 2014, 55, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, A.; Panzella, L.; Monfrecola, G.; d’Ischia, M. Pheomelanin-induced oxidative stress: Bright and dark chemistry bridging red hair phenotype and melanoma. Pigment. Cell Melanoma Res. 2014, 27, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouanet, J.; Benboubker, V.; Akil, H.; Hennino, A.; Auzeloux, P.; Besse, S.; Pereira, B.; Delorme, S.; Mansard, S.; D’Incan, M.; et al. Immune checkpoint inhibitors reverse tolerogenic mechanisms induced by melanoma targeted radionuclide therapy. Cancer Immunol. Immunother. 2020, 69, 2075–2088. [Google Scholar] [CrossRef] [PubMed]
- Ladjohounlou, R.; Lozza, C.; Pichard, A.; Constanzo, J.; Karam, J.; Le Fur, P.; Deshayes, E.; Boudousq, V.; Paillas, S.; Busson, M.; et al. Drugs that modify cholesterol metabolism alter the p38/JNK-mediated targeted and nontargeted response to alpha and auger radioimmunotherapy. Clin. Cancer Res. 2019, 25, 4775–4790. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ/Tissue | Absorbed Dose (Gy·GBq−1) | Dose for 3.7 GBq (Gy) |
|---|---|---|
| Adrenals | 0.006 | 0.023 |
| Brain | 0.007 | 0.025 |
| Gall bladder | 0.010 | 0.037 |
| Lower large intestine | 0.021 | 0.079 |
| Small Intestine | 0.048 | 0.176 |
| Stomach | 0.043 | 0.159 |
| Upper large intestine | 0.021 | 0.078 |
| Heart | 0.012 | 0.044 |
| Kidneys | 0.055 | 0.203 |
| Liver | 0.064 | 0.238 |
| Lungs | 0.028 | 0.105 |
| Muscle | 0.003 | 0.009 |
| Ovaries | 0.007 | 0.026 |
| Pancreas | 0.008 | 0.028 |
| Red Marrow | 0.003 | 0.010 |
| Skin | 0.001 | 0.004 |
| Spleen | 0.062 | 0.229 |
| Testes | 0.012 | 0.044 |
| Thymus | 0.002 | 0.007 |
| Thyroid | 0.044 | 0.163 |
| Urinary Bladder | 0.184 | 0.682 |
| Total body | 0.006 | 0.021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akil, H.; Quintana, M.; Raymond, J.H.; Billoux, T.; Benboubker, V.; Besse, S.; Auzeloux, P.; Delmas, V.; Petit, V.; Larue, L.; et al. Efficacy of Targeted Radionuclide Therapy Using [131I]ICF01012 in 3D Pigmented BRAF- and NRAS-Mutant Melanoma Models and In Vivo NRAS-Mutant Melanoma. Cancers 2021, 13, 1421. https://doi.org/10.3390/cancers13061421
Akil H, Quintana M, Raymond JH, Billoux T, Benboubker V, Besse S, Auzeloux P, Delmas V, Petit V, Larue L, et al. Efficacy of Targeted Radionuclide Therapy Using [131I]ICF01012 in 3D Pigmented BRAF- and NRAS-Mutant Melanoma Models and In Vivo NRAS-Mutant Melanoma. Cancers. 2021; 13(6):1421. https://doi.org/10.3390/cancers13061421
Chicago/Turabian StyleAkil, Hussein, Mercedes Quintana, Jérémy H. Raymond, Tommy Billoux, Valentin Benboubker, Sophie Besse, Philippe Auzeloux, Véronique Delmas, Valérie Petit, Lionel Larue, and et al. 2021. "Efficacy of Targeted Radionuclide Therapy Using [131I]ICF01012 in 3D Pigmented BRAF- and NRAS-Mutant Melanoma Models and In Vivo NRAS-Mutant Melanoma" Cancers 13, no. 6: 1421. https://doi.org/10.3390/cancers13061421