Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness



1
Biochemistry and Tumor Biology Lab, Department of Obstetrics and Gynecology, Hannover Medical School, 30625 Hannover, Germany
2
First Department of Medicine, University Hospital Schleswig-Holstein, Campus Lübeck, 23538 Lübeck, Germany
3
Department of General Surgery, Visceral, Thoracic, Transplantation and Pediatric Surgery, University Hospital Schleswig-Holstein, Campus Kiel, 24105 Kiel, Germany
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(12), 3716; https://doi.org/10.3390/cancers12123716
Submission received: 16 November 2020
/
Revised: 8 December 2020
/
Accepted: 8 December 2020
/
Published: 11 December 2020
(This article belongs to the Special Issue Stemness and Differentiation in Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
The cancer cells in solid tumors are embedded in a complex connective tissue matrix composed of various other cell types, i.e., mesenchymal stroma/stem-like cells (MSCs) and tumor-associated macrophages (TAMs). This tumor microenvironment (TME) is considered the major cause of tumor heterogeneity, which in turn accounts for treatment failure in current cancer therapies. Physical and chemical signals from the TME as well as factors secreted by MSCs and TAMs can induce epigenetic alterations in the cancer cells that alter their phenotypic plasticity, eventually resulting in the generation of cancer stem cells (CSCs). Phenotype switching of CSCs involves processes such as epithelial-mesenchymal transition, transdifferentiation, retrodifferentiation, or spontaneous cell fusion of cancer cells with stromal cells, particularly MSCs. Principally, phenotype plasticity of cancer (stem) cells may be targeted pharmacologically to reduce tumor heterogeneity and hence resistance to therapy.
Abstract
Tumor heterogeneity is considered the major cause of treatment failure in current cancer therapies. This feature of solid tumors is not only the result of clonal outgrowth of cells with genetic mutations, but also of epigenetic alterations induced by physical and chemical signals from the tumor microenvironment (TME). Besides fibroblasts, endothelial and immune cells, mesenchymal stroma/stem-like cells (MSCs) and tumor-associated macrophages (TAMs) intimately crosstalk with cancer cells and can exhibit both anti- and pro-tumorigenic effects. MSCs can alter cancer cellular phenotypes to increase cancer cell plasticity, eventually resulting in the generation of cancer stem cells (CSCs). The shift between different phenotypic states (phenotype switching) of CSCs is controlled via both genetic programs, such as epithelial-mesenchymal transdifferentiation or retrodifferentiation, and epigenetic alterations triggered by signals from the TME, like hypoxia, spatial heterogeneity or stromal cell-derived chemokines. Finally, we highlight the role of spontaneous cancer cell fusion with various types of stromal cells. i.e., MSCs in shaping CSC plasticity. A better understanding of cell plasticity and phenotype shifting in CSCs is a prerequisite for exploiting this phenomenon to reduce tumor heterogeneity, thereby improving the chance for therapy success.
1. Introduction
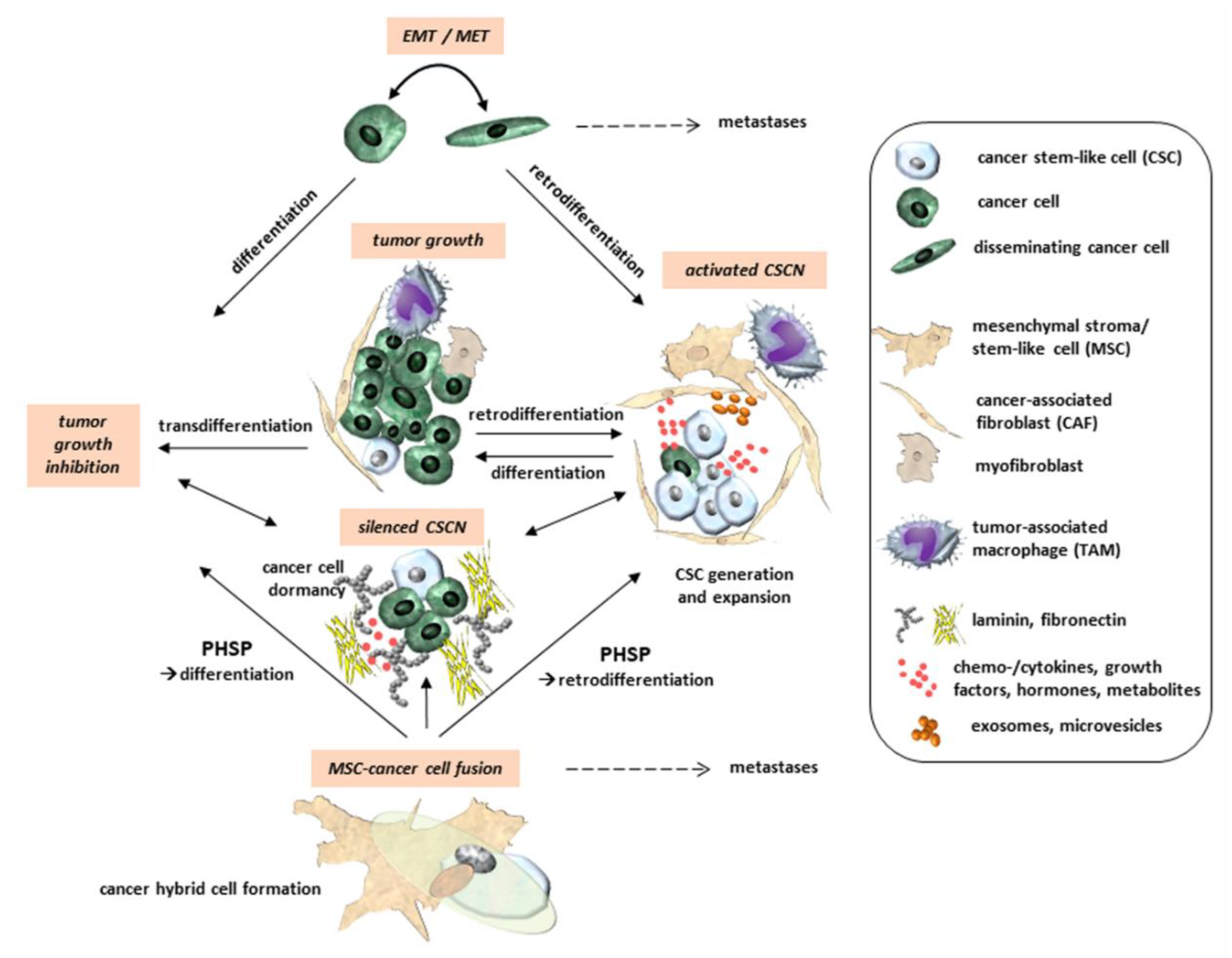
Solid tumors is composed of cancer cells interacting with a variety of non-tumorigenic cells such as immune cells (e.g., T cells, natural killer cells, macrophages), endothelial cells, adipocytes, mesenchymal stroma/stem-like cells (MSCs), and fibroblasts which are embedded in a distinct matrix of structural proteins constituting the extracellular matrix (ECM). These cellular components together with the ECM form the tumor microenvironment (TME), which promotes the development and expansion of cancer progenitor cells, tumor-initiating cells (TICs), and cancer stem cells (CSCs) [1,2]. The TME of solid tumors is subject to dynamic turnover of its structural and functional components, and this process partially accounts for the phenomenon of tumor heterogeneity. CSCs and TICs were identified and characterized in several human primary and metastatic neoplasms such as ovarian [3], prostate [4], breast [5], lung [6], and pancreatic cancer [7], melanomas [8], acute myeloid leukemia [9], glioblastoma [10,11,12], and other brain tumors [13]. TICs, CSCs, and their progeny are suggested to reside in specialized compartments termed cancer stem cell niche (CSCN) and according to their functionalities discrimination between activated and silenced CSCNs is hypothesized. Protection and progression of progenitor or CSC populations requires an activated CSCN as a distinct transient location in certain tissues (primary tumor tissue or metastatic tissue). Alternatively, CSCN-like structures in an inactivated more silenced state may also harbor quiescent CSCs or diverse CSC-derived progeny following cell cycle exit into a transiently growth-arrested G0′-like phase by entering dormancy (Figure 1 and Figure 2). Accordingly, CSCs may be distinguished by dormancy-competent, cancer-repopulating, dormancy-incompetent, and disseminated populations [14] whereby tumor dormancy can be induced by various processes including metastasis, radiation/chemotherapy, and cancer cell fusion among others [15]. This hibernation-like state of CSCs enables survival by escape from immune surveillance and maintaining treatment resistance until CSCN conversion into an activated state and reentry of CSCs into the proliferative state, eventually followed by tumor recurrence [16]. CSCNs can be reversibly established by mediators such as prostaglandin E2 signaling [17] and compartmentalized by interacting cell types involving MSCs and cancer-associated fibroblasts (CAFs) [18,19,20].
During mutual interactions within the TME the cellular partners can change their tasks. In particular, interactions of macrophages or MSCs with cancer cells play an important role in modulating tumor functions, thereby increasing tumor heterogeneity [21]. For instance, macrophages, particularly M2-type macrophages, are converted to TAMs (tumor-associated macrophages) [22,23] and MSCs can differentiate into CAFs (cancer-associated fibroblasts) [24,25] displaying mostly tumor-supportive properties (Figure 1). MSCs can also indirectly and directly interact with cancer cells by modulating their functions and contributing to cancer cell plasticity [19,20].
2. Cancer Cell Plasticity and CSCs
Cell plasticity is defined as the ability of a cell to reprogram and change its phenotypic identity, a phenomenon also known as lineage plasticity. Cell plasticity occurs in several fundamental biological processes, such as embryonic development, wound healing, tissue regeneration, or neoplastic transformation. In cancer, the reactivation of these mechanisms enables tumor cells to acquire a CSC-like phenotype with enhanced ability to escape apoptosis in hostile environments, thereby contributing to cancer initiation, progression, metastases, and therapy resistance [27,28]. Cancer are phenotypically plastic and may stochastically, or in response to environmental cues, adopt CSC and non-CSC states in a dynamic and reversible fashion, eventually giving rise to different subsets of CSCs (Figure 1). The different phenotypes of CSCs and TICs enlarge plasticity and are determined by maintenance of stemness, self-renewal capability, escape from immune surveillance, and resistance to apoptosis induced by chemotherapeutic drugs. Cancer cell plasticity is also induced by intrinsic/cell-autonomous genetic and/or epigenetic alterations and by extrinsic factors such as dynamic restructuring within the TME [29,30,31,32,33].
Besides displaying remarkable genetic/epigenetic, metabolic, and phenotypic heterogeneity, cancer cells maintain plasticity by transition along a spectrum of cellular phenotypes intermittent between the extreme epithelial or mesenchymal states in a process regulated by the TME. Deregulated/aberrant cell plasticity could be considered another hallmark of a cancer.
Together with their self-renewal capacity, immune escape, and resistance to chemotherapeutic interventions CSCs contribute to tumor maintenance and development. Thereby, CSCs can differentiate along various pathways and initiate new tumors [34]. CSCs are characterized by the expression of distinct markers. In renal cell carcinoma, expression of the stem cell protein CD133 also known as prominin-1 and the hyaluronan receptor CD44 are associated with CSCs [35]. Breast CSCs represent a combination of the GPI-anchored sialoglycoprotein CD24, aldehyde dehydrogenase-1 (ALDH-1), and CD44 in a CD24low/CD44high/ALDHhigh constellation [5]. CSCs in colorectal tumors are characterized by a set of proteins such as CD44, CD133, CD24, EpCAM, LGR5, and ALDH [36]. Detection of CSCs by the expression of corresponding markers in other tumors include pancreatic carcinoma (epithelial-specific antigen (ESA), CD24, CD44) [7], medulloblastomas and gliomas (CD133) [13], epithelial ovarian cancers (CD117 (c-kit), CD44) [3], malignant melanoma (ATP-binding cassette sub-family B member 5 (ABCB5)) [8], prostate cancer (CD133, α2β1 integrin, CD44) [4], lung cancer (CD133) [6], among others. These and more different CSC marker profiles suggest the appearance of tumor type- and tumor tissue-specific CSC populations.
CSCs may also differ from TICs as the cell of tumor origin in their phenotypic and molecular characteristics [37,38,39,40,41]. The cell of tumor origin acquires the first cancer-initiating mutational hit, i.e., TICs [38]. According to the hierarchical model this tumor-originating cell could be a normal native/lineage stem cell [41], or a committed progenitor or differentiated cell as hypothesized by the stochastic model [18,37]. Examples of cells of origin include the Lgr5+ stem cell type of the intestinal crypt for colorectal cancers [42] or acinar cells that convert through a process termed acinar-to-ductal metaplasia (ADM), to pancreatic ductal adenocarcinoma (PDAC) [43]. ADM is a prominent example for the process of transdifferentiation, the direct conversion of a one differentiated cell into a functionally altered type of differentiated cell without passing through an intermittent retrodifferentiation and progenitor/stem-like state.
Alternatively, committed progenitors or differentiated cells which undergo a retrodifferentiation program reacquire stem cell features by losing previously functional identities resulting in a CSC phenotype [27,28,39,44,45]. Retrodifferentiation is characterized by a reversion of maturated properties and expression patterns of a differentiated phenotype to a precursor cell or stem-like cell [46,47,48]. In addition, retrodifferentiation to a stem-like state includes retrograde senescence, providing longevity or rejuvenation at the cellular level [49]. CSCs which are generated by retrodifferentiation from differentiated cells regain capacity for self-renewal and may thus be able to maintain tumorigenicity [50,51]. When cells exhibit plasticity, they converge on signaling processes that induce cellular retrodifferentiation (Figure 1 and Figure 2). Activation of these programs, in turn, enables the initiation and progression of carcinogenesis and underlies resistance to therapy [52]. Interactions of cancer cells with MSCs or CAFs can induce a retrodifferentiation program to form CSCs [53,54]. This enables new differentiation pathways, whereby CSCs maturate along an altered lineage [51]. Whereas MSCs contribute to the establishment of CSCs to maintain and promote CSC growth [18], intrinsic processes such as EMT or pharmacological interference with (trans)differentiation of TICs or CSCs may induce maturation and growth reduction followed by reduced tumorigenicity [55].
Accumulating evidence suggests that certain cancer cells can adopt a CSC state associated with hybrid/partial EMT, a higher transdifferentiation potential, and increased resistance to chemo- or radiotherapy [56,57,58]. At the molecular level, resistance is acquired by several different mechanisms, including upregulation/activation of multidrug efflux pumps, enhanced DNA repair, or maintenance of a slow cycling, or a quiescent state [59,60]. The process of tumor re-initiation or recurrence by stem-like cells presumably involves differentiation of quiescent or dormant CSCs into rapidly proliferating tumor cells. Slow cycling or quiescent stem-like cells in the tumor contribute to resistance against conventional chemo- and radiotherapies because these treatments are usually directed towards rapidly dividing cells, substantiating the hypothesis of activated and silenced CSCNs (Figure 1 and Figure 2).
CSCs also exhibit a remarkable ability to reprogram their cellular metabolism in response to signals they receive from the TME [30,61]. Although the metabolic phenotypes of CSCs have been insufficiently characterized so far, the stem-like features resulting from altered metabolic pathways is another emerging hallmark of cancer, that contributes to CSC plasticity [62,63]. Various cell-intrinsic and cell-extrinsic factors may modulate this “metabostemness” and CSCs can transit from one metabolic state to another in response to various conditions in the TME, like pH, hypoxia, or nutrient supply [61,64]. A better understanding of the association between metabolic phenotype and plasticity of CSCs is required in order to exploit the underlying mechanisms for effectively targeting these cells.
3. Epigenetic Reprogramming of CSCs
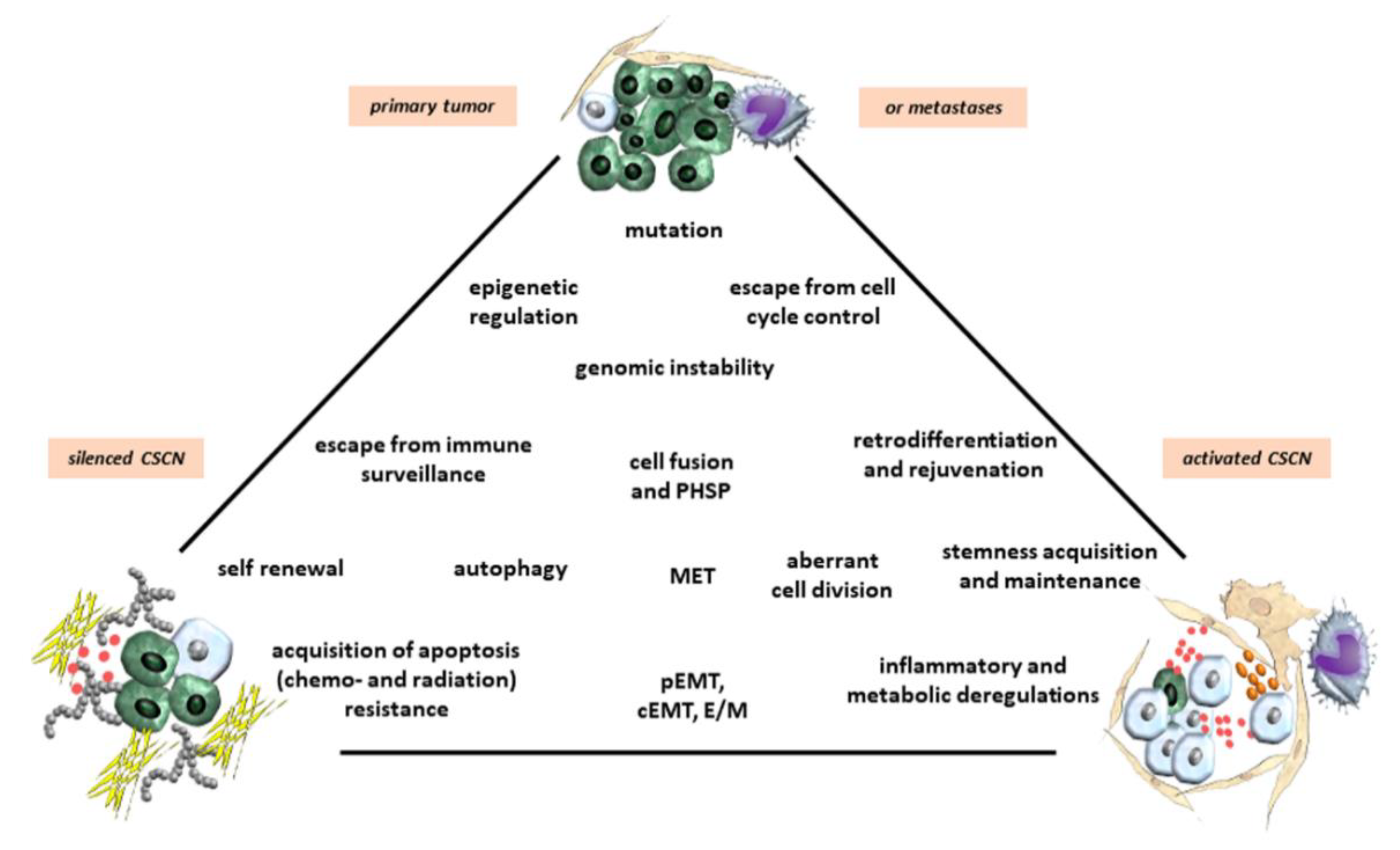
Cancer cell phenotype shifting and CSC generation are largely controlled by epigenetic mechanisms that render the chromatin restrictive or permissive for specific transcriptional programs involved in cell differentiation or reprogramming [28,34,58,65] (Figure 2). Signature patterns of “active” chromatin marks active promoters, transcribed regions, and candidate enhancers, whereas other modifications reveal distinct modes of chromatin repression, such as those mediated by the Polycomb repressor complex 2 (PRC2). In CSCs, a bivalent chromatin state may enhance plasticity and phenotypic switching [28,34,58,65]. For instance, the promoter hyper-methylation-induced silencing of HOXC8 (a homeobox gene), in non-tumorigenic mammary epithelial cells has been found to be associated with an increase in the number of CSCs, heightened self-renewal and a transformed phenotype [66]. A histone modifier, enhancer of zeste homolog 2 (EZH2), the catalytic subunit of PRC2, induces transcriptional repression of target genes via trimethylation of lysine-27 in histone H3 (H3K27me3) [67]. In CSCs of various malignant tumors EZH2 is expressed at markedly elevated levels and exhibits a crucial function in CSC maintenance and progression [68]. In breast cancer (BC) cells, EZH2 promotes expansion of TICs and mammosphere formation through activation of RAF1/β-catenin signaling [69,70], while in glioblastoma loss of H3K27me3 can lead to aberrant Wnt signaling activation, which is necessary for maintenance of CSCs [71]. In contrast, missense mutations in the genes encoding histone H3.3 and H3.1. in pediatric glioblastomas have lower overall amounts of H3K27me3 due to inhibition of the enzymatic activity of PRC2 through interaction with the EZH2 subunit, an epigenetic dysregulation that may promote gliomagenesis [72].
Epigenetic alterations can drive oncogenic programs even in the absence of mutations in classical oncogenes or tumor suppressor genes [73]. In addition, certain chromatin structures can inhibit differentiation and prevent appropriate induction of tumor suppressor programs. By contrast, permissive or “plastic” states, i.e., via enhancer landscape reprogramming during tumorigenesis [74] may allow random oncogene activation or non-physiologic cell fate transitions and therapy resistance in cancer cells [65]. Both TME-driven and epigenetic reprogramming promote such dynamic mechanisms, favoring cancer cell plasticity and tumor heterogeneity [39,75]. Current evidence points to a complex interplay between the genes, epigenetic changes and the TME in cell reprogramming, cancer cell plasticity, and tumor heterogeneity [39,76]. These observations suggest that epigenetic changes triggered by interactions with the TME modulate cancer cell phenotypes and properties, and shape tumor architecture [77]. Moreover, CSCs are capable of exploiting the reversible nature of epigenetic modifications to adjust their plastic state. Similar to the diverse EMT phenotypes, this reversibility may be harnessed for therapeutic targeting.
4. Regulatory Effects of the TME and Its Cellular Components on CSCs
4.1. The Effect of Inflammation and Hypoxia on CSC Plasticity
Chronic inflammation represents a hallmark of many cancers and inflammatory signals contribute to tumor initiation, progression and cell plasticity [34]. The tissue healing process is accompanied by remodeling of the local tissue environment, which changes the spectrum and production of various growth factors, enzymes and other signaling mediators secreted by specific niche cells and the surrounding stromal cells, such as immune cells and MSCs. Evidence for an association between inflammation and cell plasticity comes from a variety of cancer types [45,78,79,80]. A rare population of long-lived, quiescent pancreatic cells labeled by doublecortin-like kinase-1 (Dclk1) is required in vivo for pancreatic regeneration following injury and chronic inflammation. Expression of mutant Kras in Dclk1+ cells converts these cells into potent TICs upon induction of experimental pancreatitis [80]. Another study with aged mice deficient in the homeobox gene, Nkx3.1, revealed that loss of function of Nkx3.1 accelerates inflammation-driven prostate cancer initiation potentially via aberrant cellular plasticity and impairment of cellular differentiation [81]. A hypoxic microenvironment is known to regulate various aspects of malignant progression including cellular plasticity. For instance, hypoxia was found to promote self-renewal in non-stem cells in glioblastoma by upregulating OCT4, NANOG, and c-MYC [82]. Moreover, a hypoxic TME in vivo favors the accumulation of cells with CSC-like features. The authors concluded that this was due to clonal evolution or selection, since the differential phenotypes of the tumor cells from both the hypoxic and non-hypoxic TMEs were fairly stable even when subsequently cultured in vitro under normoxic culture conditions [31]. These studies underscore the importance of orchestrating components within the TME in generating intra-tumoral heterogeneity and CSC plasticity, knowledge that is crucial for the rational design of novel and more effective therapeutic strategies. However, it still remains unclear whether CSC heterogeneity is a consequence of selection pressure exerted by the TME or whether plasticity represents an inherent feature of the cancer cells that enables them to adapt to varying signals from the TME [39,83]. Recent evidence from a study on glioblastoma suggests that the expression of CSC-associated membrane markers is based on intrinsic plasticity of tumor cells rather than on clonal entity defined by distinct functional properties or transcriptomic profiles. These authors showed that phenotypic heterogeneity arose from non-hierarchical, reversible state transitions that were triggered by the TME. They went on to conclude that tumorigenic potential is not coupled to CSC multipotency but to intrinsic plasticity and, hence, that therapies directed against CSC-associated membrane epitopes may fail [83].
The physical and chemical composition of the TME with parameters such as pH, oxygen content, ion concentrations, nutrient availability, and mechanic rigidity of the ECM also plays a significant role in establishing a CSCN and regulating CSC behavior [84,85,86]. The dynamic restructuring of the tumor stroma affects CSCN establishment and maintenance. Activated or silenced CSCN structures can be dismantled at certain tumor sites and newly build up at other more favorable places within the tumor tissue, suggesting a variety of simultaneous opportunities for CSCNs to provide a CSC compartment (Figure 1 and Figure 2). This reversible construction and degradation of a CSCN depends on the form and the stability of the local TME compartment and associated tissue composition, but also applies to primary tumors and metastases. For example, CSCNs of tumor metastases in the hypoxic bone marrow are more protected and stabilized in rigid and spongy bone cavities as compared to CSCNs in metabolically-exposed tissues such as primary organ-associated tumor tissues or soft-tissue lymph node metastases. Consequently, bone marrow-associated compartments may represent a preferable location for silenced CSCNs and CSC dormancy which is supported by long-term residing cancer cells in bone metastases.
A fundamental feature of the TME is spatial heterogeneity [87,88], which is able to impact cellular phenotypes. For instance, glioblastoma cells residing in hypoxic regions of a tumor overexpress EGFR (epidermal growth factor receptor), while vascular regions were enriched in platelet-derived growth factor receptor α [89]. Similarly, a hypoxic TME induced by anti-angiogenic agents can increase breast CSCs [90]. Thus, spatial heterogeneity of TME can give rise to generation of cancer subpopulations at different locations within an individual tumor. Spatial heterogeneity in primary tumors has also been reported with respect to cells with EMT [91,92,93]. Cells at the invasive front of the primary tumor as well as metastases were deficient in the expression of membrane-bound E-cadherin but express high levels of nuclear β-catenin, suggestive of an EMT (Figure 1). In contrast, the more centrally located cells in the primary tumor and in metastases were rich in membranous E-cadherin and cytoplasmic β-catenin, probably indicative of mesenchymal-epithelial transition (MET) [94]. Likewise, mesenchymal breast CSCs were found at the invasive edge of the tumor, while the more epithelial or hybrid epithelial/mesenchymal (E/M) CSCs were localized in its central regions [93]. In oral squamous carcinoma, budding cells exhibited a particular gene expression signature when compared to cells from the central parts of tumors that comprise factors involved in EMT and activated TGF-β signaling. ZEB1 was upregulated concomitantly with the decreased expression of MET-associated transcription factors (TFs), e.g., OVOL1, Krüppel-like factors and Grainyhead-like factors. Moreover, microRNA-200 family members were found to be downregulated in budding tumor cells [95]. Using a mechanism-based dynamical model it was shown that the more mesenchymal CSCs lie at the invasive front, while the hybrid E/M CSCs reside in the central regions of the tumor. The mathematical simulations also revealed that the diffusion of TGF-β (a strong EMT-inducer) along with Notch signaling-mediated control of EMT can provide an explanation for the differential localization of CSC subpopulations with varying EMT phenotypes in the tumor [32]. Spatial heterogeneity of tumors may be used as a prognostic marker for treatment response across different cancer types [88]. Moreover, TGF-β-activated SMAD TFs contribute to a fibrogenic EMT program involving Ras-responsive element binding protein 1, a transcriptional effector of activated HRAS and KRAS. This complex induces SNAIL expression which can promote subsequent intratumoral fibrosis and tumor growth [96].
4.2. The Effects of Stromal Cells of the TME on Cancer Cell EMT and Plasticity
4.2.1. CAFs and TAMs in EMT and Tumors
Accumulating evidence suggests that cancer cell/stromal cell intercellular communications strongly impact stem-like behavior and phenotypic plasticity of cancer cells [97,98,99]. As mentioned above, CAFs are a major component of the TME and play a fundamental role in various aspects of tumor progression [100]. These cells were found to regulate the plasticity of TICs in hepatocellular carcinomas through c-Met/FRA1/HEY1 signaling [101], in PDAC through p125FAK signaling [102], and in lung cancer by insulin-like growth factor receptor signaling [103].
A series of studies on various cancers highlight the contribution of CCL chemokine expression to the activation of EMT programs [104]. CCL2-mediated monocyte/macrophage trafficking was also observed in the inducible KrasG12Dp53-null PDAC mouse model [33]. Subsequently, TGF-β secreted by recruited TAMs induces tumor cells to adopt a mesenchymal phenotype. This enabled them to survive extinction of oncogenic Kras, indicating a significant role of the CCL2-TGF-β/EMT signaling pathway in the resistance to Kras-targeted therapy [33].
In response to chemotherapy, macrophages can secrete Oncostatin-M (OSM), an IL-6 family cytokine, which—via activation of STAT3/SMAD3 signaling [105]—induces the retrodifferentiation of triple-negative breast cancer cells into aggressive stem cells [106]. OSM is also secreted by cancer-associated adipocytes, which are likewise able to promote stemness [107]. Li and colleagues studied how differently polarized M1 or M2 macrophages communicate with epithelial-mesenchymal plasticity of cancer cells, and vice versa, how cancer cells in an epithelial or mesenchymal state can influence the polarization of macrophages [108]. Using in silico co-culture models it was found that the interactions between cancer cells and macrophages can give rise to multiple stable steady-states, with each steady-state being stable against external perturbations. More recently, in a transgenic mouse model of ovarian carcinoma, it was demonstrated that the chemoresistance-promoting functions of TAMs require expression of Zeb1 by TAMs with the release of CCL2 by the cancer cells [109]. As discussed above, expression of Zeb1 by cancer cells endows them with a more aggressive phenotype, including enhanced invasive capacities, therapeutic resistance, and stemness, resulting in poor clinical outcomes in a variety of human cancer types [110]. Clinical trials are currently designed with cancer cell-expressed ZEB1 as a potential molecular target. However, the above data suggest that effective inhibition of tumor growth and improved response to chemotherapy would also require targeting ZEB1 in TAMs [109]. Similar contributions by TAMs to the resistance to cytostatic drugs via EMT induction have been observed in other cancer types, including pancreatic and colorectal cancers [111,112]. Hence, strategies targeting TAM function, infiltration, or activation can be exploited for therapeutic purposes.
4.2.2. MSC Origin and Role in Tumors
MSCs represent a heterogeneous mixture of subpopulations also termed multipotent mesenchymal stromal cells or medicinal signaling cells [113,114,115]. While the underlying basis of MSC heterogeneity remains unclear, previous work suggested that this mixed population may be explained by mutual interdependency of different stromal cell clones. These subclones adapt their availability by clonal convergence or expansion to maintain growth potential of the entire population and to provide the various MSC properties within progressively changing environments such as tumor stroma [20]. Different organs also exhibit a tissue-specific environment for MSCs which adds to their variable characteristics. Primary origins of MSCs are localized in perivascular regions of various adult tissues [116,117]. In addition, MSCs can also be isolated from neonatal tissues such as placenta or umbilical cord in large quantities. These MSC populations originating from birth-associated tissues exhibit superior in vitro growth potential and regenerative capacity [118].
Together with other subpopulations displaying stem-like characteristics, MSCs are functionally involved in tissue repair and regenerative activities [115,119,120,121]. Primary MSC cultures enable in vitro maintenance for a limited time as compared to constitutively proliferating MSC-like cells, representing a cell source with permanently reproducible properties [122,123]. Following recruitment to cancer cell-induced lesions, the regenerative potential of MSCs operates in promoting tissue repair during invasive tumor growth. MSCs thereby, develop both tumor-inhibiting [124] and tumor-promoting properties [19,125,126] during crosstalk with cancer cells and other neighboring tumor-associated populations within the TME [127]. MSCs interact with a variety of immune cells and exhibit immune-modulatory functions. They suppress the cytotoxic capacity of NK cells [128], inhibit T cell activation, and contribute to a conversion of inflammation-associated M1 to repair-oriented M2(a–d) macrophages by altering immune cell functions and favoring immune suppression [129,130,131,132].
Activation of MSC’s paracrine capabilities produces a variety of chemokines, growth factors, and metabolites which are secreted into the TME, e.g., by induction of endothelial cells to support tumor vasculogenesis [133]. Conversely, the phenotypes of cancer cells are altered under the influence of MSCs, e.g., via release of TGF-β to promote tumor growth by acquisition of novel properties and to mediate differentiation of endothelial cells for enhanced tumor angiogenesis [134]. Moreover, delivery of TGF-β can also induce an epithelial-mesenchymal transition (EMT) in cancer cells, eventually promoting the generation of new CSCs by retrodifferentiation (Figure 1). EMT cells can also be reversed by the mesenchymal-epithelial transition (MET) program, i.e., during outgrowth of macrometastases at distant sites (see below).
Besides these more indirect, paracrine communication pathways, MSCs directly interact with cancer cells, for example via gap junctional intercellular communication [125]. Connexins as the molecular monomers can form a homo- or heterohexameric hemichannel in the plasma membrane which can assemble to a corresponding hemichannel of an adjacent cell to build a gap junction. These structures play an essential role in the transcellular exchange of ions, metabolites, and second messengers and also affect cytoskeletal signaling. A variety of different connexins are distributed among cancer cells modulating proper communication and contributing to heterogeneity by tumor inhibition or tumor progression. Since tumor-type- and stage-specific compositions of connexins can be determined, these membrane proteins possess tumor prognostic value and are a useful molecular target for therapeutic interventions [135]. Further examples for direct interactions between MSCs and cancer cells include the formation of F-actin-rich tunneling nanotubes or trogocytosis to exchange molecules, small organelles or cell membrane patches [19,53]. Moreover, notch receptor signaling [125] is involved in maintaining self-renewal and amplification of CSCs. Previous work has demonstrated that the range of intracellular Notch1 signaling in MSC-derived dermal fibroblasts governs the capability of these cells to regulate melanoma aggressiveness, stemness, and phenotypic plasticity [136].
Following close cellular interactions spontaneous cell fusion between MSCs and cancer cells with the generation of new hybrid cancer cell populations represents a rare event in tumors which can also display CSC characteristics [137,138,139,140,141] (Figure 1). Several distinct molecular mechanisms can contribute to cell fusion in a cell type-specific manner with tight membrane approaches as a prerequisite to enable a fusogenic environment. Fused hybrid cells undergo a post-hybrid selection process (PHSP) to enable chromosomal rearrangements for successful cell cycle progression and a renewed accurate cell metabolism [142] (see below).
4.3. Cancer Cell Fusion and CSC Plasticity
Besides mutual communication via soluble and physical means, close cellular interactions eventually result in spontaneous cell fusion, for example between macrophages/TAMs and cancer cells [143,144,145], or between MSCs/CAFs and cancer cells [137,141,146,147]. During fusion, the newly formed hybrid cancer cells express different DNA profiles and acquire diverse functional characteristics from the parental cells. For example, the macrophage-specific factors DAP12 [148] and CD163 [149], both of which are not detectable in breast cancer cells, are expressed in fused hybrid breast cancer cells [150].
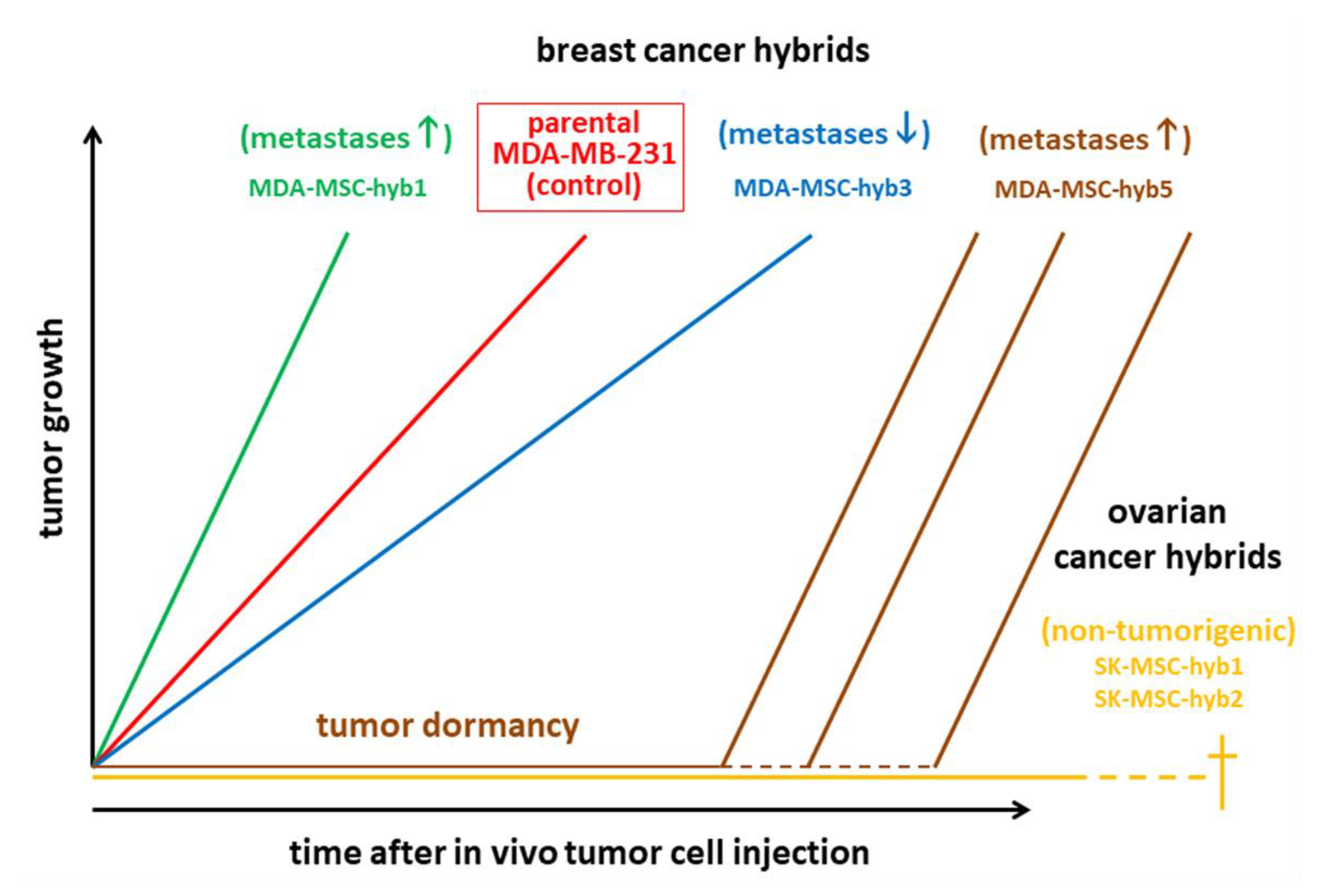
Cancer cell fusion with the generation of new hybrid cancer cell populations increases tumor plasticity by the generation of subpopulations with CSC-like traits [21,137,138,139,141,151]. Hybrid cancer cells, however, can either enhance tumorigenicity and metastatic capacity [152] or reduce neoplastic behavior [138,153] (Figure 1, Figure 2 and Figure 3). The acquisition of novel functionalities and increased tumor plasticity is determined by a PHSP leading to hybrid cancer cells with different tumorigenic and metastatic behavior [142]. This selection program is required since fusion-derived cancer hybrid cells generate aneuploid or polyploid giant cells displaying uncoordinated nuclear and DNA communication [154]. Further processes underlying the generation of aneuploidy include endoreplication, endomitosis, aging, or engulfment by entosis [155], or cannibalism [156]. The accompanying chromosomal instabilities require appropriate reduction to a (meta)stabilized level that is regulated during a PHSP. This multistep program of PHSP displays a clonal convergence of the initial hybrid population by elimination, silencing, or stabilizing surviving hybrids [142]. Thereafter, PHSP-generated hybrid cancer cells enhance tumor plasticity by newly acquired functionalities including CSC-like properties, which affect tumor growth and metastatic spreading (Figure 3). Indeed, cancer cell fusion contributes to the development of CSC subtypes [157]. The resulting tumor heterogeneity complicates therapeutic regimen, suggesting unfavorable patient outcome (Figure 1, Figure 2 and Figure 3).
Cancer cell fusion with MSCs can also reduce tumorigenic properties as demonstrated by MDA-MSC-hyb3 and –hyb4 breast cancer cells [138] and SK-MSC-hyb1 and –hyb2 ovarian cancer hybrids [153] (Figure 3). Moreover, the aggressive tumor-promoting hybrids MDA-MSC-hyb1 and –hyb2 demonstrate increased vulnerability to various chemotherapeutic compounds such as taxol, cisplatin, methotrexate, epirubicin, and foretinib [152], suggesting that fusion of cancer cells with MSCs causes distinct therapy-oriented effects, that are not observed during cancer cell fusion with macrophages.
These findings underscore the need for a better mechanistic understanding of the fusion process and subsequent PHSP to predict and potentially regulate the outcome and functionality of hybrid cells. Cancer cell fusion in different cancer cell types, however, involves different molecular mechanisms. For instance, MSC fusion with neoplastic MCF-10A mammary epithelial cells can be promoted by tumor necrosis factor α (TNFα) and blocked by inhibition of TNF receptor [137]. Alternatively, occurrence and progression of a rhabdomyoblastoma by cancer cell fusion was suppressed by inhibition of the interleukin-4 receptor [158]. This mechanistic heterogeneity among different tumor types complicates common therapeutic strategies and successful interventions.
5. Exploiting Cancer Cell Plasticity for Improving Therapeutic Success
In the past few years, cell plasticity has emerged as a mode of escape from targeted and non-targeted therapies in various cancers. However, our understanding of this phenomenon has also expanded, raising hope that vulnerabilities associated with tumor cell plasticity may be harnessed for the development of novel and innovative therapeutic concepts. Alone or in combination with existing anticancer treatments, these could lead to more complete and longer-lasting clinical responses. In this last section, we comment on translational aspects citing a selection of preclinical and clinical studies mainly in malignant melanoma providing some proof that targeting tumor cell plasticity is a viable therapeutic option. Further in-depth discussions on this issue are provided by two recent reviews [26,29].
Experimental animal models: Melanoma plasticity is linked to phenotype switching, where the TME induces switches between invasive vs. proliferative states. In a zebrafish model, melanoma cells following extravasation activate genes of melanocyte differentiation and become pigmented. After metastatic dissemination, the TME provides signals in the form of EDN3 to promote phenotype switching, which can induce a state that is both proliferative and differentiated and associated with decreased animal survival [159]. The smoothened inhibitor, vismodegib/GDC0449, has been shown to induce tumor shrinkage of basal cell carcinoma (BCC) by promoting tumor cell differentiation. However, a small subpopulation of tumor cells survives and accounts for tumor relapse following treatment interruption, a situation seen also in humans. In both mouse and human BCC, this persisting, slow-cycling subpopulation is characterized by expression of the stem cell marker, LGR5, and active Wnt signaling. Employing GEMMs of BCC, Sánchez-Danés and coworkers showed that the combination of vismodegib treatment with Lgr5 lineage ablation, or an inhibitor of Wnt signaling, led to tumor eradication, demonstrating that the synergy between Wnt and Smoothened inhibitors is a clinically feasible strategy to overcome tumor recurrence in BCC [160].
Patient-derived biological material: Human melanoma cells can display profound transcriptional variability at the single-cell level, which involves high-level transcription in a very small percentage of cells of a number of genes encoding resistance markers. This set of marker genes and the related gene expression signatures reveal the prognostic relevance for defining transcriptional phenotypes/states that in turn predict, which cells will ultimately resist drug treatment [161]. Another study highlights the potential of therapies directed towards minimal residual disease (MRD). In malignant cells isolated from BRAF mutant melanoma PDXs exposed to RAF/MEK inhibitors, varying combinations of distinct drug-tolerant transcriptional states were identified, which co-existed within MRDs from PDXs and biopsies of patients under therapy. Among these, the authors were able to identify a novel neural crest stem cell (NCSC) transcriptional program and the nuclear receptor RXRG as key drivers of resistance by showing that inhibiting RXRG prevented the accumulation of NCSCs in MRD and delayed the generation of resistant cells [162].
Continuous BRAF inhibition of BRAF mutant melanomas is known to induce a series of phenotypic changes in the cancer cells that result in therapy resistance and escape from immune control before genetic fixation of the acquired resistant state. This is due to activation of certain signaling networks shortly after BRAF inhibition, but before the appearance of drug-resistant phenotypes. Drug targeting those networks, in combination with BRAF inhibition, halted the adaptive transition and led to prolonged growth inhibition in multiple patient-derived cell lines [163]. Also in melanoma, AXLhigh cells are resistant, while AXLlow cells are sensitive to MAPK pathway inhibitors, rationalizing a differential therapeutic approach. To achieve this goal, Boshuizen and colleagues developed an antibody-drug conjugate, AXL-107-MMAE that as a single agent displayed potent in vivo anti-tumor activity in PDXs derived from melanoma, lung, pancreatic and cervical tumors. Moreover, in combination with MAPK inhibitors, AXL-107-MMAE eliminated distinct populations in heterogeneous melanoma cell pools and inhibited tumor growth. These findings provide proof-of-concept that rationalized combinatorial targeting of distinct populations in heterogeneous tumors can improve therapeutic efficiency [164].
Clinical trials: In melanoma, MITF and its upstream activator, PAX3, are drivers of an early non-mutational and reversible drug-tolerant state. Nelfinavir has been identified as a potent suppressor of PAX3 and MITF expression and sensitizer of BRAF and NRAS mutant melanoma cells to MAPK pathway inhibitors. Moreover, nelfinavir is effective in BRAF and NRAS mutant melanoma cells isolated from patients progressed on MAPK inhibitor therapy and in BRAF/NRAS/PTEN mutant tumors [165]. As discussed above, PRC2 has been shown to play a major role in transcriptional silencing in part by methylation of H3K27 and deregulation of its function correlates with poor prognosis in certain cancers. Vaswani et al. have identified CPI-1205, a highly potent and selective inhibitor of EZH2, the active subunit of PRC2, that displayed robust antitumor effects in a xenograft models and is currently evaluated in phase 1 and 1b/2 clinical trials, alone and in combination [166], (https://www.constellationpharma.com/constellation-pharmaceuticals-announces-first-patient-dosed-phase-1b-2-prostar-combination-study-cpi-1205-advanced-form-prostate-cancer/). Two other selective EZH2 inhibitors, Tazemetostat and GSK2816126, are currently subject to phase-1 clinical testing. While the Tazemetostat trial is being conducted in patients with relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumors [167], GSK2816126 is being performed with relapsed/refractory diffuse large B cell lymphoma, transformed follicular lymphoma, other Non-Hodgkin’s lymphomas, multiple myeloma, and castrate-resistant prostate cancer (https://www.clinicaltrials.gov/ct2/show/NCT02082977). The above referenced studies impressively show that therapeutic manipulation intended to increase or decrease tumor heterogeneity and cancer cell plasticity, either alone or in combination with conventional therapies, holds great promise in overcoming therapy resistance and improving patient outcomes.
6. Conclusions
Enhanced recruitment of MSCs or TAMs to the tumor tissue increases the chance of mutual interactions with the cancer cells via soluble mediators, physical interactions or even cell fusion, eventually resulting in cancer cell EMT, retro-/transdifferentiation, and the generation of various CSC phenotypes besides their contribution to establish CSCNs. These cellular programs or the signaling peculiarities that distinguish hybrid cancer populations from their parental counterparts and the intermediate states during a PHSP represent potential therapeutic targets to direct cancer cell development along a more differentiated and less tumorigenic path [129,153,168,169]. Transfer of chromosomes from non-tumor-associated MSCs during fusion with cancer cells may provide more tumor-reducing or anti-tumor properties in contrast to cancer cell fusion with macrophages. Deciphering the underlying mechanisms of cancer cell fusion, CSC development, maintenance in activated or silenced (dormant) CSCNs, and repopulating/differentiating capabilities is crucial for a better understanding of tumor plasticity. This will pave the ground for novel therapeutic strategies that alone or in combination with conventional therapies will hopefully overcome therapy resistance and improve patient outcomes.
Author Contributions
R.H. and H.U. drafted the manuscript, J.v.d.O. contributed initial figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no financial, personal, or professional conflict of interest.
Abbreviations
| ADM | acinar-to-ductal metaplasia |
| CAFs | cancer-associated fibroblasts |
| CSCs | cancer stem cells |
| CSCN | cancer stem cell niche |
| CTCs | circulating tumor cells |
| ECM | extracellular matrix |
| E/M | epithelial/mesenchymal |
| EZH2 | enhancer of zeste homolog 2 |
| EMT | epithelial-mesenchymal transition |
| MET | mesenchymal-epithelial transition |
| MSC | mesenchymal stroma/stem-like cells |
| PDAC | pancreatic ductal adenocarcinoma |
| PHSP | post-hybrid selection process |
| PRC2 | polycomb repressor complex 2 |
| TAMs | tumor-associated macrophages |
| TFs | transcription factors |
| TICs | tumor-initiating cells |
| TME | tumor microenvironment |
| TNF | tumor necrosis factor |
| Zeb1/2 | zinc finger E-box-binding 1/2 |
References
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitland, N.J.; Collins, A.T. Prostate cancer stem cells: A new target for therapy. J. Clin. Oncol. 2008, 26, 2862–2870. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Tabatabai, G.; Weller, M. Glioblastoma stem cells. Cell Tissue Res. 2011, 343, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Tabatabai, G.; Wick, W.; Weller, M. Stem cell-mediated gene therapies for malignant gliomas: A promising targeted therapeutic approach? Discov. Med. 2011, 11, 529–536. [Google Scholar] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [PubMed]
- Bu, Y.; Cao, D. The origin of cancer stem cells. Front. Biosci. Schol. Ed. 2012, 4, 819–830. [Google Scholar] [PubMed] [Green Version]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Hass, R. Role of MSC in the Tumor Microenvironment. Cancers 2020, 12, 2107. [Google Scholar] [CrossRef]
- Shabo, I.; Svanvik, J.; Lindstrom, A.; Lechertier, T.; Trabulo, S.; Hulit, J.; Sparey, T.; Pawelek, J. Roles of cell fusion, hybridization and polyploid cell formation in cancer metastasis. World J. Clin. Oncol. 2020, 11, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [Green Version]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to be exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef]
- Le Magnen, C.; Shen, M.M.; Abate-Shen, C. Lineage Plasticity in Cancer Progression and Treatment. Annu. Rev. Cancer Biol. 2018, 2, 271–289. [Google Scholar] [CrossRef]
- Varga, J.; Greten, F.R. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 2017, 19, 1133–1141. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, B.; Zhao, X.; Liu, Z.; Wang, X.; Yao, X.; Dong, X.; Chi, J. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J. Surg. Oncol. 2013, 108, 414–419. [Google Scholar] [CrossRef]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef] [Green Version]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Poli, V.; Fagnocchi, L.; Zippo, A. Tumorigenic Cell Reprogramming and Cancer Plasticity: Interplay between Signaling, Microenvironment, and Epigenetics. Stem Cells Int. 2018, 2018, 4598195. [Google Scholar] [CrossRef]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. How does multistep tumorigenesis really proceed? Cancer Discov. 2015, 5, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Hass, R. Retrodifferentiation--an alternative biological pathway in human leukemia cells. Eur. J. Cell Biol. 1992, 58, 1–11. [Google Scholar]
- Hass, R. Retrodifferentiation and cell death. Crit. Rev. Oncog. 1994, 5, 359–371. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [Green Version]
- Hass, R. Rejuvenation in distinct cell populations—What does it mean? Exp. Gerontol. 2009, 44, 634–638. [Google Scholar] [CrossRef] [Green Version]
- Cabillic, F.; Corlu, A. Regulation of Transdifferentiation and Retrodifferentiation by Inflammatory Cytokines in Hepatocellular Carcinoma. Gastroenterology 2016, 151, 607–615. [Google Scholar] [CrossRef]
- Hass, R. Retrodifferentiation—A mechanism for cellular regeneration? Biol. Chem. 2009, 390, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. Concise Review: Crosstalk of Mesenchymal Stroma/Stem-Like Cells with Cancer Cells Provides Therapeutic Potential. Stem Cells 2018, 36, 951–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekir, K.; Dubois-Pot-Schneider, H.; Desert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; et al. Retrodifferentiation of Human Tumor Hepatocytes to Stem Cells Leads to Metabolic Reprogramming and Chemoresistance. Cancer Res. 2019, 79, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Wang, L.; Tang, J.; Cao, P.; Luo, Z.; Sun, J.; Kiflu, A.; Sai, B.; Zhang, M.; Wang, F.; et al. Activation of anaphase-promoting complex by p53 induces a state of dormancy in cancer cells against chemotherapeutic stress. Oncotarget 2016, 7, 25478–25492. [Google Scholar] [CrossRef]
- Kapse-Mistry, S.; Govender, T.; Srivastava, R.; Yergeri, M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front. Pharmacol. 2014, 5, 159. [Google Scholar]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Alarcon, T. Metabostemness: A new cancer hallmark. Front. Oncol. 2014, 4, 262. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, 6348. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Cardenas, R.; Wang, B.; Persson, J.; Mongan, N.P.; Grabowska, A.; Allegrucci, C. HOXC8 regulates self-renewal, differentiation and transformation of breast cancer stem cells. Mol. Cancer 2017, 16, 38. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hung, M.C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. 2014, 46, 209–222. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Cai, J.; Hou, Y.; Huang, Z.; Wang, Z. Role of EZH2 in cancer stem cells: From biological insight to a therapeutic target. Oncotarget 2017, 8, 37974–37990. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Suva, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Fagnocchi, L.; Poli, V.; Zippo, A. Enhancer reprogramming in tumor progression: A new route towards cancer cell plasticity. Cell. Mol. Life. Sci. 2018, 75, 2537–2555. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S. Cell plasticity in cancer: A complex interplay of genetic, epigenetic mechanisms and tumor micro-environment. Surg. Oncol. 2020, 34, 154–162. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Kwon, O.J.; Zhang, L.; Ittmann, M.M.; Xin, L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc. Natl. Acad. Sci. USA 2014, 111, E592–E600. [Google Scholar] [CrossRef] [Green Version]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Le Magnen, C.; Virk, R.K.; Dutta, A.; Kim, J.Y.; Panja, S.; Lopez-Bujanda, Z.A.; Califano, A.; Drake, C.G.; Mitrofanova, A.; Abate-Shen, C. Cooperation of loss of NKX3.1 and inflammation in prostate cancer initiation. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, W.; Liu, Z.; Lei, W.; Sun, L.; Yang, H.; Wang, Y.; Ramdas, S.; Dong, X.; Xu, R.; Cai, H.; et al. Mutation landscape and intra-tumor heterogeneity of two MANECs of the esophagus revealed by multi-region sequencing. Oncotarget 2017, 8, 69610–69621. [Google Scholar] [CrossRef]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef] [Green Version]
- Little, S.E.; Popov, S.; Jury, A.; Bax, D.A.; Doey, L.; Al-Sarraj, S.; Jurgensmeier, J.M.; Jones, C. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012, 72, 1614–1620. [Google Scholar] [CrossRef] [Green Version]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor Budding: The Name is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef]
- Liu, W.; Vivian, C.J.; Brinker, A.E.; Hampton, K.R.; Lianidou, E.; Welch, D.R. Microenvironmental Influences on Metastasis Suppressor Expression and Function during a Metastatic Cell’s Journey. Cancer Microenviron. 2014, 7, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, D.H.; Dabelsteen, E.; Specht, L.; Fiehn, A.M.; Therkildsen, M.H.; Jonson, L.; Vikesaa, J.; Nielsen, F.C.; von Buchwald, C. Molecular profiling of tumour budding implicates TGFbeta-mediated epithelial-mesenchymal transition as a therapeutic target in oral squamous cell carcinoma. J. Pathol. 2015, 236, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, M.C.; Hollingsworth, R.E.; Hurt, E.M. Cancer stem cell plasticity and tumor hierarchy. World J. Stem Cells. 2015, 7, 27–36. [Google Scholar] [CrossRef]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions With Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, M.; Han, Z.; Jiang, F.; Xu, C.; Qin, Y.; Ding, N.; Liu, Y.; Zhang, T.; An, Z.; et al. Clinical significance of FAP-alpha on microvessel and lymphatic vessel density in lung squamous cell carcinoma. J. Clin. Pathol. 2018, 71, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, B.; Eades, G.; Zhou, Q. Adipocyte activation of cancer stem cell signaling in breast cancer. World J. Biol. Chem. 2015, 6, 39–47. [Google Scholar] [CrossRef]
- Li, X.; Jolly, M.K.; George, J.T.; Pienta, K.J.; Levine, H. Computational Modeling of the Crosstalk Between Macrophage Polarization and Tumor Cell Plasticity in the Tumor Microenvironment. Front. Oncol. 2019, 9, 10. [Google Scholar] [CrossRef]
- Cortes, M.; Sanchez-Moral, L.; de Barrios, O.; Fernandez-Acenero, M.J.; Martinez-Campanario, M.C.; Esteve-Codina, A.; Darling, D.S.; Gyorffy, B.; Lawrence, T.; Dean, D.C.; et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J. 2017, 36, 3336–3355. [Google Scholar] [CrossRef]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Xiong, B. M2 macrophages confer resistance to 5-fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling. Onco. Targets Ther. 2019, 12, 3051–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwada, K.; Kagawa, S.; Yoshida, R.; Sakamoto, S.; Ito, A.; Watanabe, M.; Ieda, T.; Kuroda, S.; Kikuchi, S.; Tazawa, H.; et al. The epithelial-to-mesenchymal transition induced by tumor-associated macrophages confers chemoresistance in peritoneally disseminated pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 307. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majore, I.; Moretti, P.; Hass, R.; Kasper, C. Identification of subpopulations in mesenchymal stem cell-like cultures from human umbilical cord. Cell Commun. Signal. 2009, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT(R)) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar] [PubMed]
- Corselli, M.; Chen, C.W.; Sun, B.; Yap, S.; Rubin, J.P.; Peault, B. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev. 2012, 21, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Chapel, A.; Bertho, J.M.; Bensidhoum, M.; Fouillard, L.; Young, R.G.; Frick, J.; Demarquay, C.; Cuvelier, F.; Mathieu, E.; Trompier, F.; et al. Mesenchymal stem cells home to injured tissues when co-infused with hematopoietic cells to treat a radiation-induced multi-organ failure syndrome. J. Gene. Med. 2003, 5, 1028–1038. [Google Scholar] [CrossRef]
- Bianco, P. “Mesenchymal” stem cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 677–704. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Bucan, V.; Reimers, K.; Hass, R. Mesenchymal stem cells maintain long-term in vitro stemness during explant culture. Tissue Eng. Part C Methods 2013, 19, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Jacobs, R.; Dittmar, T.; Pich, A.; von der Ohe, J.; Yang, Y.; Hass, R. Reversible Growth-Arrest of a Spontaneously-Derived Human MSC-Like Cell Line. Int. J. Mol. Sci. 2020, 21, 4752. [Google Scholar] [CrossRef] [PubMed]
- Gauthaman, K.; Yee, F.C.; Cheyyatraivendran, S.; Biswas, A.; Choolani, M.; Bongso, A. Human umbilical cord Wharton’s jelly stem cell (hWJSC) extracts inhibit cancer cell growth in vitro. J. Cell. Biochem. 2012, 113, 2027–2039. [Google Scholar] [CrossRef]
- Mandel, K.; Yang, Y.; Schambach, A.; Glage, S.; Otte, A.; Hass, R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013, 22, 3114–3127. [Google Scholar] [CrossRef]
- Yang, Y.; Bucan, V.; Baehre, H.; von der Ohe, J.; Otte, A.; Hass, R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int. J. Oncol. 2015, 47, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, J. Mesenchymal stem cells: “repair cells” that serve wounds and cancer? Sci. World J. 2010, 10, 1234–1238. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, D.; Marquardt, N.; Tufa, D.M.; Beauclair, G.; Low, H.Z.; Hatlapatka, T.; Hass, R.; Kasper, C.; von Kaisenberg, C.; Schmidt, R.E.; et al. Role of gamma-secretase in human umbilical-cord derived mesenchymal stem cell mediated suppression of NK cell cytotoxicity. Cell Commun. Signal. 2014, 12, 63. [Google Scholar] [CrossRef]
- Hass, R.; Otte, A. Mesenchymal stem cells as all-round supporters in a normal and neoplastic microenvironment. Cell Commun. Signal. 2012, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Cruz, C.M.; Shearer, J.J.; Figueiredo Neto, M.; Figueiredo, M.L. The Immunomodulatory Effects of Mesenchymal Stem Cell Polarization within the Tumor Microenvironment Niche. Stem Cells Int. 2017, 2017, 4015039. [Google Scholar] [CrossRef] [Green Version]
- De Miguel, M.P.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Pascual, C.Y.; Aller, M.A.; Arias, J.; Arnalich-Montiel, F. Immunosuppressive properties of mesenchymal stem cells: Advances and applications. Curr. Mol. Med. 2012, 12, 574–591. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sun, R.; Origuchi, M.; Kanehira, M.; Takahata, T.; Itoh, J.; Umezawa, A.; Kijima, H.; Fukuda, S.; Saijo, Y. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol. Med. 2011, 17, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.H.; Chang, M.C.; Tsai, K.S.; Hung, M.C.; Chen, H.L.; Hung, S.C. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 2013, 32, 4343–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayan, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Shao, H.; Moller, M.; Prokupets, R.; Tse, Y.T.; Liu, Z.J. Intracellular Notch1 Signaling in Cancer-Associated Fibroblasts Dictates the Plasticity and Stemness of Melanoma Stem/Initiating Cells. Stem Cells 2019, 37, 865–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. In Vitro Fusion of Normal and Neoplastic Breast Epithelial Cells with Human Mesenchymal Stroma/Stem Cells Partially Involves Tumor Necrosis Factor Receptor Signaling. Stem Cells 2018, 36, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. In vivo cell fusion between mesenchymal stroma/stem-like cells and breast cancer cells. Cancers 2019, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Chitwood, C.A.; Dietzsch, C.; Jacobs, G.; McArdle, T.; Freeman, B.T.; Banga, A.; Noubissi, F.K.; Ogle, B.M. Breast tumor cell hybrids form spontaneously in vivo and contribute to breast tumor metastases. APL Bioeng. 2018, 2, 031907. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Ohe, J.V.; Hass, R. Altered Tumor Plasticity after Different Cancer Cell Fusions with MSC. Int. J. Mol. Sci. 2020, 21, 8347. [Google Scholar] [CrossRef] [PubMed]
- Busund, L.T.; Killie, M.K.; Bartnes, K.; Seljelid, R. Spontaneously formed tumorigenic hybrids of Meth A sarcoma and macrophages grow faster and are better vascularized than the parental tumor. Int. J. Cancer 2002, 100, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelek, J.M.; Chakraborty, A.K. The cancer cell—Leukocyte fusion theory of metastasis. Adv. Cancer Res. 2008, 101, 397–444. [Google Scholar]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, D.; Dai, X.; Chen, J.; Rong, X.; Wang, H.; Wang, A.; Li, M.; Dong, J.; Huang, Q.; et al. Fusion of cancer stem cells and mesenchymal stem cells contributes to glioma neovascularization. Oncol. Rep. 2015, 34, 2022–2030. [Google Scholar] [CrossRef] [Green Version]
- Shabo, I.; Olsson, H.; Stal, O.; Svanvik, J. Breast cancer expression of DAP12 is associated with skeletal and liver metastases and poor survival. Clin. Breast Cancer 2013, 13, 371–377. [Google Scholar] [CrossRef]
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef]
- Shabo, I.; Midtbo, K.; Andersson, H.; Akerlund, E.; Olsson, H.; Wegman, P.; Gunnarsson, C.; Lindstrom, A. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Dittmar, T. Minocycline impairs TNF-alpha-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-kappaB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 2018, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. MSC stimulate ovarian tumor growth during intercellular communication but reduce tumorigenicity after fusion with ovarian cancer cells. Cell Commun. Signal. 2018, 16, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, B.; Zanker, K.S.; Dittmar, T. Cell fusion is a potent inducer of aneuploidy and drug resistance in tumor cell/ normal cell hybrids. Crit. Rev. Oncog. 2013, 18, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Sottile, F.; Aulicino, F.; Theka, I.; Cosma, M.P. Mesenchymal stem cells generate distinct functional hybrids in vitro via cell fusion or entosis. Sci. Rep. 2016, 6, 36863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartosh, T.J.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D.J. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagler, C.; Zanker, K.S.; Dittmar, T. Cell Fusion, Drug Resistance and Recurrence CSCs. Adv. Exp. Med. Biol. 2011, 714, 173–182. [Google Scholar]
- Li, G.; Kikuchi, K.; Radka, M.; Abraham, J.; Rubin, B.P.; Keller, C. IL-4 receptor blockade abrogates satellite cell: Rhabdomyosarcoma fusion and prevents tumor establishment. Stem Cells 2013, 31, 2304–2312. [Google Scholar] [CrossRef]
- Kim, I.S.; Heilmann, S.; Kansler, E.R.; Zhang, Y.; Zimmer, M.; Ratnakumar, K.; Bowman, R.L.; Simon-Vermot, T.; Fennell, M.; Garippa, R.; et al. Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat. Commun. 2017, 8, 14343. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Danes, A.; Larsimont, J.C.; Liagre, M.; Munoz-Couselo, E.; Lapouge, G.; Brisebarre, A.; Dubois, C.; Suppa, M.; Sukumaran, V.; Del Marmol, V.; et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature 2018, 562, 434–438. [Google Scholar] [CrossRef]
- Skopenko, E.V.; Bratus, N.I.; Parkhomets, T.I.; Vasil’ev, A.N.; Kucherenko, N.E. Metabolism of cGMP with K+ concentration increase in the medium causing depolarization of cortical cell membranes in normal and irradiated rats. Ukr. Biokhim. Zh. 1985, 57, 76–79. [Google Scholar] [PubMed]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Homet Moreno, B.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Vaswani, R.G.; Gehling, V.S.; Dakin, L.A.; Cook, A.S.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 2016, 59, 9928–9941. [Google Scholar]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Yang, Y.; Otte, A.; Hass, R. Human mesenchymal stroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev. 2015, 24, 1205–1222. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Tumor plasticity comprises several genetic and epigenetic programs including epithelial-mesenchymal transition (EMT)/ mesenchymal-epithelial transition (MET) or cancer cell fusion. These contribute to the enrichment of cancer cell progenitors or cancer stem cells (CSC) populations within an activated CSCN (cancer stem cell niche) for CSC expansion or a silenced CSCN for maintenance of dormant CSCs. The multiple underlying programs such as differentiation, trans- or retrodifferentiation, metastases formation, or PHSP (post-hybrid selection process) may take place simultaneously in distinct compartments of the tumor tissue (adapted from [18,26]).
Figure 1.
Tumor plasticity comprises several genetic and epigenetic programs including epithelial-mesenchymal transition (EMT)/ mesenchymal-epithelial transition (MET) or cancer cell fusion. These contribute to the enrichment of cancer cell progenitors or cancer stem cells (CSC) populations within an activated CSCN (cancer stem cell niche) for CSC expansion or a silenced CSCN for maintenance of dormant CSCs. The multiple underlying programs such as differentiation, trans- or retrodifferentiation, metastases formation, or PHSP (post-hybrid selection process) may take place simultaneously in distinct compartments of the tumor tissue (adapted from [18,26]).

Figure 2.
Various pathways and mechanisms can contribute to cancer cell plasticity during shuttling between (I) an activated cancer stem cell niche (CSCN) with cancer cell progenitor and CSC expansion, (II) a silenced CSCN displaying cancer cell dormancy, and (III) the dynamic alterations in the primary tumor tissue and metastases. Symbols are described in Figure 1.
Figure 2.
Various pathways and mechanisms can contribute to cancer cell plasticity during shuttling between (I) an activated cancer stem cell niche (CSCN) with cancer cell progenitor and CSC expansion, (II) a silenced CSCN displaying cancer cell dormancy, and (III) the dynamic alterations in the primary tumor tissue and metastases. Symbols are described in Figure 1.

Figure 3.
Tumor development of different human breast cancer hybrid cells demonstrates enhanced (green) and reduced (blue) growth properties as compared to the parental breast cancer cells (red) before fusion with mesenchymal stroma/stem-like cells (MSCs) and subsequent PHSP. A third form of tumor growth is characterized by an initial phase of dormancy. The two presented ovarian cancer hybrid populations displayed no detectable tumor development and the animals eventually died of old age.
Figure 3.
Tumor development of different human breast cancer hybrid cells demonstrates enhanced (green) and reduced (blue) growth properties as compared to the parental breast cancer cells (red) before fusion with mesenchymal stroma/stem-like cells (MSCs) and subsequent PHSP. A third form of tumor growth is characterized by an initial phase of dormancy. The two presented ovarian cancer hybrid populations displayed no detectable tumor development and the animals eventually died of old age.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers 2020, 12, 3716. https://doi.org/10.3390/cancers12123716
AMA Style
Hass R, von der Ohe J, Ungefroren H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers. 2020; 12(12):3716. https://doi.org/10.3390/cancers12123716
Chicago/Turabian StyleHass, Ralf, Juliane von der Ohe, and Hendrik Ungefroren. 2020. "Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness" Cancers 12, no. 12: 3716. https://doi.org/10.3390/cancers12123716
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.