Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations

Abstract
:Simple Summary
Abstract
1. Introduction
2. Hypoxia
2.1. Molecular Markers
2.2. Immune Cells
2.3. Tumor and Stromal Cells
2.4. Angiogenesis
2.5. Targeting Hypoxia
3. Oxidative Stress
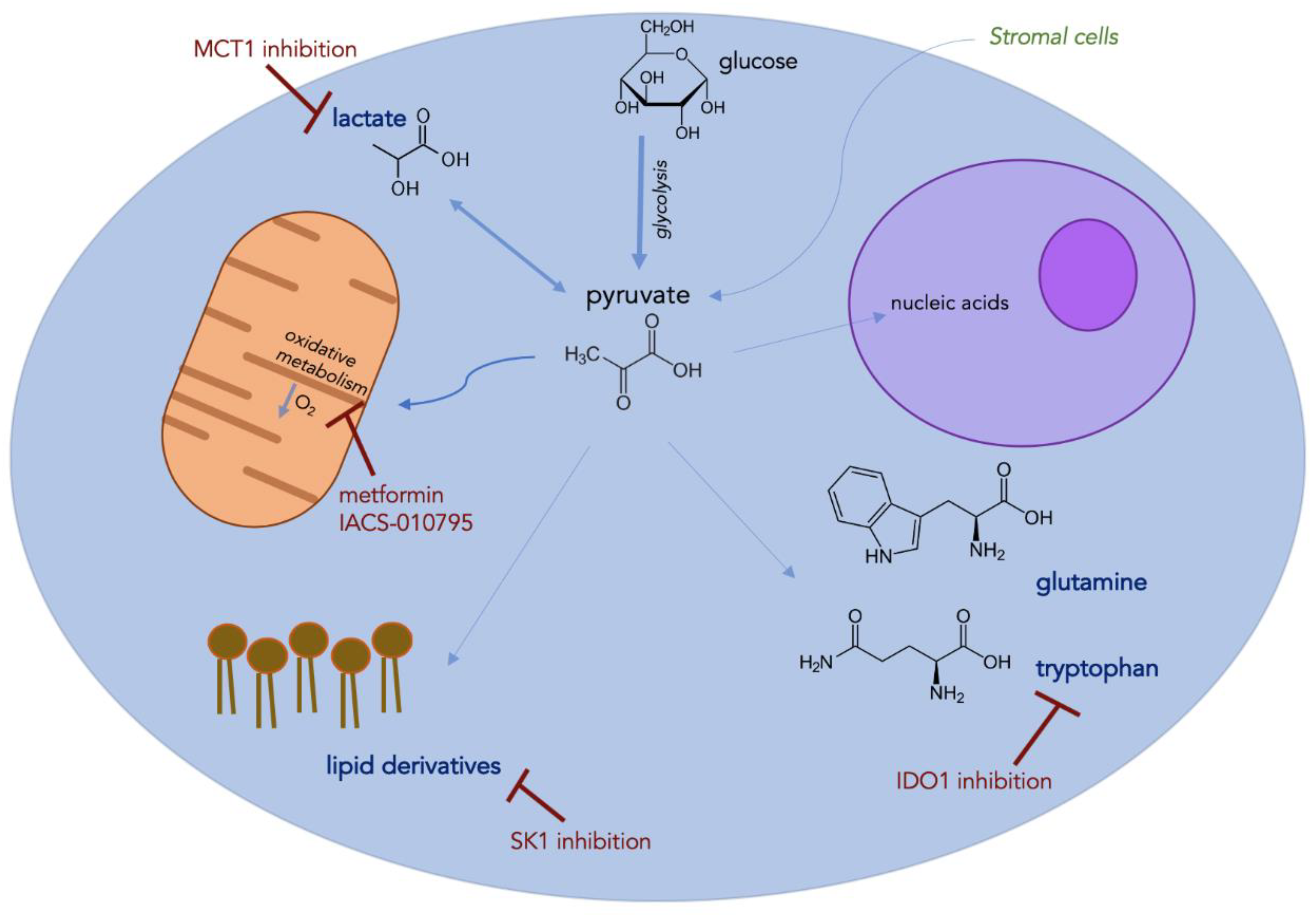
4. Metabolic Alterations
4.1. Glycolysis and Oxidative Phosphorylation
4.2. Amino Acids
4.3. Lipids
4.4. Lactate
5. Conclusions
Funding
Conflicts of Interest
References
- Wang, Y.-P.; Li, J.-T.; Qu, J.; Yin, M.; Lei, Q.-Y. Metabolite sensing and signaling in Cancer. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Wei, F.; Wang, D.; Wei, J.; Tang, N.; Tang, L.; Xiong, F.; Guo, C.; Zhou, M.; Li, X.; Li, G.; et al. Metabolic crosstalk in the tumor microenvironment regulates antitumor immunosuppression and immunotherapy resistance. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles Garces, A.H.; Au, L.; Mason, R.; Thomas, J.; Larkin, J. Building on the anti-PD1/PD-L1 backbone: Combination immunotherapy for cancer. Expert Opin. Investig. Drugs 2019, 28, 695–708. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Brown, J.M. Exploiting tumour hypoxia and overcoming mutant p53 with tirapazamine. Br. J. Cancer 1998, 77 (Suppl. 4), 12–14. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Hasmim, M.; Lequeux, A.; Xiao, M.; Duhem, C.; Chouaib, S.; Berchem, G.; Janji, B. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: New opportunities and challenges. Cells 2019, 8, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusi, A.; Festino, L.; Botti, G.; Masucci, G.; Melero, I.; Lorigan, P.; Ascierto, P.A. PD-L1 expression as a potential predictive biomarker. Lancet Oncol. 2015, 16, 1285–1287. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Li, J.; Sarde, A.; Lines, J.L.; Lee, Y.-C.; Qian, D.C.; Pechenick, D.A.; Manivanh, R.; Le Mercier, I.; Lowrey, C.H.; et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol. Res. 2019, 7, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [Green Version]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L.; et al. CD47 blockade as an adjuvant immunotherapy for resectable pancreatic cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Yaghi, L.; Poras, I.; Simoes, R.T.; Donadi, E.A.; Tost, J.; Daunay, A.; de Almeida, B.S.; Carosella, E.D.; Moreau, P. Hypoxia inducible factor-1 mediates the expression of the immune checkpoint HLA-G in glioma cells through hypoxia response element located in exon 2. Oncotarget 2016, 7, 63690–63707. [Google Scholar] [CrossRef] [Green Version]
- Ambade, A.; Satishchandran, A.; Saha, B.; Gyongyosi, B.; Lowe, P.; Kodys, K.; Catalano, D.; Szabo, G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10. Oncoimmunology 2016, 5, e1221557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Jagannathan, N. Tumor associated macrophage: A review on the phenotypes, traits and functions. Iran. J. Cancer Prev. 2014, 7, 1–8. [Google Scholar]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Najjar, Y.G.; Rayman, P.; Jia, X.; Pavicic, P.G.; Rini, B.I.; Tannenbaum, C.; Ko, J.; Haywood, S.; Cohen, P.; Hamilton, T.; et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and Mip-1α. Clin. Cancer Res. 2017, 23, 2346–2355. [Google Scholar] [CrossRef] [Green Version]
- Najjar, Y.G.; Finke, J.H. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front. Oncol. 2013, 3, 49. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Deng, G. Tumor-infiltrating regulatory T cells: Origins and features. Am. J. Clin. Exp. Immunol. 2018, 7, 81–87. [Google Scholar]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef] [Green Version]
- de Silly, V.R.; Dietrich, P.-Y.; Walker, P.R. Hypoxia and antitumor CD8+ T cells: An incompatible alliance? Oncoimmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.G.; Hill, R.P.; Tannock, I.F. Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieran, M.W.; Kalluri, R.; Cho, Y.-J. The VEGF pathway in cancer and disease: Responses, resistance, and the path forward. Cold Spring Harb. Perspect. Med. 2012, 2, a006593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Scientific rationale for combined immunotherapy with PD-1/PD-L1 antibodies and VEGF inhibitors in advanced hepatocellular carcinoma. Cancers 2020, 12, 1089. [Google Scholar] [CrossRef]
- A Study of Atezolizumab in Combination with Bevacizumab Compared with Sorafenib in Patients with Untreated Locally Advanced or Metastatic Hepatocellular Carcinoma [IMbrave150]—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03434379 (accessed on 16 July 2020).
- A Phase I/II Study of Regorafenib Plus Avelumab in Solid Tumors—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03475953 (accessed on 16 July 2020).
- Study of Cabozantinib in Combination With Atezolizumab to Subjects with Locally Advanced or Metastatic Solid Tumors—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03170960 (accessed on 16 July 2020).
- Safety and Efficacy Study of Pembrolizumab (MK-3475) Combined with Lenvatinib (MK-7902/E7080) as First-line Intervention in Adults with Advance Melanoma (MK-7902-003/E7080-G000-312/LEAP-003)—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03820986 (accessed on 5 August 2020).
- Arance Fernandez, A.M.; Ascierto, P.A.; Carlino, M.S.; Daud, A.; Eggermont, A.M.; Hauschild, A.; Kluger, H.M.; Taylor, M.H.; Smith, A.; Chen, K.; et al. Lenvatinib (len) plus pembrolizumab (pembro) in patients (pts) with advanced melanoma previously exposed to anti–PD-1/PD-L1 agents: Phase 2 LEAP-004 study. JCO 2019, 37, TPS9594. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. KEYNOTE-426 investigators pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T cell-inflamed versus non-T cell-inflamed tumors: A conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: New approaches for combinational immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Hunter, F.W.; Wouters, B.G.; Wilson, W.R. Hypoxia-activated prodrugs: Paths forward in the era of personalised medicine. Br. J. Cancer 2016, 114, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical advances of hypoxia-activated prodrugs in combination with radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Borad, M.J.; Reddy, S.G.; Bahary, N.; Uronis, H.E.; Sigal, D.; Cohn, A.L.; Schelman, W.R.; Stephenson, J.; Chiorean, E.G.; Rosen, P.J.; et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Lenz, H.-J.; Furuse, J.; Tabernero, J.; Heinemann, V.; Ioka, T.; Bazin, I.; Ueno, M.; Csõszi, T.; Wasan, H.; et al. MAESTRO: A randomized, double-blind phase III study of evofosfamide (Evo) in combination with gemcitabine (Gem) in previously untreated patients (pts) with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2016, 34, 4007. [Google Scholar] [CrossRef]
- Ai, M.; Budhani, P.; Sheng, J.; Balasubramanyam, S.; Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Haria, D.D.; Curran, M.A. Tumor hypoxia drives immune suppression and immunotherapy resistance. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.-K.; Moniz, R.J.; Xu, Z.; Sun, J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Human anti-CAIX antibodies mediate immune cell inhibition of renal cell carcinoma in vitro and in a humanized mouse model in vivo. Mol. Cancer 2015, 14, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamie, K.; Donin, N.M.; Klöpfer, P.; Bevan, P.; Fall, B.; Wilhelm, O.; Störkel, S.; Said, J.; Gambla, M.; Hawkins, R.E.; et al. Adjuvant weekly girentuximab following nephrectomy for high-risk renal cell carcinoma: The ARISER randomized clinical trial. JAMA Oncol. 2017, 3, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-R.; Deng, W.-W.; Liu, J.-F.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef] [PubMed]
- Phase 1/1b Study to Evaluate the Safety and Tolerability of Ciforadenant Alone and in Combination with Atezolizumab in Advanced Cancers—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2016-00227&r=1 (accessed on 5 August 2020).
- CPI-006 Alone and in Combination with Ciforadenant and with Pembrolizumab for Patients With Advanced Cancers—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2018-02296&r=1 (accessed on 5 August 2020).
- Fallah, J.; Rini, B.I. HIF inhibitors: Status of current clinical development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive oxygen species and antitumor immunity-from surveillance to evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Sierra, C.; Laranjeira, P.; Domingues, M.R.; Paiva, A. Lipoxidation and cancer immunity. Redox Biol. 2019, 23, 101103. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Wall, S.B.; Kramer, P.A.; Ravi, S.; Mitchell, T.; Johnson, M.S.; Wilson, L.; Barnes, S.; Landar, A.; Darley-Usmar, V.M. Pleiotropic effects of 4-hydroxynonenal on oxidative burst and phagocytosis in neutrophils. Redox Biol. 2016, 9, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-R.; Chang, S.-Y.; Hong, E.-H.; Kwon, B.-E.; Kim, H.M.; Kim, Y.-J.; Lee, J.; Cho, H.-J.; Cheon, J.-H.; Ko, H.-J. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget 2014, 5, 12331–12345. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, C.; Cuomo, A.; Spadoni, I.; Magni, E.; Silvola, A.; Conte, A.; Sigismund, S.; Ravenda, P.S.; Bonaldi, T.; Zampino, M.G.; et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 2016, 22, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Anufreichik, V.; Rodvold, J.J.; Chiu, K.T.; Sepulveda, H.; Zanetti, M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8+ T cell priming. PLoS ONE 2012, 7, e51845. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, A.C.S.; Victorino, V.J.; Campos, F.C.; Verenitach, B.D.; Lemos, L.T.; Aranome, A.M.F.; Oliveira, S.R.; Cecchini, A.L.; Simão, A.N.C.; Abdelhay, E.; et al. Impact of tumor removal on the systemic oxidative profile of patients with breast cancer discloses lipid peroxidation at diagnosis as a putative marker of disease recurrence. Clin. Breast. Cancer 2014, 14, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Devic, S. Warburg effect—A consequence or the cause of carcinogenesis? J. Cancer 2016, 7, 817–822. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015, 282, 3892–3898. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- A Trial of Pembrolizumab and Metformin Versus Pembrolizumab Alone in Advanced Melanoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03311308 (accessed on 23 June 2020).
- Fischer, G.M.; Jalali, A.; Kircher, D.A.; Lee, W.-C.; McQuade, J.L.; Haydu, L.E.; Joon, A.Y.; Reuben, A.; de Macedo, M.P.; Carapeto, F.C.L.; et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 2019, 9, 628–645. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Rodon Ahnert, J.; Piha-Paul, S.A.; Fu, S.; Janku, F.; Karp, D.D.; Naing, A.; Ileana Dumbrava, E.E.; Pant, S.; Subbiah, V.; et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3014. [Google Scholar] [CrossRef]
- Oxidative Phosphorylation Inhibitor IACS-010759 in Treating Patients With Relapsed or Refractory Acute Myeloid Leukemia—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02882321 (accessed on 6 August 2020).
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Tham, M.; Tan, K.W.; Keeble, J.; Wang, X.; Hubert, S.; Barron, L.; Tan, N.S.; Kato, M.; Prevost-Blondel, A.; Angeli, V.; et al. Melanoma-initiating cells exploit M2 macrophage TGFβ and arginase pathway for survival and proliferation. Oncotarget 2014, 5, 12027–12042. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Chang, M.Y.; Parker, K.H.; Beury, D.W.; DuHadaway, J.B.; Flick, H.E.; Boulden, J.; Sutanto-Ward, E.; Soler, A.P.; Laury-Kleintop, L.D.; et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012, 2, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.P.; Ferreira, A.C.F.; Pinho, M.P.; de Moraes, C.J.; Bergami-Santos, P.C.; Barbuto, J.A.M. Tolerogenic IDO(+) dendritic cells are induced by PD-1-expressing mast cells. Front. Immunol. 2016, 7, 9. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label Phase I/II trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Tiwary, S.; Berzofsky, J.A.; Terabe, M. Altered lipid tumor environment and its potential effects on NKT cell function in tumor immunity. Front. Immunol. 2019, 10, 2187. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.-J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Y.; Li, C.-F.; Kuo, C.-C.; Tsai, K.K.; Hou, M.-F.; Hung, W.-C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef] [Green Version]
- Heusinkveld, M.; de Vos van Steenwijk, P.J.; Goedemans, R.; Ramwadhdoebe, T.H.; Gorter, A.; Welters, M.J.P.; van Hall, T.; van der Burg, S.H. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J. Immunol. 2011, 187, 1157–1165. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Lloyd, F.L.; Trifan, O.C.; Knapp, B.; Rizzo, M.T. Potential involvement of the cyclooxygenase-2 pathway in the regulation of tumor-associated angiogenesis and growth in pancreatic cancer. Mol. Cancer Ther. 2003, 2, 1–7. [Google Scholar] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef]
- Raynor, A.; Jantscheff, P.; Ross, T.; Schlesinger, M.; Wilde, M.; Haasis, S.; Dreckmann, T.; Bendas, G.; Massing, U. Saturated and mono-unsaturated lysophosphatidylcholine metabolism in tumour cells: A potential therapeutic target for preventing metastases. Lipids Health Dis. 2015, 14, 69. [Google Scholar] [CrossRef] [Green Version]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Bilal, F.; Montfort, A.; Gilhodes, J.; Garcia, V.; Riond, J.; Carpentier, S.; Filleron, T.; Colacios, C.; Levade, T.; Daher, A.; et al. Sphingomyelin synthase 1 (SMS1) downregulation is associated with sphingolipid reprogramming and a worse prognosis in melanoma. Front. Pharmacol. 2019, 10, 443. [Google Scholar] [CrossRef] [Green Version]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Toti, A.; Giannoni, E.; Bianchini, F.; Margheri, F.; Del Rosso, M.; Calorini, L. Metformin is also effective on lactic acidosis-exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle 2016, 15, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Beloueche-Babari, M.; Casals Galobart, T.; Delgado-Goni, T.; Wantuch, S.; Parkes, H.G.; Tandy, D.; Harker, J.A.; Leach, M.O. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br. J. Cancer 2020, 122, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.-M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058.e4. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Drug | Mechanism | Clinical Investigation |
|---|---|---|
| Evofosfamide (TH-302) | Pro-drug, converting to alkylating cytotoxic agent in hypoxic conditions | Phase I, evofosfamide plus ipilimumab in advanced solid cancers (NCT03098160) Phase I/II, evofosfamide plus bortezomib in relapsed/refractory MM (NCT01522872) |
| Girentuximab | Anti-CAIX antibody (upregulated via HIF-1) | Phase III, girentuximab vs. placebo in localized high-risk ccRCC (NCT00087022) |
| Ciforadenant (CPI-444) | Selective inhibitor of adenosine A2A receptor (A2AR) | Phase I/Ib, ciforadenant plus atezolizumab in advanced solid tumors (NCT02655822) Phase I/Ib, anti-CD73 plus ciforadenant plus pembrolizumab in advanced solid tumors (NCT03454451) |
| 2ME2 | Multifactorial inhibitor of HIF-1α and angiogenesis | Phase I, 2ME2 in advanced solid tumors (NCT00028821) Phase II, 2ME2 plus sunitinib in metastatic RCC (NCT00444314) |
| 17-AAG | Inhibitor of HSP-90, increases degradation of HIF-1α | Phase II, 17-AAG in advanced melanoma (NCT00087386) Phase I, 17-AAG plus sorafenib in advanced solid tumors (NCT00121264) |
| Vorinostat | Inhibitor of histone deacetylase (HDAC), leads to HIF-1α degradation | Phase II, vorinostat plus pembrolizumab in recurrent HNSCC (NCT02538510) |
| PT2977 | HIF-2α inhibitor (second generation) | Phase II, PT2977 plus cabozantinib in advanced RCC (NCT03634540) |
| EZN-2208 | Metabolite of irinotecan, suppresses HIF-1 mRNA expression | Phase II, EZN-2208 plus cetuximab in metastatic CRC (NCT00931840) |
| CRLX101 | Nanoparticle conjugate with camptothecin, inhibits HIF-1α | Phase II, CRLX101 plus bevacizumab in smetastatic RCC (NCT02187302) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Augustin, R.C.; Delgoffe, G.M.; Najjar, Y.G. Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations. Cancers 2020, 12, 3802. https://doi.org/10.3390/cancers12123802
Augustin RC, Delgoffe GM, Najjar YG. Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations. Cancers. 2020; 12(12):3802. https://doi.org/10.3390/cancers12123802
Chicago/Turabian StyleAugustin, Ryan C., Greg M. Delgoffe, and Yana G. Najjar. 2020. "Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations" Cancers 12, no. 12: 3802. https://doi.org/10.3390/cancers12123802