PET Imaging Agents (FES, FFNP, and FDHT) for Estrogen, Androgen, and Progesterone Receptors to Improve Management of Breast and Prostate Cancers by Functional Imaging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
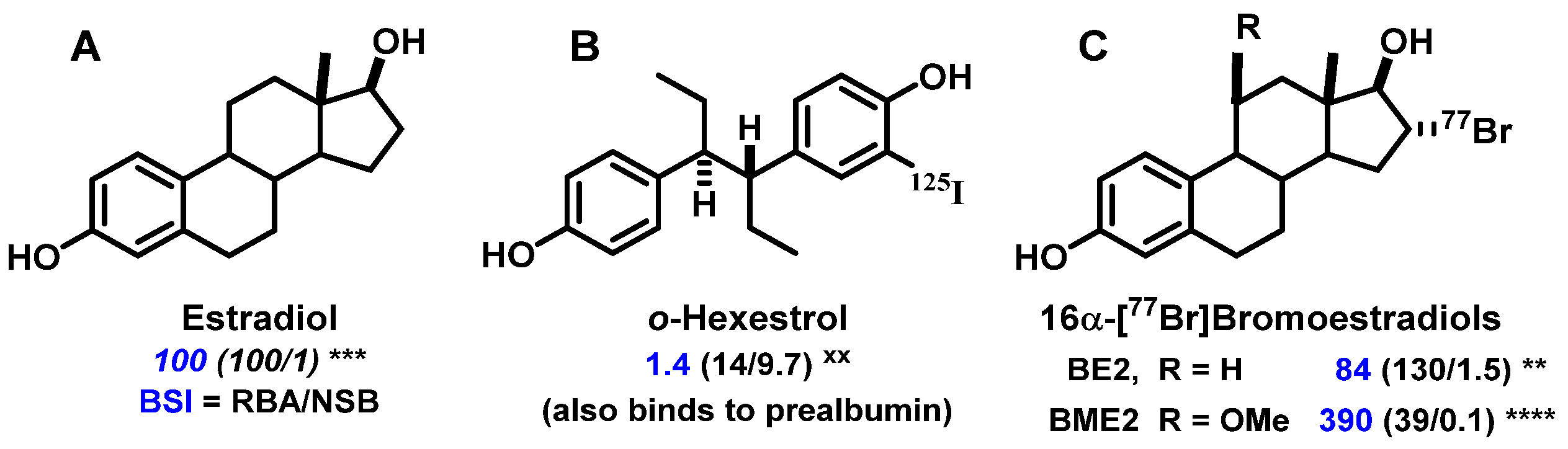
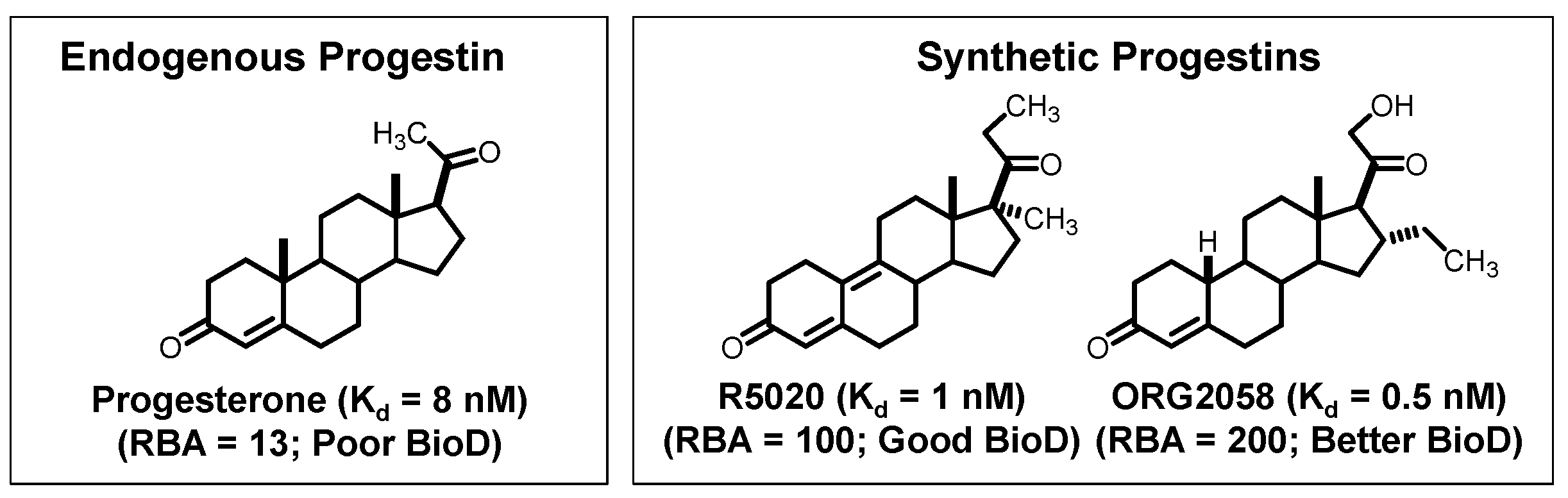
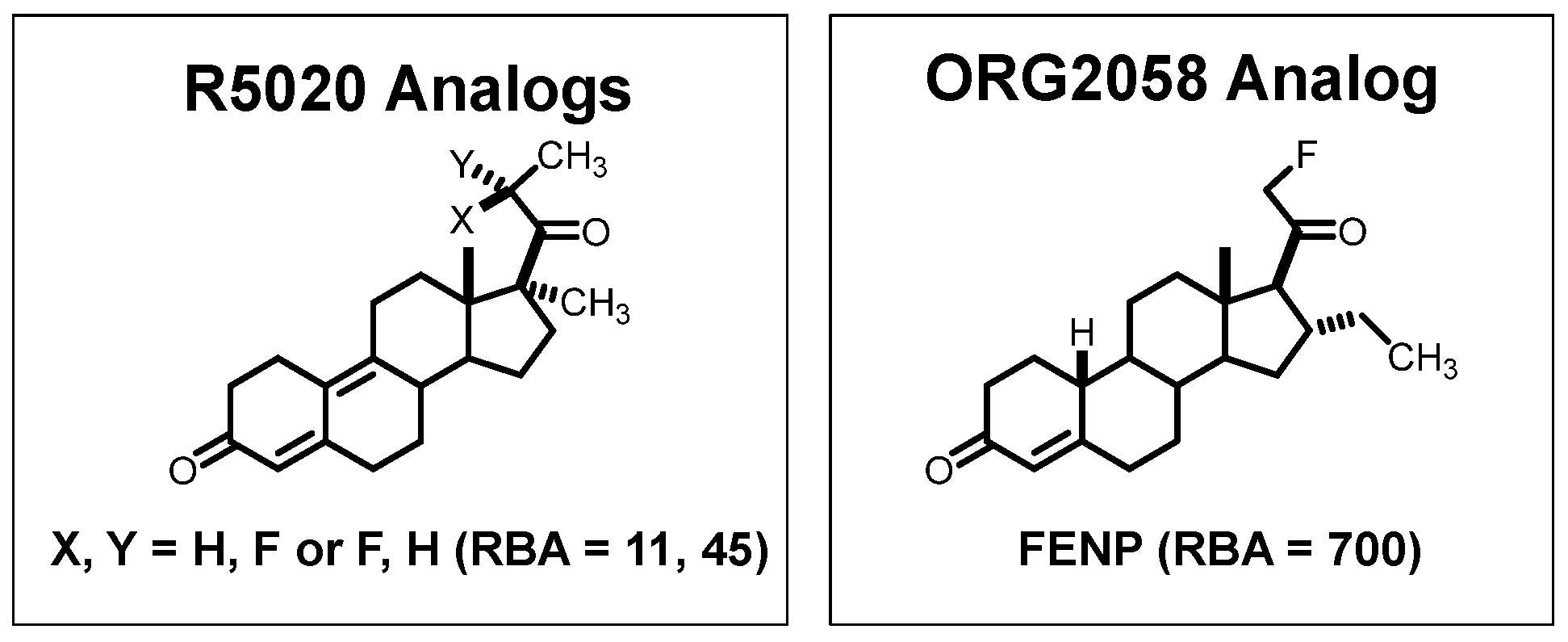
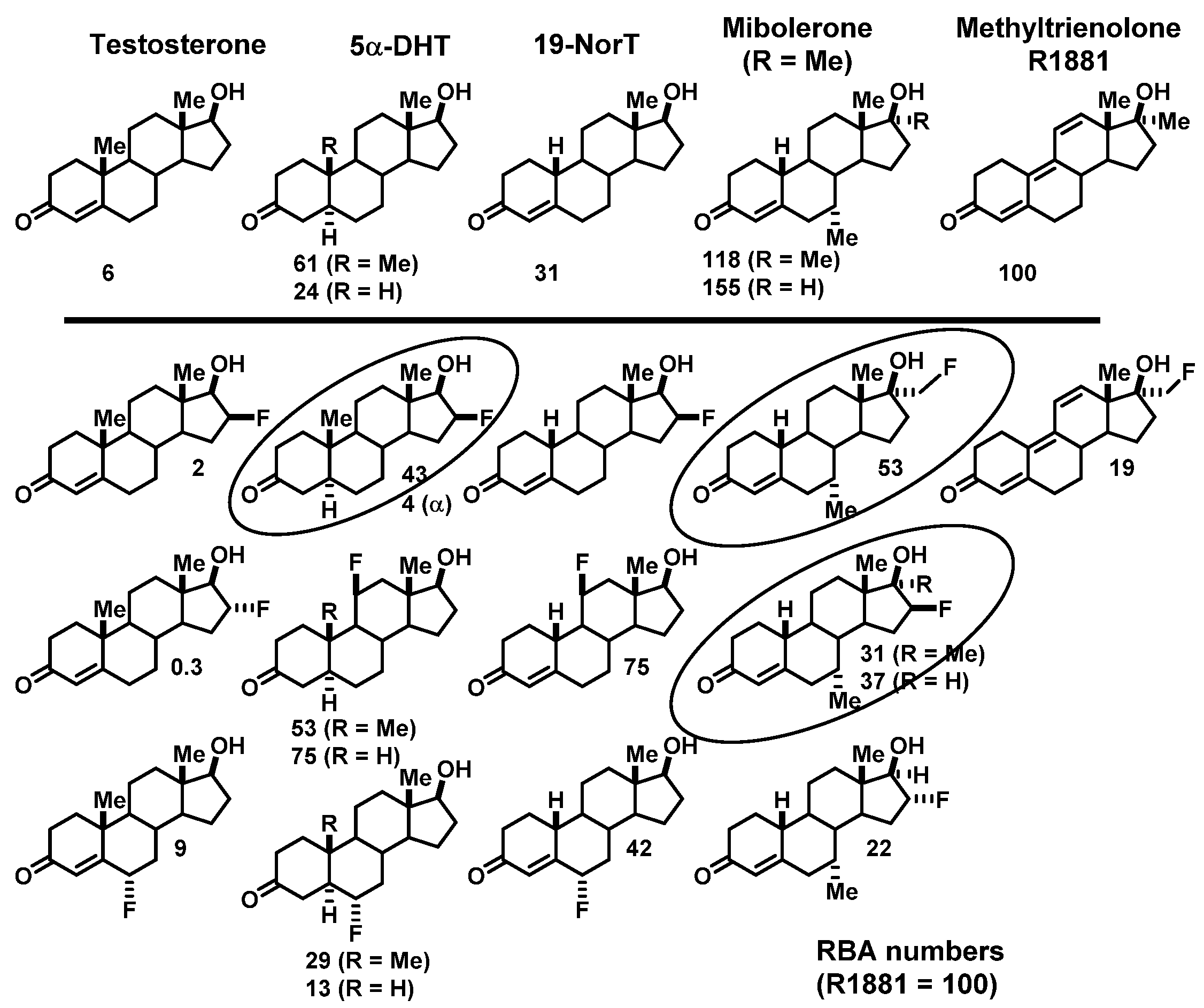
- RBA and Ki: Affinity for the intended target is expressed relative to a standard ligand, typically a tritiated version of a known high-affinity ligand used as a tracer in convenient radiometric competitive binding assays, such as [3H]estradiol for ER [5]. Thus, for ER, the standard, estradiol (which has a Kd value of 0.2 nM), is assigned a relative binding affinity (RBA) value of 100, with lower affinity candidate tracers having RBA values below 100, and higher affinity ones above 100. If of interest, Ki values can be calculated from RBA values: Ki (in nM) = (0.2 × 100)/RBA. The standard compound for PgR is R5020, with a Kd of 0.4 nM [6,7], and for AR is methyltrienolone (R1881), with a Kd of 0.2 nM [6].
- NSB and BSI: Nonspecific binding (NSB) is high-capacity, low-affinity, off-target binding, to proteins such as serum albumin and possibly lipid phases, and it is also measured relative to that of the standard ligand, but in this case, the standard is assigned an NSB value of 1. As a measure of target binding selectivity, we defined the ratio of specific to nonspecific binding as the binding selectivity index, BSI: BSI = RBA/NSB [8,9]. Thus, the standard ligand has a BSI of 100 (i.e., 100/1), and candidate tracers with higher selectivity have BSI values above 100, whereas those with lower selectivity have BSI values below 100. Notably, unlike RBA, BSI is sensitive to both target binding affinity and nonspecific binding.
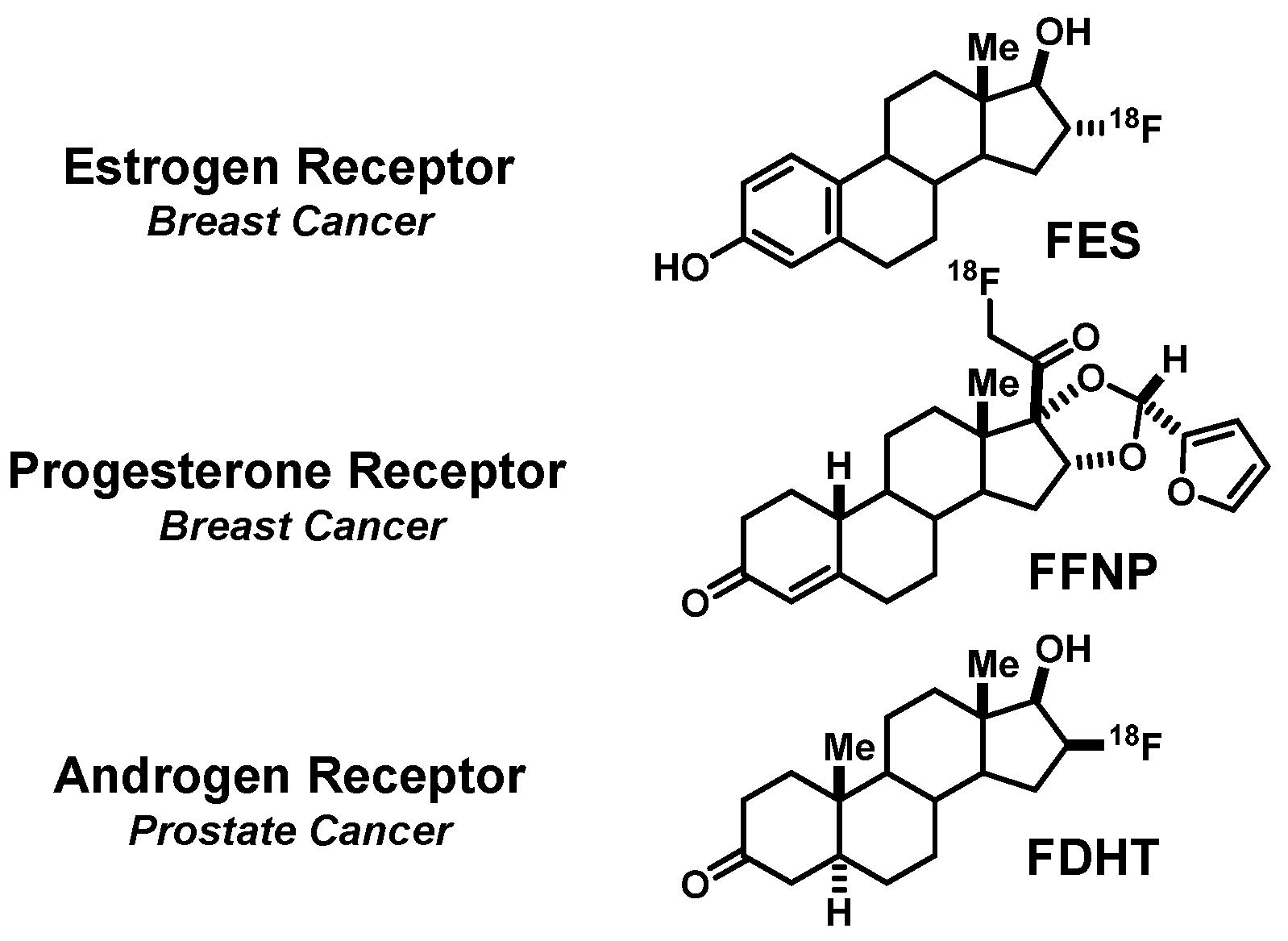
2. FES, a PET Imaging Agent for Estrogen Receptors in Breast Cancers
2.1. A Prelude to In Vivo Imaging of ER: Finding a Ligand (Steroidal vs. Nonsteroidal) and a Radionuclide (Iodine, Bromine, or Fluorine) Enabling Effective and Selective Uptake by ER-Target Tissues
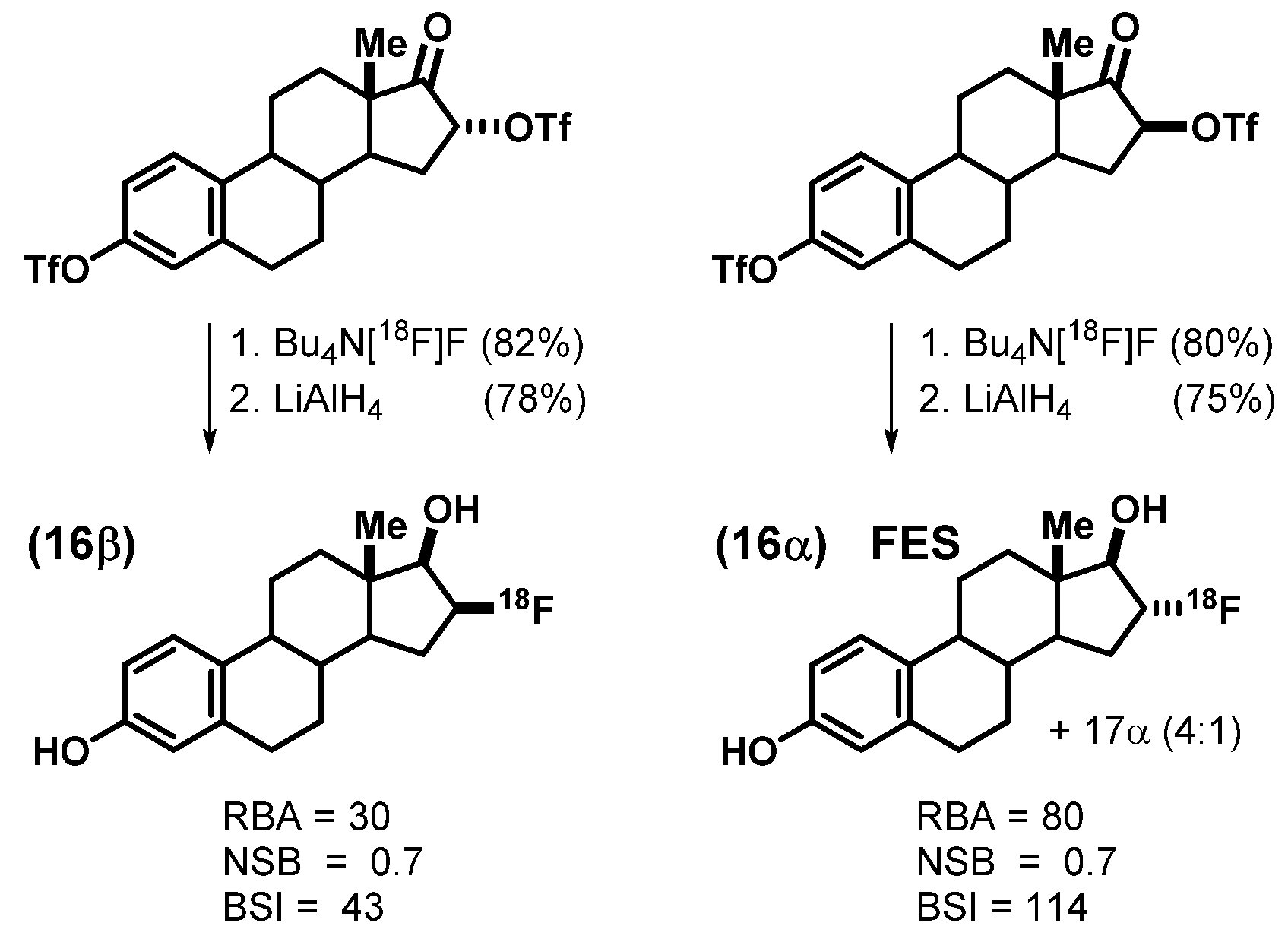
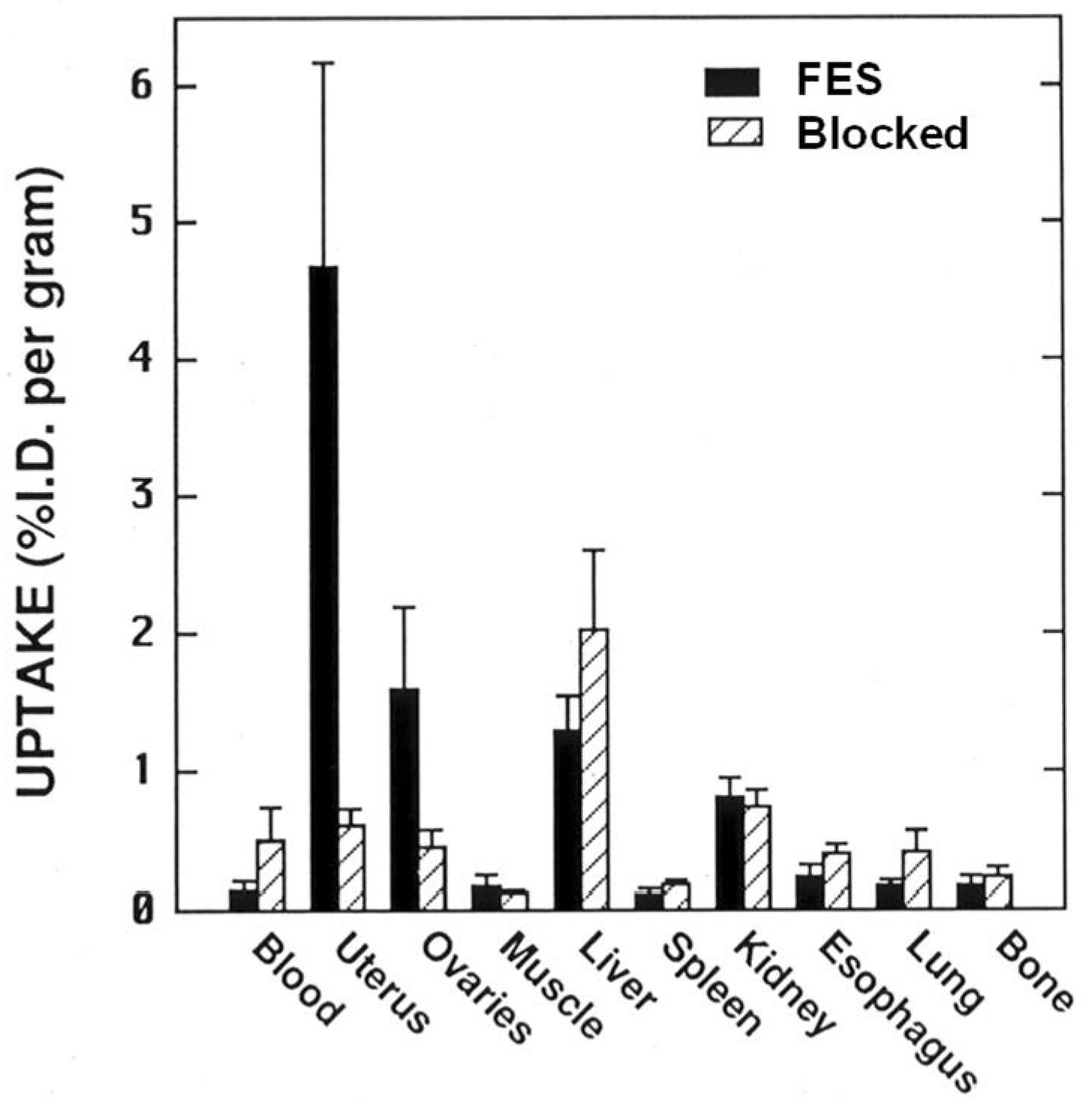
2.2. An Exploration of Various [18F]Estrogens and the Birth of FES
3. FFNP, a PET Imaging Agent for Progesterone Receptors in Breast Cancers
3.1. Bumps on the Road to the Discovery of a PET Radiotracer for PgR in Breast Cancer
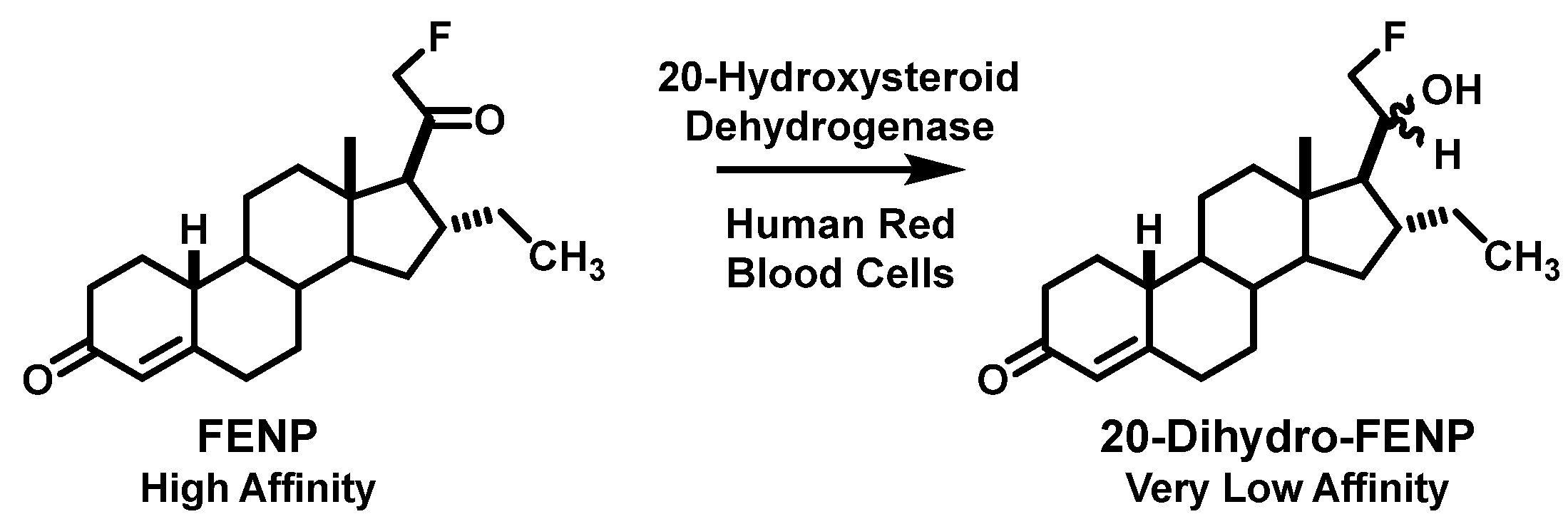
3.2. Defeating a Metabolic Inactivation in Human Blood Led to the Discovery of FFNP as an Effective PET Imaging Agent for PgR in Breast Cancer
3.3. Alternative Approaches and Efforts to Improve PET Imaging Agents for PgR
4. FDHT, a PET Imaging Agent for Androgen Receptors in Prostate Cancers
4.1. A Prelude to In Vivo Imaging of AR: Optimizing the Binding Characteristics and Pharmacokinetic Properties of Radiotracers Leading to FDHT
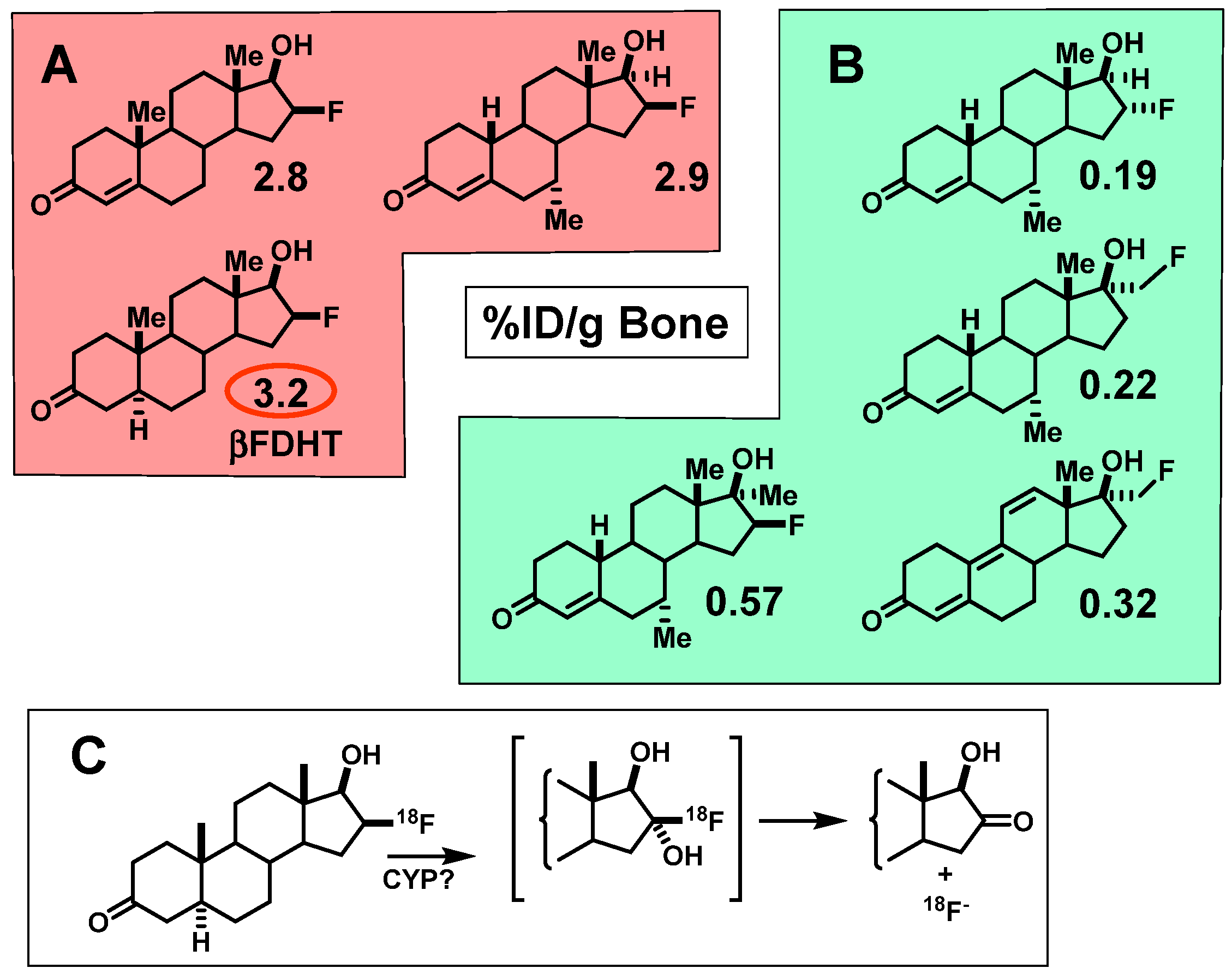
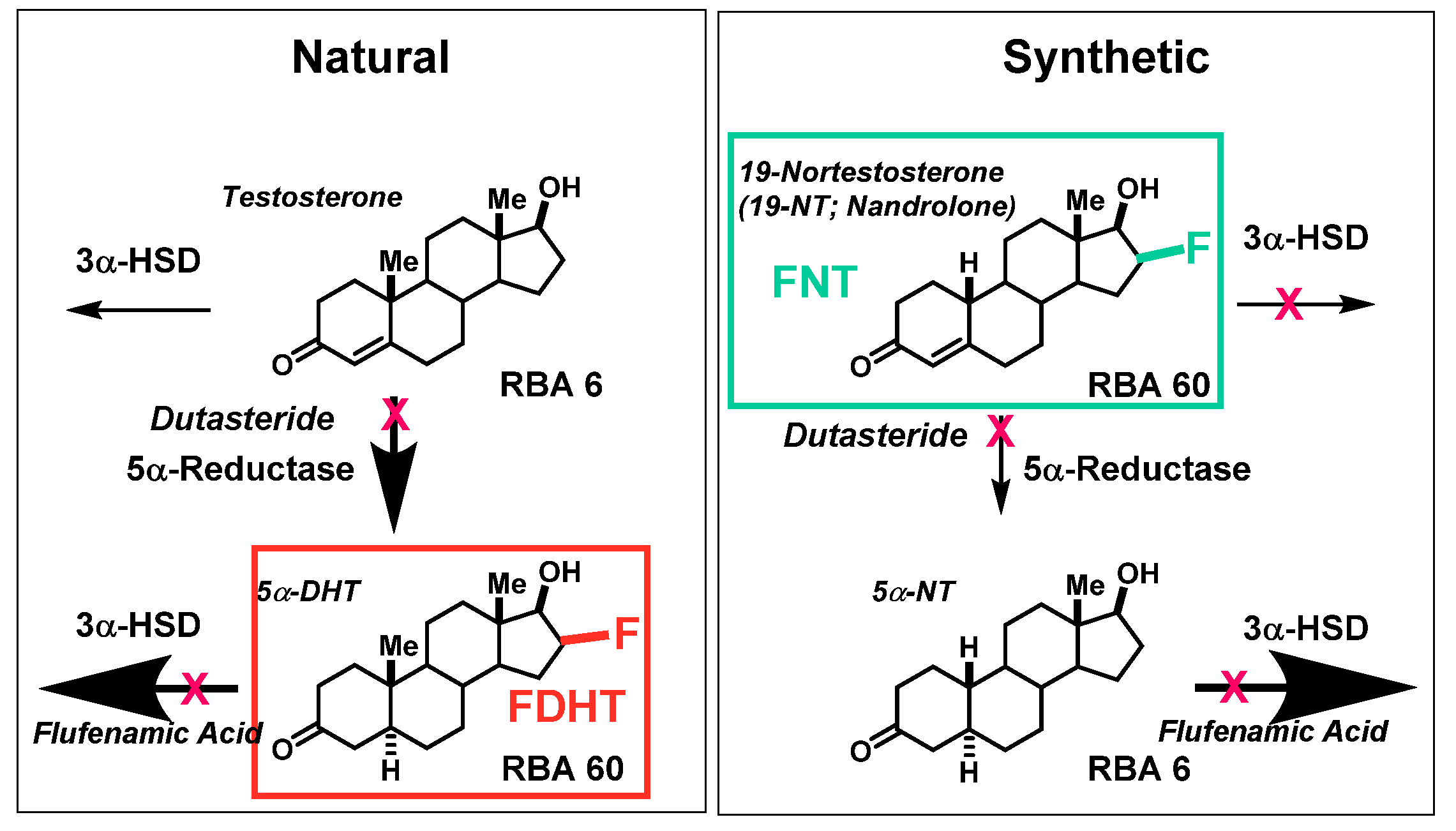
4.2. Opportunities and Efforts to Improve the Imaging Characteristics of FDHT in Prostate Cancer
5. Considerations for How PET Imaging of Steroid Receptors Might Best be Used to Improve the Management of Breast and Prostate Cancers
5.1. Utility of FES-PET Imaging of ER in Breast Cancers for Guiding Endocrine Therapy Selection
5.2. Clinical Utility of FFNP-PET Imaging of PgR in Breast Cancers: PET Imaging-Based Hormone-Challenge Tests
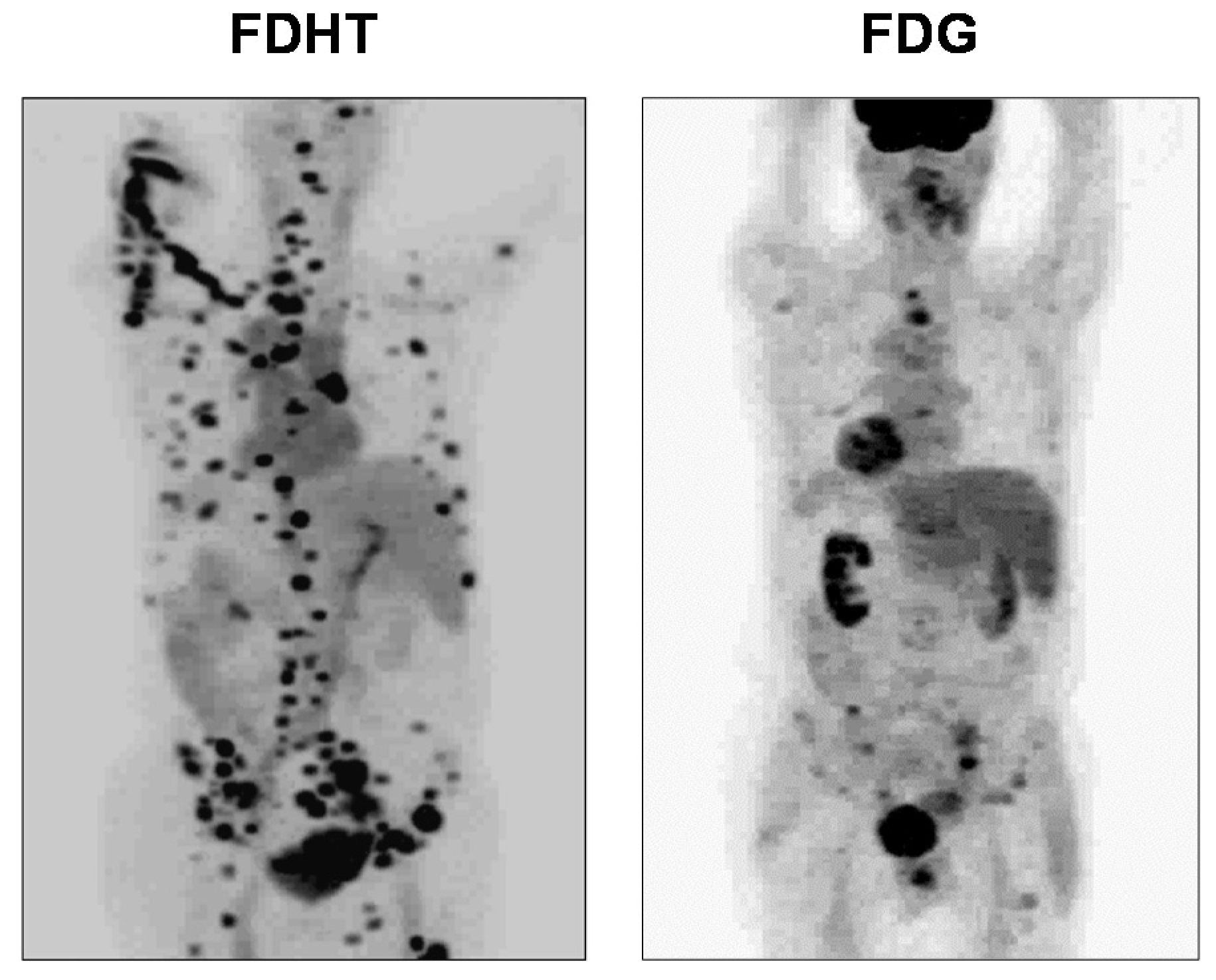
5.3. Clinical Studies of FDHT-PET in Prostate Cancer
6. Closing Comments
Funding
Acknowledgments
Conflicts of Interest
References
- Kiesewetter, D.O.; Katzenellenbogen, J.A.; Kilbourn, M.R.; Welch, M.J. Synthesis of 16-fluoroestrogens by unusually facile fluoride ion displacement reactions: Prospects for the preparation of fluorine-18 labeled estrogens. J. Org. Chem. 1984, 49, 4900–4905. [Google Scholar] [CrossRef]
- Kiesewetter, D.O.; Kilbourn, M.R.; Landvatter, S.W.; Heiman, D.F.; Katzenellenbogen, J.A.; Welch, M.J. Preparation of four fluorine-18-labeled estrogens and their selective uptakes in target tissues of immature rats. J. Nucl. Med. 1984, 25, 1212–1221. [Google Scholar] [PubMed]
- Dehdashti, F.; Picus, J.; Michalski, J.M.; Dence, C.S.; Siegel, B.A.; Katzenellenbogen, J.A.; Welch, M.J. Positron tomographic assessment of androgen receptors in prostatic carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Dehdashti, F.; Laforest, R.; Gao, F.; Aft, R.L.; Dence, C.S.; Zhou, D.; Shoghi, K.I.; Siegel, B.A.; Katzenellenbogen, J.A.; Welch, M.J. Assessment of progesterone receptors in breast carcinoma by PET with 21-18F-fluoro-16alpha,17alpha-[(R)-(1’-alpha-furylmethylidene)dioxy]-19-norpregn- 4-ene-3,20-dione. J. Nucl. Med. 2012, 53, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Katzenellenbogen, J.A.; Johnson, H.J., Jr.; Myers, H.N. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry 1973, 12, 4085–4092. [Google Scholar] [CrossRef] [PubMed]
- Brandes, S.J.; Katzenellenbogen, J.A. Fluorinated androgens and progestins: Molecular probes for androgen and progesterone receptors with potential use in positron emission tomography. Mol. Pharmacol. 1987, 32, 391–403. [Google Scholar]
- Kochanny, M.J.; VanBrocklin, H.F.; Kym, P.R.; Carlson, K.E.; O’Neil, J.P.; Bonasera, T.A.; Welch, M.J.; Katzenellenbogen, J.A. Fluorine-18-labeled progestin ketals: Synthesis and target tissue uptake selectivity of potential imaging agents for receptor-positive breast tumors. J. Med. Chem. 1993, 36, 1120–1127. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Carlson, K.E.; Heiman, D.F.; Lloyd, J.E. Receptor binding as a basis for radiopharmaceutical design. In Radiopharmaceuticals, Structure-Activity Relationships; Grune and Stratton: New York, NY, USA, 1981; pp. 23–86. [Google Scholar]
- Katzenellenbogen, J.A.; Heiman, D.F.; Senderoff, S.G.; Landvatter, S.W.; Carlson, K.E.; Goswami, R.; Lloyd, J.E.; McElvany, K.D. Estrogen receptor-based agents for imaging breast tumors: Binding selectivity as a basis for design and optimization. In Applications of Nuclear and Radiochemistry; Pergamon: Elmsford, NY, USA, 1982; pp. 311–323. [Google Scholar]
- Pomper, M.G.; VanBrocklin, H.; Thieme, A.M.; Thomas, R.D.; Kiesewetter, D.O.; Carlson, K.E.; Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A. 11. beta.-methoxy-, 11. beta.-ethyl, and 17. alpha.-ethynyl-substituted 16. alpha.-fluoroestradiols: Receptor-based imaging agents with enhanced uptake efficiency and selectivity. J. Med. Chem. 1990, 33, 3143–3155. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Hsiung, H.M. Iodohexestrols. I. Synthesis and photoreactivity of iodinated hexestrol derivatives. Biochemistry 1975, 14, 1736–1741. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Hsiung, H.M.; Carlson, K.E.; McGuire, W.L.; Kraay, R.J.; Katzenellenbogen, B.S. Iodohexestrols. II. Characterization of the binding and estrogenic activity of iodinated hexestrol derivatives, in vitro and in vivo. Biochemistry 1975, 14, 1742–1750. [Google Scholar] [CrossRef]
- Jensen, E.; Jacobson, H. Recent Progr. Hormone Res. 1962, 18, 387. [Google Scholar]
- Jensen, E.; Jacobson, H.; Flesher, J.; Saha, N.; Gupta, G.; Smith, S.; Colucci, V.; Shiplacoff, D.; Neumann, H.; DeSombre, E. Estrogen receptors in target tissues. Steroid Dyn. 1966, 133–157. [Google Scholar]
- McElvany, K.D.; Carlson, K.E.; Katzenellenbogen, J.A.; Welch, M.J. Factors affecting the target site uptake selectivity of estrogen radiopharmaceuticals: Serum binding and endogenous estrogens. J. Steroid Biochem. 1983, 18, 635–641. [Google Scholar] [CrossRef]
- Peterson, L.M.; Kurland, B.F.; Link, J.M.; Schubert, E.K.; Stekhova, S.; Linden, H.M.; Mankoff, D.A. Factors influencing the uptake of 18F-fluoroestradiol in patients with estrogen receptor positive breast cancer. Nucl. Med. Biol. 2011, 38, 969–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonasera, T.A.; O’Neil, J.P.; Xu, M.; Dobkin, J.A.; Cutler, P.D.; Lich, L.L.; Choe, Y.S.; Katzenellenbogen, J.A.; Welch, M.J. Preclinical evaluation of fluorine-18-labeled androgen receptor ligands in baboons. J. Nucl. Med. 1996, 37, 1009–1015. [Google Scholar] [PubMed]
- Jonson, S.D.; Bonasera, T.A.; Dehdashti, F.; Cristel, M.E.; Katzenellenbogen, J.A.; Welch, M.J. Comparative breast tumor imaging and comparative in vitro metabolism of 16alpha-[18F]fluoroestradiol-17beta and 16beta-[18F]fluoromoxestrol in isolated hepatocytes. Nucl. Med. Biol. 1999, 26, 123–130. [Google Scholar] [CrossRef]
- Hochberg, R.B.; Rosner, W. Interaction of 16 alpha-[125I] iodo-estradiol with estrogen receptor and other steroid-binding proteins. Proc. Natl. Acad. Sci. USA 1980, 77, 328. [Google Scholar] [CrossRef] [Green Version]
- McManaway, M.; Jagoda, E.; Eckelman, W.; Larson, S.; Francis, B.; Gibson, R.; Reba, R.; Lippman, M.E. Binding characteristics and biological activity of 17α-[125I] iodovinyl-11β-methoxyestradiol, an estrogen receptor-binding radiopharmaceutical, in human breast cancer cells (MCF-7). Cancer Res. 1986, 46, 2386–2389. [Google Scholar]
- Kabalka, G.W.; Gooch, E.E.; Hsu, H.C.; Washburn, L.C.; Sun, T.T.; Hayes, R.L. Rapid and mild syntheses of radioiodinated estrogen derivatives via organoborane technology. In Applications of Nuclear and Radiochemistry; Pergamon: Elmsford, NY, USA, 1982; pp. 197–203. [Google Scholar]
- Hanson, R.N.; Seitz, D.E.; Botarro, J.C. E-17-[125 I] Iodovinylestradiol: An estrogen-receptor-seeking radiopharmaceutical. J. Nucl. Med. 1982, 23, 436. [Google Scholar]
- DeSombre, E.R.; Mease, R.C.; Sanghavl, J.; Singh, T.; Seevers, R.H.; Hughes, A. Estrogen receptor binding affinity and uterotrophic activity of triphenylhaloethylenes. J. Steroid Biochem. 1988, 29, 583–590. [Google Scholar] [CrossRef]
- Ali, H.; Rousseau, J.; Van Lier, J. Synthesis of (17α, 20E/Z) iodovinyl testosterone and 19-nortestosterone derivatives as potential radioligands for androgen and progesterone receptors. J. Steroid Biochem. Mol. Biol. 1994, 49, 15–29. [Google Scholar] [CrossRef]
- Rijks, L.J.; Bakker, P.J.; van Tienhoven, G.; Noorduyn, L.A.; Boer, G.J.; Rietbroek, R.C.; Taat, C.W.; Janssen, A.G.; Veenhof, C.H.; van Royen, E.A. Imaging of estrogen receptors in primary and metastatic breast cancer patients with iodine-123-labeled Z-MIVE. J. Clin. Oncol. 1997, 15, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Heiman, D.F.; Senderoff, S.G.; Katzenellenbogen, J.A.; Neeley, R.J. Estrogen receptor-based imaging agents. 1. Synthesis and receptor binding affinity of some aromatic and D-ring halogenated estrogens. J. Med. Chem. 1980, 23, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, J.A.; Senderoff, S.G.; McElvany, K.D.; O’Brien, H.; Welch, M.J. 16α-[77Br] bromoestradiol-17β: A high specific-activity, gamma-emitting tracer with uptake in rat uterus and induced mammary tumors. J. Nucl. Med. 1981, 22, 42–47. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; McElvany, K.D.; Senderoff, S.G.; Carlson, K.E.; Landvatter, S.W.; Welch, M.J. 16 alpha-[77Br]bromo-11 beta-methoxyestradiol-17 beta: A gamma-emitting estrogen imaging agent with high uptake and retention by target organs. J. Nucl. Med. 1982, 23, 411–419. [Google Scholar]
- Senderoff, S.G.; McElvany, K.D.; Carlson, K.E.; Heiman, D.F.; Katzenellenbogen, J.A.; Welch, M.J. Methodology for the synthesis and specific activity determination of 16α-[77Br]-Bromoestradiol-17 β and 16α-[77Br]-11 β-methoxyestradiol-17 β, Two estrogen receptor-binding radiopharmaceuticals. Int. J. Appl. Radiat. Isot. 1982, 33, 545–551. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A. Estrogen and progestin radiopharmaceuticals for imaging breast cancer. In Estrogens, Progestins, and Their Antagonists; Pavlik, E.J., Ed.; Birkhäuser: Boston, MS, USA, 1997; Volume 2, pp. 197–242. [Google Scholar]
- Krohn, K.A.; Vera, D.R. Concepts for design and analysis of receptor radiopharmaceuticals: The Receptor-Binding Radiotracers series of meetings provided the foundation. Nucl. Med. Biol. 2020. [Google Scholar] [CrossRef]
- McElvany, K.D.; Katzenellenbogen, J.A.; Shafer, K.E.; Siegel, B.A.; Senderoff, S.G.; Welch, M.J. 16α-[77Br] bromoestradiol: Dosimetry and preliminary clinical studies. J. Nucl. Med. 1982, 23, 425–430. [Google Scholar]
- Tewson, T.J.; Welch, M.J.; Raichle, M.E. [18F]-labeled 3-deoxy-3-fluoro-D-glucose: Synthesis and preliminary biodistribution data. J. Nucl. Med. 1978, 19, 1339–1345. [Google Scholar] [CrossRef]
- Welch, M.J.; Kilbourn, M.R.; Mathias, C.J.; Mintun, M.A.; Raichle, M.E. Comparison in animal models of 18F-spiroperidol and 18F-haloperidol: Potential agents for imaging the dopamine receptor. Life Sci. 1983, 33, 1687–1693. [Google Scholar] [CrossRef]
- Berridge, M.; Apana, S.; Hersh, J. Teflon radiolysis as the major source of carrier in fluorine-18. J. Labelled Compd. Radiopharm. 2009, 52, 543–548. [Google Scholar] [CrossRef]
- Bergmann, K.E.; Landvatter, S.W.; Rocque, P.G.; Carlson, K.E.; Welch, M.J.; Katzenellenbogen, J.A. Oxohexestrol derivatives labeled with fluorine-18. Synthesis, receptor binding and in vivo distribution of two non-steroidal estrogens as potential breast tumor imaging agents. Nucl. Med. Biol. 1994, 21, 25–39. [Google Scholar] [CrossRef]
- Landvatter, S.W.; Katzenellenbogen, J.A. Nonsteroidal estrogens: Synthesis and estrogen receptor binding affinity of derivatives of (3R*, 4S*)-3, 4-bis (4-hydroxyphenyl) hexane (hexestrol) and (2R*, 3S*)-2, 3-bis (4-hydroxyphenyl) pentane (norhexestrol) functionalized on the side chain. J. Med. Chem. 1982, 25, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Landvatter, S.W.; Kiesewetter, D.O.; Kilbourn, M.R.; Katzenellenbogen, J.A.; Welch, M.J. (2R∗, 3S∗)-1-[18fluoro-2, 3-bis (4-hydroxyphenyl) pentane ([18F] fluoronorhexestrol), A positron-emitting estrogen that shows highly-selective, receptor-mediated uptake by target tissues in vivo. Life Sci. 1983, 33, 1933–1938. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A. The pharmacology of steroid radiopharmaceuticals: Specific and non-specific binding and uptake selectivity. In Radiopharmaceuticals: Chemistry and Pharmacology; Nunn, A.D., Ed.; M Dekker: New York, NY, USA, 1992; pp. 297–331. [Google Scholar]
- Katzenellenbogen, J.; Heiman, D.; Carlson, K.; Lloyd, J. In vivo and in vitro steroid receptor assays in the design of estrogen radiopharmaceuticals. In Receptor-Binding Radiotracers I; Eckelman, W.C., Ed.; CRC Press: Boca Raton, FL, USA, 1982; Volume I, pp. 93–126. [Google Scholar]
- Lim, J.L.; Zheng, L.; Berridge, M.S.; Tewson, T.J. The use of 3-methoxymethyl-16 beta, 17 beta-epiestriol-O-cyclic sulfone as the precursor in the synthesis of F-18 16 alpha-fluoroestradiol. Nucl. Med. Biol. 1996, 23, 911–915. [Google Scholar] [CrossRef]
- Kil, H.S.; Cho, H.Y.; Lee, S.J.; Oh, S.J.; Chi, D.Y. Alternative synthesis for the preparation of 16alpha-[(18) F]fluoroestradiol. J. Label. Comp. Radiopharm 2013, 56, 619–626. [Google Scholar] [CrossRef]
- Zhou, D.; Lin, M.; Yasui, N.; Al-Qahtani, M.H.; Dence, C.S.; Schwarz, S.; Katzenellenbogen, J.A. Optimization of the preparation of fluorine-18-labeled steroid receptor ligands 16alpha-[18F] fluoroestradiol (FES),[18F] fluoro furanyl norprogesterone (FFNP), and 16beta-[18F] fluoro-5alpha-dihydrotestosterone (FDHT) as radiopharmaceuticals. J. Label. Compd. Radiopharm. 2014, 57, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, O.; Nikolaeva, V.; Krasikova, R. Automated SPE-based synthesis of 16alpha-[(18)F]fluoroestradiol without HPLC purification step. Appl. Radiat. Isot. 2018, 141, 57–63. [Google Scholar] [CrossRef]
- Shi, J.; Afari, G.; Bhattacharyya, S. Rapid synthesis of [18F]fluoroestradiol: Remarkable advantage of microwaving over conventional heating. J. Label. Comp. Radiopharm 2014, 57, 730–736. [Google Scholar] [CrossRef]
- Liang, S.; Lan, X.; Zhang, Y.; Xu, X.; Li, B. Fully automatic synthesis of [(1)(8)F]FES for reporter gene hERL expression imaging. Nucl. Med. Commun. 2012, 33, 29–33. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A. The quest for improving the management of breast cancer by functional imaging: The discovery and development of 16α-[18F] fluoroestradiol (FES), a PET radiotracer for the estrogen receptor, a historical review. Nucl. Med. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Phoenix, S.; Ouellet, R.; Langlois, R.; van Lier, J.E.; Turcotte, E.E.; Benard, F.; Lecomte, R. Assessment of the novel estrogen receptor PET tracer 4-fluoro-11beta-methoxy-16alpha-[(18)F]fluoroestradiol (4FMFES) by PET imaging in a breast cancer murine model. Mol. Imaging Biol. 2013, 15, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A.; Brodack, J.W.; Kilbourn, M.R.; Carlson, K.E.; Kiesewetter, D.O. Characterization of the uptake of 16 alpha-([18F]fluoro)-17 beta-estradiol in DMBA-induced mammary tumors. Int. J. Rad. Appl. Instrum. B 1987, 14, 15–25. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Mathias, C.J.; VanBrocklin, H.F.; Brodack, J.W.; Welch, M.J. Titration of the in vivo uptake of 16 alpha-[18F]fluoroestradiol by target tissues in the rat: Competition by tamoxifen, and implications for quantitating estrogen receptors in vivo and the use of animal models in receptor-binding radiopharmaceutical development. Nucl. Med. Biol. 1993, 20, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.; Welch, M.; Siegel, B.; Mathias, C.; Brodack, J.; McGuire, A.; Katzenellenbogen, J. Breast cancer: PET imaging of estrogen receptors. Radiology 1988, 169, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.N., Jr. SNM Highlights as History: 1987. J. Nucl. Med. 2004, 45, N27–N33. [Google Scholar]
- Bennink, R.J.; Rijks, L.J.; van Tienhoven, G.; Noorduyn, L.A.; Janssen, A.G.; Sloof, G.W. Estrogen receptor status in primary breast cancer: Iodine 123-labeled cis-11beta-methoxy-17alpha-iodovinyl estradiol scintigraphy. Radiology 2001, 220, 774–779. [Google Scholar] [CrossRef]
- Bennink, R.J.; van Tienhoven, G.; Rijks, L.J.; Noorduyn, A.L.; Janssen, A.G.; Sloof, G.W. In vivo prediction of response to antiestrogen treatment in estrogen receptor-positive breast cancer. J. Nucl. Med. 2004, 45, 1–7. [Google Scholar]
- Bénard, F.; Turcotte, É. Imaging in breast cancer: Single-photon computed tomography and positron-emission tomography. Breast Cancer Res. 2005, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Desombre, E.R.; Pribish, J.; Hughes, A. Comparison of the distribution of radioiodinated di-and tri-hydroxyphenylethylene estrogens in the immature female rat. Nucl. Med. Biol. 1995, 22, 679–687. [Google Scholar] [CrossRef]
- Yasui, L.S.; Hughes, A.; Desombre, E.R. DNA damage induction by 125I-estrogen. Acta Oncol. 1996, 35, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Hanson, R.N.; Tongcharoensirikul, P.; Barnsley, K.; Ondrechen, M.J.; Hughes, A.; DeSombre, E.R. Synthesis and evaluation of 2-halogenated-1, 1-bis (4-hydroxyphenyl)-2-(3-hydroxyphenyl)-ethylenes as potential estrogen receptor-targeted radiodiagnostic and radiotherapeutic agents. Steroids 2015, 96, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allott, L.; Smith, G.; Aboagye, E.O.; Carroll, L. PET imaging of steroid hormone receptor expression. Mol. Imaging 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitznagle, L.; Kasina, S.; Marino, C. Structure-distribution studies with fluorine-18 labeled pregnenolones: Effect of structure on adrenal uptake and elimination. J. Label. Compd. Radiopharm. 1982, 19, 1512–1513. [Google Scholar]
- Spitznagle, L.; Marino, C.; Kasina, S. Design of radiopharmaceuticals for structure distribution studies: Some fluorine-18 labeled pregnane derivatives. Nucl. Med. Biol. 1981, 182, 28. [Google Scholar]
- Spitznagle, L.A.; Marino, C.A. Synthesis of fluorine-18 labeled 21-fluoroprogesterone. Steroids 1977, 30, 435–438. [Google Scholar] [CrossRef]
- Grill, H.; Heubner, A.; Pollow, K.; Manz, B.; Schulze, P.; Hofmeister, H.; Laurent, H.; Wiechert, R.; Elger, W. (Z)-17 beta-hydroxy-17 alpha-(2-[125I] iodovinyl)-4-estren-3-one: A new specific gamma-emitting ligand for determination of progesterone receptor. J. Clin. Chem. Clin. Biochem. Z. Fur Klin. Chem. Und Klin. Biochem. 1987, 25, 107–112. [Google Scholar]
- Hoyte, R.; Rosner, W.; Johnson, I.; Zielinski, J.; Hochberg, R. Synthesis and evaluation of potential radioligands for the progesterone receptor. J. Med. Chem. 1985, 28, 1695–1699. [Google Scholar] [CrossRef]
- Hochberg, R.B.; Hoyte, R.M.; Rosner, W. E-17 alpha-(2-[125I]iodovinyl)-19-nortestosterone: The synthesis of a gamma-emitting ligand for the progesterone receptor. Endocrinology 1985, 117, 2550–2552. [Google Scholar] [CrossRef]
- Salman, M.; Chamness, G.C. A potential radioiodinated ligand for androgen receptor: 7. alpha.-methyl-17. alpha.-[2’-(E)-iodovinyl]-19-nortestosterone. J. Med. Chem. 1991, 34, 1019–1024. [Google Scholar] [CrossRef]
- Van den Bos, J.C.; Rijks, L.J.; van Doremalen, P.A.; de Bruin, K.; Janssen, A.G.; van Royen, E.A. New iodinated progestins as potential ligands for progesterone receptor imaging in breast cancer. Part 1: Synthesis and in vitro pharmacological characterization. Nucl. Med. Biol. 1998, 25, 781–789. [Google Scholar] [CrossRef]
- Rijks, L.J.; van den Bos, J.C.; van Doremalen, P.A.; Boer, G.J.; de Bruin, K.; Janssen, A.G.; van Royen, E.A. New iodinated progestins as potential ligands for progesterone receptor imaging in breast cancer. Part 2: In vivo pharmacological characterization. Nucl. Med. Biol. 1998, 25, 791–798. [Google Scholar] [CrossRef]
- Brandes, S.J.; Katzenellenbogen, J.A. Fundamental considerations in the design of fluorine-18 labeled progestins and androgens as imaging agents for receptor-positive tumors of the breast and prostate. Int. J. Rad. Appl. Instrum. B 1988, 15, 53–67. [Google Scholar] [CrossRef]
- Ojasoo, T.; Raynaud, J.-P. Unique steroid congeners for receptor studies. Cancer Res. 1978, 38, 4186–4198. [Google Scholar] [PubMed]
- Zeelen, F.; van den Broek, A. Synthesis of 16α-ethyl-21-hydroxy-19-norpregn-4-ene-3, 20-dione (Org 2058). Recl. Des Trav. Chim. Des Pays Bas 1985, 104, 239–242. [Google Scholar] [CrossRef]
- Carlson, K.E.; Brandes, S.J.; Pomper, M.G.; Katzenellenbogen, J.A. Uptake of three [3H] progestins by target tissues in vivo: Implications for the design of diagnostic imaging agents. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1988, 15, 403–408. [Google Scholar] [CrossRef]
- Kontula, K.; Jänne, O.; Vihko, R.; de Jager, E.; de Visser, J.; Zeelen, F. Progesterone-binding proteins: In vitro binding and biological activity of different steroidal ligands. Eur. J. Endocrinol. 1975, 78, 574–592. [Google Scholar] [CrossRef]
- Pomper, M.G.; Katzenellenbogen, J.A.; Welch, M.J.; Brodack, J.W.; Mathias, C.J. 21-[18F]fluoro-16 alpha-ethyl-19-norprogesterone: Synthesis and target tissue selective uptake of a progestin receptor based radiotracer for positron emission tomography. J. Med. Chem. 1988, 31, 1360–1363. [Google Scholar] [CrossRef]
- Pomper, M.G.; Pinney, K.G.; Carlson, K.E.; Van Brocklin, H.; Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A. Target tissue uptake selectivity of three fluorine-substituted progestins: Potential imaging agents for receptor-positive breast tumors. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1990, 17, 309–319. [Google Scholar] [CrossRef]
- Dehdashti, F.; McGuire, A.H.; Van Brocklin, H.F.; Siegel, B.A.; Andriole, D.P.; Griffeth, L.K.; Pomper, M.G.; Katzenellenbogen, J.A.; Welch, M.J. Assessment of 21-[18F]fluoro-16 alpha-ethyl-19-norprogesterone as a positron-emitting radiopharmaceutical for the detection of progestin receptors in human breast carcinomas. J. Nucl. Med. 1991, 32, 1532–1537. [Google Scholar]
- Verhagen, A.; Elsinga, P.H.; de Groot, T.J.; Paans, A.M.; de Goeij, C.C.; Sluyser, M.; Vaalburg, W. A fluorine-18 labeled progestin as an imaging agent for progestin receptor positive tumors with positron emission tomography. Cancer Res. 1991, 51, 1930–1933. [Google Scholar] [PubMed]
- Verhagen, A.; Luurtsema, G.; Pesser, J.; De Groot, T.; Wouda, S.; Oosterhuis, J.; Vaalburg, W. Preclinical evaluation of a positron emitting progestin ([18F] fluoro-16α-methyl-19-norprogesterone) for imaging progesterone receptor positive tumours with positron emission tomography. Cancer Lett. 1991, 59, 125–132. [Google Scholar] [CrossRef]
- Verhagen, A.; Studeny, M.; Luurtsema, G.; Visser, G.M.; De Goeij, C.C.; Sluyser, M.; Nieweg, O.E.; Van der Ploeg, E.; Go, K.G.; Vaalburg, W. Metabolism of a [18F]fluorine labeled progestin (21-[18F]fluoro-16 alpha-ethyl-19-norprogesterone) in humans: A clue for future investigations. Nucl. Med. Biol. 1994, 21, 941–952. [Google Scholar] [CrossRef]
- Buckman, B.O.; Bonasera, T.A.; Kirschbaum, K.S.; Welch, M.J.; Katzenellenbogen, J.A. Fluorine-18-labeled progestin 16 alpha, 17 alpha-dioxolanes: Development of high-affinity ligands for the progesterone receptor with high in vivo target site selectivity. J. Med. Chem. 1995, 38, 328–337. [Google Scholar] [CrossRef]
- Fried, J.; Sabo, E.; Grabowich, P.; Lerner, L.; Kessler, W.; Brennan, D.; Borman, A. Progestationally Active Acetals and Ketals of 16-Alpha,17-Alpha-Dihydroprogesterone. Chem. Ind. London 1961, 15, 465–466. [Google Scholar]
- Lerner, L.J.; Brennan, D.M.; Borman, A. Biological activities of 16α, 17α dihydroxyprogesterone derivatives. Proc. Soc. Exp. Biol. Med. 1961, 106, 231–234. [Google Scholar] [CrossRef]
- Kym, P.R.; Carlson, K.E.; Katzenellenbogen, J.A. Progestin 16. alpha., 17. alpha.-dioxolane ketals as molecular probes for the progesterone receptor: Synthesis, binding affinity, and photochemical evaluation. J. Med. Chem. 1993, 36, 1111–1119. [Google Scholar] [CrossRef]
- Chan, S.R.; Fowler, A.M.; Allen, J.A.; Zhou, D.; Dence, C.S.; Sharp, T.L.; Fettig, N.M.; Dehdashti, F.; Katzenellenbogen, J.A. Longitudinal noninvasive imaging of progesterone receptor as a predictive biomarker of tumor responsiveness to estrogen deprivation therapy. Clin. Cancer Res. 2015, 21, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Fowler, A.M.; Chan, S.R.; Sharp, T.L.; Fettig, N.M.; Zhou, D.; Dence, C.S.; Carlson, K.E.; Jeyakumar, M.; Katzenellenbogen, J.A.; Schreiber, R.D.; et al. Small-animal PET of steroid hormone receptors predicts tumor response to endocrine therapy using a preclinical model of breast cancer. J. Nucl. Med. 2012, 53, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Basuli, F.; Zhang, X.; Blackman, B.; White, M.E.; Jagoda, E.M.; Choyke, P.L.; Swenson, R.E. Fluorine-18 Labeled Fluorofuranylnorprogesterone ([18F] FFNP) and Dihydrotestosterone ([18F] FDHT) Prepared by “Fluorination on Sep-Pak” Method. Molecules 2019, 24, 2389. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Zhou, H.-b.; Dence, C.S.; Carlson, K.E.; Welch, M.J.; Katzenellenbogen, J.A. Development of [F-18] fluorine-substituted Tanaproget as a progesterone receptor imaging agent for positron emission tomography. Bioconjug. Chem. 2010, 21, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Pomper, M.G.; Kochanny, M.J.; Thieme, A.M.; Carlson, K.E.; Vanbrocklin, H.F.; Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A. Fluorine-substituted corticosteroids: Synthesis and evaluation as potential receptor-based imaging agents for positron emission tomography of the brain. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1992, 19, 461–480. [Google Scholar] [CrossRef]
- Volden, P.A.; Conzen, S.D. The influence of glucocorticoid signaling on tumor progression. Brain. Behav. Immun. 2013, 30, S26–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fensome, A.; Bender, R.; Chopra, R.; Cohen, J.; Collins, M.A.; Hudak, V.; Malakian, K.; Lockhead, S.; Olland, A.; Svenson, K. Synthesis and Structure− Activity Relationship of Novel 6-Aryl-1, 4-dihydrobenzo [d][1,3] oxazine-2-thiones as Progesterone Receptor Modulators Leading to the Potent and Selective Nonsteroidal Progesterone Receptor Agonist Tanaproget. J. Med. Chem. 2005, 48, 5092–5095. [Google Scholar] [CrossRef]
- Zhou, H.-B.; Lee, J.H.; Mayne, C.G.; Carlson, K.E.; Katzenellenbogen, J.A. Imaging progesterone receptor in breast tumors: Synthesis and receptor binding affinity of fluoroalkyl-substituted analogues of tanaproget. J. Med. Chem. 2010, 53, 3349–3360. [Google Scholar] [CrossRef] [Green Version]
- Allott, L.; Miranda, C.; Hayes, A.; Raynaud, F.; Cawthorne, C.; Smith, G. Synthesis of a benzoxazinthione derivative of tanaproget and pharmacological evaluation for PET imaging of PR expression. EJNMMI Radiopharm. Chem. 2019, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Merchant, S.; Allott, L.; Carroll, L.; Tittrea, V.; Kealey, S.; Witney, T.H.; Miller, P.W.; Smith, G.; Aboagye, E.O. Synthesis and pre-clinical evaluation of a [18 F] fluoromethyl-tanaproget derivative for imaging of progesterone receptor expression. RSC Adv. 2016, 6, 57569–57579. [Google Scholar] [CrossRef]
- Zhou, D.; Carlson, K.E.; Katzenellenbogen, J.A.; Welch, M.J. Bromine-and iodine-substituted 16α, 17α-dioxolane progestins for breast tumor imaging and radiotherapy: Synthesis and receptor binding affinity. J. Med. Chem. 2006, 49, 4737–4744. [Google Scholar] [CrossRef]
- Auwerx, J.; Baulieu, E.; Beato, M.; Becker-Andre, M.; Burbach, P.; Camerino, G.; Chambon, P.; Cooney, A.; Dejean, A.; Dreyer, C. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar]
- Leo, J.C.; Guo, C.; Woon, C.T.; Aw, S.E.; Lin, V.C. Glucocorticoid and mineralocorticoid cross-talk with progesterone receptor to induce focal adhesion and growth inhibition in breast cancer cells. Endocrinology 2004, 145, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.R.; Harkless, R.V.; Oh, A.; Bowie, K.R.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R. Glucocorticoid receptor modulation decreases ER-positive breast cancer cell proliferation and suppresses wild-type and mutant ER chromatin association. Breast Cancer Res. 2019, 21, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiniger, B.; Kniess, T.; Bergmann, R.; Pietzsch, J.; Wuest, F.R. Radiolabeled glucocorticoids as molecular probes for imaging brain glucocorticoid receptors by means of positron emission tomography (PET). Mini Rev. Med. Chem. 2008, 8, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Kniess, T.; Henry, B.; Peeters, B.W.; Wiegerinck, P.H.; Pietzsch, J.; Bergmann, R. Radiosynthesis and radiopharmacological evaluation of [N-methyl-11C] Org 34850 as a glucocorticoid receptor (GR)-binding radiotracer. Appl. Radiat. Isot. 2009, 67, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Hoyte, R.M.; Labaree, D.C.; Fede, J.-M.; Harris, C.; Hochberg, R.B. Iodinated and fluorinated steroid 2′-aryl-[3, 2-c] pyrazoles as potential glucocorticoid receptor imaging agents. Steroids 1998, 63, 595–602. [Google Scholar] [CrossRef]
- Wuest, F.; Carlson, K.E.; Katzenellenbogen, J.A. Expeditious synthesis of steroids containing a 2-methylsulfanyl-acetyl side chain as potential glucocorticoid receptor imaging agents. Steroids 2008, 73, 69–76. [Google Scholar] [CrossRef]
- Eakins, M.; Waters, S. The synthesis of 77Br-labelled 5α-dihydrotestosterone and a comparison of its distribution in rats with 77Br-bromide. Int. J. Appl. Radiat. Isot. 1979, 30, 701–703. [Google Scholar] [CrossRef]
- Ghanadian, R.; Waters, S.; Chisholm, G. Investigations into the use of 77 Br labelled 5α-dihydrotestosterone for scanning the prostate. Eur. J. Nucl. Med. 1977, 2, 155–157. [Google Scholar] [CrossRef]
- Hoyte, R.; Rosner, W.; Hochberg, R. Synthesis of 16α-[125I] iodo-5α-dihydrotestosterone and evaluation of its affinity for the androgen receptor. J. Steroid Biochem. 1982, 16, 621–628. [Google Scholar] [CrossRef]
- Tarle, M.; Padovan, R.; Spaventi, Š. The uptake of radioiodinated 5α-dihydrotestosterone by the prostate of intact and castrated rats. Eur. J. Nucl. Med. 1981, 6, 79–83. [Google Scholar] [CrossRef]
- Hoyte, R.M.; MacLusky, N.J.; Hochberg, R.B. The synthesis and testing of E-17α-(2-iodovinyl)-5α-dihydrotestosterone and Z-17α-(2-iodovinyl)-5α-dihydrotestosterone as γ-emitting ligands for the androgen receptor. J. Steroid Biochem. 1990, 36, 125–132. [Google Scholar] [CrossRef]
- Ali, H.; Rousseau, J.; Ahmed, N.; Guertin, V.; Hochberg, R.B.; van Lier, J.E. Synthesis of the 7α-cyano-(17α, 20E/Z)-[125I] iodovinyl-19-nortestosterones: Potential radioligands for androgen and progesterone receptors. Steroids 2003, 68, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Schänzer, W. Metabolism of anabolic androgenic steroids. Clin. Chem. 1996, 42, 1001–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Carlson, K.E.; Katzenellenbogen, J.A. Synthesis of high-affinity fluorine-substituted ligands for the androgen receptor. Potential agents for imaging prostatic cancer by positron emission tomography. J. Med. Chem. 1992, 35, 2113–2129. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.S.; Katzenellenbogen, J.A. Synthesis of C-6 fluoroandrogens: Evaluation of ligands for tumor receptor imaging. Steroids 1995, 60, 414–422. [Google Scholar] [CrossRef]
- Liu, A.; Katzenellenbogen, J.A.; VanBrocklin, H.F.; Mathias, C.J.; Welch, M.J. 20-[18F] fluoromibolerone, a positron-emitting radiotracer for androgen receptors: Synthesis and tissue distribution studies. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1991, 32, 81–88. [Google Scholar]
- Choe, Y.S.; Lidstroem, P.J.; Chi, D.Y.; Bonasera, T.A.; Welch, M.J.; Katzenellenbogen, J.A. Synthesis of 11. beta.-[18F] Fluoro-5. alpha.-dihydrotestosterone and 11. beta.-[18F] Fluoro-19-nor-5. alpha.-dihydrotestosterone: Preparation via halofluorination-reduction, receptor binding, and tissue distribution. J. Med. Chem. 1995, 38, 816–825. [Google Scholar] [CrossRef]
- Carlson, K.E.; Katzenellenbogen, J.A. A comparative study of the selectivity and efficiency of target tissue uptake of five tritium-labeled androgens in the rat. J. Steroid Biochem. 1990, 36, 549–561. [Google Scholar] [CrossRef]
- Liu, A.; Dence, C.S.; Welch, M.J.; Katzenellenbogen, J.A. Fluorine-18-labeled androgens: Radiochemical synthesis and tissue distribution studies on six fluorine-substituted androgens, potential imaging agents for prostatic cancer. J. Nucl. Med. 1992, 33, 724–734. [Google Scholar]
- Larson, S.M.; Morris, M.; Gunther, I.; Beattie, B.; Humm, J.L.; Akhurst, T.A.; Finn, R.D.; Erdi, Y.; Pentlow, K.; Dyke, J. Tumor localization of 16β-18F-fluoro-5α-dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J. Nucl. Med. 2004, 45, 366–373. [Google Scholar]
- Beattie, B.J.; Smith-Jones, P.M.; Jhanwar, Y.S.; Schöder, H.; Schmidtlein, C.R.; Morris, M.J.; Zanzonico, P.; Squire, O.; Meirelles, G.S.; Finn, R. Pharmacokinetic assessment of the uptake of 16β-18F-fluoro-5α-dihydrotestosterone (FDHT) in prostate tumors as measured by PET. J. Nucl. Med. 2010, 51, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Pippione, A.C.; Carnovale, I.M.; Bonanni, D.; Sini, M.; Goyal, P.; Marini, E.; Pors, K.; Adinolfi, S.; Zonari, D.; Festuccia, C. Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: Application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur. J. Med. Chem. 2018, 150, 930–945. [Google Scholar] [CrossRef] [PubMed]
- Naslund, M.J.; Miner, M. A review of the clinical efficacy and safety of 5α-reductase inhibitors for the enlarged prostate. Clin. Ther. 2007, 29, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Downer, J.B.; Jones, L.A.; Engelbach, J.A.; Lich, L.L.; Mao, W.; Carlson, K.E.; Katzenellenbogen, J.A.; Welch, M.J. Comparison of animal models for the evaluation of radiolabeled androgens. Nucl. Med. Biol. 2001, 28, 613–626. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [Green Version]
- Teutsch, G.; Goubet, F.; Battmann, T.; Bonfils, A.; Bouchoux, F.; Cerede, E.; Gofflo, D.; Gaillard-Kelly, M.; Philibert, D. Non-steroidal antiandrogens: Synthesis and biological profile of high-affinity ligands for the androgen receptor. J. Steroid Biochem. Mol. Biol. 1994, 48, 111–119. [Google Scholar] [CrossRef]
- Parent, E.E.; Dence, C.S.; Jenks, C.; Sharp, T.L.; Welch, M.J.; Katzenellenbogen, J.A. Synthesis and biological evaluation of [18F] bicalutamide, 4-[76Br] bromobicalutamide, and 4-[76Br] bromo-thiobicalutamide as non-steroidal androgens for prostate cancer imaging. J. Med. Chem. 2007, 50, 1028–1040. [Google Scholar] [CrossRef]
- Parent, E.E.; Jenks, C.; Sharp, T.; Welch, M.J.; Katzenellenbogen, J.A. Synthesis and biological evaluation of a nonsteroidal bromine-76-labeled androgen receptor ligand 3-[76Br] bromo-hydroxyflutamide. Nucl. Med. Biol. 2006, 33, 705–713. [Google Scholar] [CrossRef]
- Parent, E.E.; Dence, C.S.; Sharp, T.L.; Welch, M.J.; Katzenellenbogen, J.A. Synthesis and biological evaluation of a fluorine-18-labeled nonsteroidal androgen receptor antagonist, N-(3-[18F] fluoro-4-nitronaphthyl)-cis-5-norbornene-endo-2, 3-dicarboxylic imide. Nucl. Med. Biol. 2006, 33, 615–624. [Google Scholar] [CrossRef]
- Linden, H.M.; Kurland, B.F.; Peterson, L.M.; Schubert, E.K.; Gralow, J.R.; Specht, J.M.; Ellis, G.K.; Lawton, T.J.; Livingston, R.B.; Petra, P.H.; et al. Fluoroestradiol positron emission tomography reveals differences in pharmacodynamics of aromatase inhibitors, tamoxifen, and fulvestrant in patients with metastatic breast cancer. Clin. Cancer Res. 2011, 17, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- van Kruchten, M.; de Vries, E.G.; Glaudemans, A.W.; van Lanschot, M.C.; van Faassen, M.; Kema, I.P.; Brown, M.; Schröder, C.P.; de Vries, E.F.; Hospers, G.A. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 2015, 5, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Heidari, P.; Deng, F.; Esfahani, S.A.; Leece, A.K.; Shoup, T.M.; Vasdev, N.; Mahmood, U. Pharmacodynamic imaging guides dosing of a selective estrogen receptor degrader. Clin. Cancer Res. 2015, 21, 1340–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathkopf, D.E.; Morris, M.J.; Fox, J.J.; Danila, D.C.; Slovin, S.F.; Hager, J.H.; Rix, P.J.; Maneval, E.C.; Chen, I.; Gönen, M. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J. Clin. Oncol. 2013, 31, 3525. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.-E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Evan, Y.Y.; Alumkal, J. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Beatson, G.T. Meeting IX.—May 20, 1896: On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases. Trans. Med. Chir. Soc. Edinb. 1896, 15, 153. [Google Scholar]
- Clarke, M.J. Ovarian ablation in breast cancer, 1896 to 1998: Milestones along hierarchy of evidence from case report to Cochrane review. BMJ 1998, 317, 1246–1248. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, M. A review of endocrine options for the treatment of advanced breast cancer. Oncology 1997, 54, 2–5. [Google Scholar] [CrossRef]
- Kumar, M.; Salem, K.; Tevaarwerk, A.J.; Strigel, R.M.; Fowler, A.M. Recent Advances in Imaging Steroid Hormone Receptors in Breast Cancer. J. Nucl. Med. 2020, 61, 172–176. [Google Scholar] [CrossRef]
- van Kruchten, M.; de Vries, E.G.; Brown, M.; de Vries, E.F.; Glaudemans, A.W.; Dierckx, R.A.; Schröder, C.P.; Hospers, G.A. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncol. 2013, 14, e465–e475. [Google Scholar] [CrossRef]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef]
- King, W.J.; DeSombre, E.R.; Jensen, E.V.; Greene, G.L. Comparison of immunocytochemical and steroid-binding assays for estrogen receptor in human breast tumors. Cancer Res. 1985, 45, 293–304. [Google Scholar]
- Fowler, A.M.; Clark, A.S.; Katzenellenbogen, J.A.; Linden, H.M.; Dehdashti, F. Imaging Diagnostic and Therapeutic Targets-Steroid Receptors in Breast Cancer. J. Nucl. Med. 2016, 57, 75S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertych, A.; Mohan, S.; Maclary, S.; Mohanty, S.; Wawrowsky, K.; Mirocha, J.; Balzer, B.; Knudsen, B.S. Effects of tissue decalcification on the quantification of breast cancer biomarkers by digital image analysis. Diagn. Pathol. 2014, 9, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurland, B.F.; Peterson, L.M.; Lee, J.H.; Linden, H.M.; Schubert, E.K.; Dunnwald, L.K.; Link, J.M.; Krohn, K.A.; Mankoff, D.A. Between-patient and within-patient (site-to-site) variability in estrogen receptor binding, measured in vivo by 18F-fluoroestradiol PET. J. Nucl. Med. 2011, 52, 1541–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nienhuis, H.H.; van Kruchten, M.; Elias, S.G.; Glaudemans, A.; de Vries, E.F.J.; Bongaerts, A.H.H.; Schroder, C.P.; de Vries, E.G.E.; Hospers, G.A.P. (18)F-Fluoroestradiol Tumor Uptake Is Heterogeneous and Influenced by Site of Metastasis in Breast Cancer Patients. J. Nucl. Med. 2018, 59, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Amir, E.; Miller, N.; Geddie, W.; Freedman, O.; Kassam, F.; Simmons, C.; Oldfield, M.; Dranitsaris, G.; Tomlinson, G.; Laupacis, A. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. 2012, 30, 587. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- McGuire, A.H.; Dehdashti, F.; Siegel, B.A.; Lyss, A.P.; Brodack, J.W.; Mathias, C.J.; Mintun, M.A.; Katzenellenbogen, J.A.; Welch, M.J. Positron tomographic assessment of 16 alpha-[18F] fluoro-17 beta-estradiol uptake in metastatic breast carcinoma. J. Nucl. Med. 1991, 32, 1526–1531. [Google Scholar]
- Dehdashti, F.; Mortimer, J.E.; Siegel, B.A.; Griffeth, L.K.; Bonasera, T.J.; Fusselman, M.J.; Detert, D.D.; Cutler, P.D.; Katzenellenbogen, J.A.; Welch, M.J. Positron tomographic assessment of estrogen receptors in breast cancer: Comparison with FDG-PET and in vitro receptor assays. J. Nucl. Med. 1995, 36, 1766–1774. [Google Scholar]
- Mortimer, J.E.; Dehdashti, F.; Siegel, B.A.; Katzenellenbogen, J.A.; Fracasso, P.; Welch, M.J. Positron emission tomography with 2-[18F]Fluoro-2-deoxy-D-glucose and 16alpha-[18F]fluoro-17beta-estradiol in breast cancer: Correlation with estrogen receptor status and response to systemic therapy. Clin. Cancer Res. 1996, 2, 933–939. [Google Scholar]
- Park, J.H.; Kang, M.J.; Ahn, J.-H.; Kim, J.E.; Jung, K.H.; Gong, G.; Lee, H.J.; Son, B.-H.; Ahn, S.-H.; Kim, H.-H. Phase II trial of neoadjuvant letrozole and lapatinib in Asian postmenopausal women with estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2)-positive breast cancer [Neo-ALL-IN]: Highlighting the TILs, ER expressional change after neoadjuvant treatment, and FES-PET as potential significant biomarkers. Cancer Chemother. Pharmacol. 2016, 78, 685–695. [Google Scholar]
- Kurland, B.F.; Peterson, L.M.; Lee, J.H.; Schubert, E.K.; Currin, E.R.; Link, J.M.; Krohn, K.A.; Mankoff, D.A.; Linden, H.M. Estrogen Receptor Binding (18F-FES PET) and Glycolytic Activity (18F-FDG PET) Predict Progression-Free Survival on Endocrine Therapy in Patients with ER+ Breast Cancer. Clin. Cancer Res. 2017, 23, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kruchten, M.; Glaudemans, A.W.; de Vries, E.F.; Beets-Tan, R.G.; Schroder, C.P.; Dierckx, R.A.; de Vries, E.G.; Hospers, G.A. PET imaging of estrogen receptors as a diagnostic tool for breast cancer patients presenting with a clinical dilemma. J. Nucl. Med. 2012, 53, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Linden, H.M.; Peterson, L.M.; Fowler, A.M. Clinical Potential of Estrogen and Progesterone Receptor Imaging. PET Clin. 2018, 13, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.J.; Clark, A.S.; Schubert, E.K.; Mankoff, D.A. 18F-Fluoroestradiol PET: Current Status and Potential Future Clinical Applications. J. Nucl. Med. 2016, 57, 1269–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelista, L.; Guarneri, V.; Conte, P.F. 18F-Fluoroestradiol Positron Emission Tomography in Breast Cancer Patients: Systematic Review of the Literature & Meta-Analysis. Curr. Radiopharm. 2016, 9, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.Y.; Kim, S.-B.; Ahn, S.H.; Kim, H.O.; Yoon, D.H.; Ahn, J.-H.; Jung, K.H.; Han, S.; Oh, S.J.; Lee, S.J. A randomized feasibility study of 18F-Fluoroestradiol PET to predict pathologic response to neoadjuvant therapy in estrogen receptor–rich postmenopausal breast cancer. J. Nucl. Med. 2017, 58, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Venema, C.M.; Apollonio, G.; Hospers, G.A.; Schröder, C.P.; Dierckx, R.A.; de Vries, E.F.; Glaudemans, A.W. Recommendations and technical aspects of 16α-[18F] fluoro-17β-estradiol PET to image the estrogen receptor in vivo: The Groningen experience. Clin. Nucl. Med. 2016, 41, 844–851. [Google Scholar] [CrossRef]
- Koleva-Kolarova, R.; Greuter, M.; Van Kruchten, M.; Vermeulen, K.; Feenstra, T.; Buskens, E.; Glaudemans, A.; De Vries, E.; De Vries, E.; Hospers, G. The value of PET/CT with FES or FDG tracers in metastatic breast cancer: A computer simulation study in ER-positive patients. Br. J. Cancer 2015, 112, 1617–1625. [Google Scholar] [CrossRef] [Green Version]
- Kurland, B.F.; Wiggins, J.R.; Coche, A.; Fontan, C.; Bouvet, Y.; Webner, P.; Divgi, C.; Linden, H.M. Whole-Body Characterization of Estrogen Receptor Status in Metastatic Breast Cancer with 16α-18F-Fluoro-17β-Estradiol Positron Emission Tomography: Meta-Analysis and Recommendations for Integration into Clinical Applications. Oncologist 2020, 25, 1–10. [Google Scholar] [CrossRef]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Albain, K.S.; Saphner, T.J.; Badve, S.S.; Wagner, L.I.; Kaklamani, V.G.; Keane, M.M.; Gomez, H.L.; et al. Clinical Outcomes in Early Breast Cancer With a High 21-Gene Recurrence Score of 26 to 100 Assigned to Adjuvant Chemotherapy Plus Endocrine Therapy: A Secondary Analysis of the TAILORx Randomized Clinical Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Mouridsen, H.; Palshof, T.; Patterson, J.; Battersby, L. Tamoxifen in advanced breast cancer. Cancer Treat. Rev. 1978, 5, 131–141. [Google Scholar] [CrossRef]
- Wittliff, J.L. Steroid-hormone receptors in breast cancer. Cancer 1984, 53, 630–643. [Google Scholar] [CrossRef]
- Kraus, W.L.; Katzenellenbogen, B.S. Regulation of progesterone receptor gene expression and growth in the rat uterus: Modulation of estrogen actions by progesterone and sex steroid hormone antagonists. Endocrinology 1993, 132, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Katzenellenbogen, B.S. Effects of estrogens and antiestrogens on estrogen receptor dynamics and the induction of progesterone receptor in MCF-7 human breast cancer cells. Cancer Res. 1982, 42, 139–144. [Google Scholar] [PubMed]
- May, F.E.; Johnson, M.D.; Wiseman, L.R.; Wakeling, A.E.; Kastner, P.; Westley, B.R. Regulation of progesterone receptor mRNA by oestradiol and antioestrogens in breast cancer cell lines. J. Steroid Biochem. 1989, 33, 1035–1041. [Google Scholar] [CrossRef]
- Horwitz, K.B.; McGuire, W.L. Predicting response to endocrine therapy in human breast cancer: A hypothesis. Science 1975, 189, 726–727. [Google Scholar] [CrossRef]
- McGuire, W.L.; Horwitz, K.B.; Pearson, O.H.; Segaloff, A. Current status of estrogen and progesterone receptors in breast cancer. Cancer 1977, 39, 2934–2947. [Google Scholar] [CrossRef]
- Boland, M.; Ryan, É.; Dunne, E.; Aherne, T.; Bhatt, N.; Lowery, A. Meta-analysis of the impact of progesterone receptor status on oncological outcomes in oestrogen receptor-positive breast cancer. Br. J. Surg. 2020, 107, 33–43. [Google Scholar] [CrossRef]
- Purdie, C.; Quinlan, P.; Jordan, L.; Ashfield, A.; Ogston, S.; Dewar, J.; Thompson, A. Progesterone receptor expression is an independent prognostic variable in early breast cancer: A population-based study. Br. J. Cancer 2014, 110, 565. [Google Scholar] [CrossRef] [Green Version]
- Inda, M.A.; Blok, E.J.; Kuppen, P.J.; Charehbili, A.; den Biezen-Timmermans, E.C.; van Brussel, A.; Fruytier, S.E.; Kranenbarg, E.M.-K.; Kloet, S.L.; van der Burg, B. Estrogen Receptor pathway activity score to predict clinical response or resistance to neo-adjuvant endocrine therapy in primary breast cancer. Mol. Cancer Ther. 2019. [Google Scholar]
- Wu, J.-R.; Zhao, Y.; Zhou, X.-P.; Qin, X. Estrogen receptor 1 and progesterone receptor are distinct biomarkers and prognostic factors in estrogen receptor-positive breast cancer: Evidence from a bioinformatic analysis. Biomed. Pharmacother. 2020, 121, 109647. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, D.; Yin, X.; Zhang, X.; Huang, J.; Wu, Y.; Wang, M.; Yi, Z.; Li, H.; Li, H. Clinicopathological Characteristics and Breast Cancer–Specific Survival of Patients With Single Hormone Receptor–Positive Breast Cancer. JAMA Netw. Open 2020, 3, e1918160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J. Estrogen and Progesterone Receptor Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Guideline Update. Arch. Pathol. Lab. Med. 2020, 144, 545–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.R.; Vermi, W.; Luo, J.; Lucini, L.; Rickert, C.; Fowler, A.M.; Lonardi, S.; Arthur, C.; Young, L.J.; Levy, D.E. STAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas. Breast Cancer Res. 2012, 14, R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, K.; Kumar, M.; Yan, Y.; Jeffery, J.J.; Kloepping, K.C.; Michel, C.J.; Powers, G.L.; Mahajan, A.M.; Fowler, A.M. Sensitivity and isoform specificity of 18F-fluorofuranylnorprogesterone for measuring progesterone receptor protein response to estradiol challenge in breast cancer. J. Nucl. Med. 2019, 60, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Haddow, A.; Watkinson, J.M.; Paterson, E.; Koller, P.C. Influence of Synthetic Oestrogens on Advanced Malignant Disease. Br. Med. J. 1944, 2, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.C.; Sedransk, N.; Kelley, R.M.; Ansfield, F.J.; Ravdin, R.G.; Talley, R.W.; Potter, N.R. Diethylstilbestrol: Recommended dosages for different categories of breast cancer patients: Report of the Cooperative Breast Cancer Group. JAMA 1977, 237, 2079–2085. [Google Scholar] [CrossRef]
- Ingle, J.N.; Ahmann, D.L.; Green, S.J.; Edmonson, J.H.; Bisel, H.F.; Kvols, L.K.; Nichols, W.C.; Creagan, E.T.; Hahn, R.G.; Rubin, J. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N. Engl. J. Med. 1981, 304, 16–21. [Google Scholar] [CrossRef]
- Lønning, P.E.; Taylor, P.D.; Anker, G.; Iddon, J.; Wie, L.; Jørgensen, L.-M.; Mella, O.; Howell, A. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res. Treat. 2001, 67, 111–116. [Google Scholar] [CrossRef]
- Ellis, M.J.; Gao, F.; Dehdashti, F.; Jeffe, D.B.; Marcom, P.K.; Carey, L.A.; Dickler, M.N.; Silverman, P.; Fleming, G.F.; Kommareddy, A. Lower-dose vs. high-dose oral estradiol therapy of hormone receptor–positive, aromatase inhibitor–resistant advanced breast cancer: A phase 2 randomized study. JAMA 2009, 302, 774–780. [Google Scholar] [CrossRef]
- Plotkin, D.; Lechner, J.J.; Jung, W.E.; Rosen, P.J. Tamoxifen flare in advanced breast cancer. JAMA 1978, 240, 2644–2646. [Google Scholar] [CrossRef]
- Reddel, R.R.; Sutherland, R.L. Tamoxifen stimulation of human breast cancer cell proliferation in vitro: A possible model for tamoxifen tumour flare. Eur. J. Cancer Clin. Oncol. 1984, 20, 1419–1424. [Google Scholar] [CrossRef]
- Katzenellenbogen, B.S.; Kendra, K.L.; Norman, M.J.; Berthois, Y. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res. 1987, 47, 4355–4360. [Google Scholar] [PubMed]
- Fabian, C.; Sternson, L.; El-Serafi, M.; Cain, L.; Hearne, E. Clinical pharmacology of tamoxifen in patients with breast cancer: Correlation with clinical data. Cancer 1981, 48, 876–882. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Coleman, R.E.; Hawkins, R.A.; Krohn, K.A.; Larson, S.M.; Mendelsohn, J.; Osborne, C.K.; Piwnica-Worms, D.; Reba, R.C.; Siegel, B.A. Tumor receptor imaging: Proceedings of the National Cancer Institute workshop, review of current work, and prospective for further investigations. Clin. Cancer Res. 1995, 1, 921–932. [Google Scholar] [PubMed]
- Mortimer, J.E.; Dehdashti, F.; Siegel, B.A.; Trinkaus, K.; Katzenellenbogen, J.A.; Welch, M.J. Metabolic flare: Indicator of hormone responsiveness in advanced breast cancer. J. Clin. Oncol. 2001, 19, 2797–2803. [Google Scholar] [CrossRef]
- Dehdashti, F.; Flanagan, F.L.; Mortimer, J.E.; Katzenellenbogen, J.A.; Welch, M.J.; Siegel, B.A. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur. J. Nucl. Med. 1999, 26, 51–56. [Google Scholar] [CrossRef]
- Dehdashti, F.; Mortimer, J.E.; Trinkaus, K.; Naughton, M.J.; Ellis, M.; Katzenellenbogen, J.A.; Welch, M.J.; Siegel, B.A. PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res. Treat. 2009, 113, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.J.; Gavane, S.C.; Blanc-Autran, E.; Nehmeh, S.; Gönen, M.; Beattie, B.; Vargas, H.A.; Schöder, H.; Humm, J.L.; Fine, S.W. Positron emission tomography/computed tomography–based assessments of androgen receptor expression and glycolytic activity as a prognostic biomarker for metastatic castration-resistant prostate cancer. JAMA Oncol. 2018, 4, 217–224. [Google Scholar] [CrossRef]
- Chaturvedi, A.P.; Dehm, S.M. Androgen Receptor Dependence. Adv. Exp. Med. Biol. 2019, 1210, 333–350. [Google Scholar] [CrossRef]
- Montironi, R.; Cimadamore, A.; Lopez-Beltran, A.; Scarpelli, M.; Aurilio, G.; Santoni, M.; Massari, F.; Cheng, L. Morphologic, Molecular and Clinical Features of Aggressive Variant Prostate Cancer. Cells 2020, 9, 1073. [Google Scholar] [CrossRef] [PubMed]
- Sheikhbahaei, S.; Afshar-Oromieh, A.; Eiber, M.; Solnes, L.B.; Javadi, M.S.; Ross, A.E.; Pienta, K.J.; Allaf, M.E.; Haberkorn, U.; Pomper, M.G. Pearls and pitfalls in clinical interpretation of prostate-specific membrane antigen (PSMA)-targeted PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2117–2136. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Woo, S.; Kim, Y.J.; Suh, C.H. Impact of 68Ga-PSMA PET on the management of patients with prostate cancer: A systematic review and meta-analysis. Eur. Urol. 2018, 74, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.; Liu, Q.; Sui, H.; Wang, R.; Jacobson, O.; Fan, X.; Zhu, Z.; Chen, X. (177)Lu-EB-PSMA radioligand therapy with escalating doses in patients with metastatic castration-resistant prostate cancer. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Shen, C.J.; Minn, I.; Hobbs, R.F.; Chen, Y.; Josefsson, A.; Brummet, M.; Banerjee, S.R.; Brayton, C.F.; Mease, R.C.; Pomper, M.G.; et al. Auger radiopharmaceutical therapy targeting prostate-specific membrane antigen in a micrometastatic model of prostate cancer. Theranostics 2020, 10, 2888–2896. [Google Scholar] [CrossRef]
- Wagner, H.N., Jr.; Conti, P.S. Advances in medical imaging for cancer diagnosis and treatment. Cancer 1991, 67, 1121–1128. [Google Scholar] [CrossRef]





















© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katzenellenbogen, J.A. PET Imaging Agents (FES, FFNP, and FDHT) for Estrogen, Androgen, and Progesterone Receptors to Improve Management of Breast and Prostate Cancers by Functional Imaging. Cancers 2020, 12, 2020. https://doi.org/10.3390/cancers12082020
Katzenellenbogen JA. PET Imaging Agents (FES, FFNP, and FDHT) for Estrogen, Androgen, and Progesterone Receptors to Improve Management of Breast and Prostate Cancers by Functional Imaging. Cancers. 2020; 12(8):2020. https://doi.org/10.3390/cancers12082020
Chicago/Turabian StyleKatzenellenbogen, John A. 2020. "PET Imaging Agents (FES, FFNP, and FDHT) for Estrogen, Androgen, and Progesterone Receptors to Improve Management of Breast and Prostate Cancers by Functional Imaging" Cancers 12, no. 8: 2020. https://doi.org/10.3390/cancers12082020