STAT3 Mutation Is Associated with STAT3 Activation in CD30+ ALK− ALCL

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. STAT3 Mutations Are Frequent in PTCLs
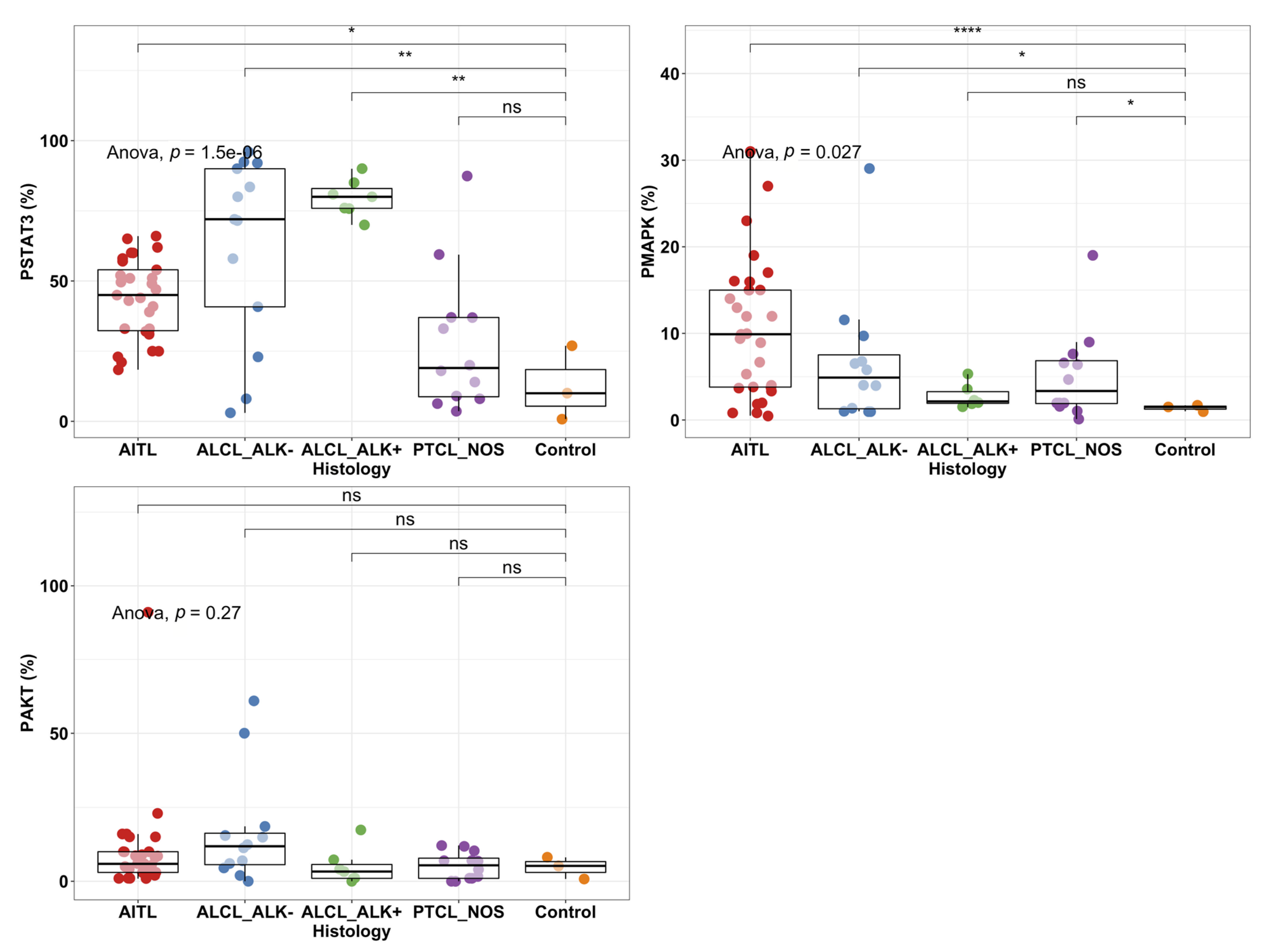
2.2. STAT3 Phosphorylation Levels Are Highest in ALK+ and ALK− ALCL Cases
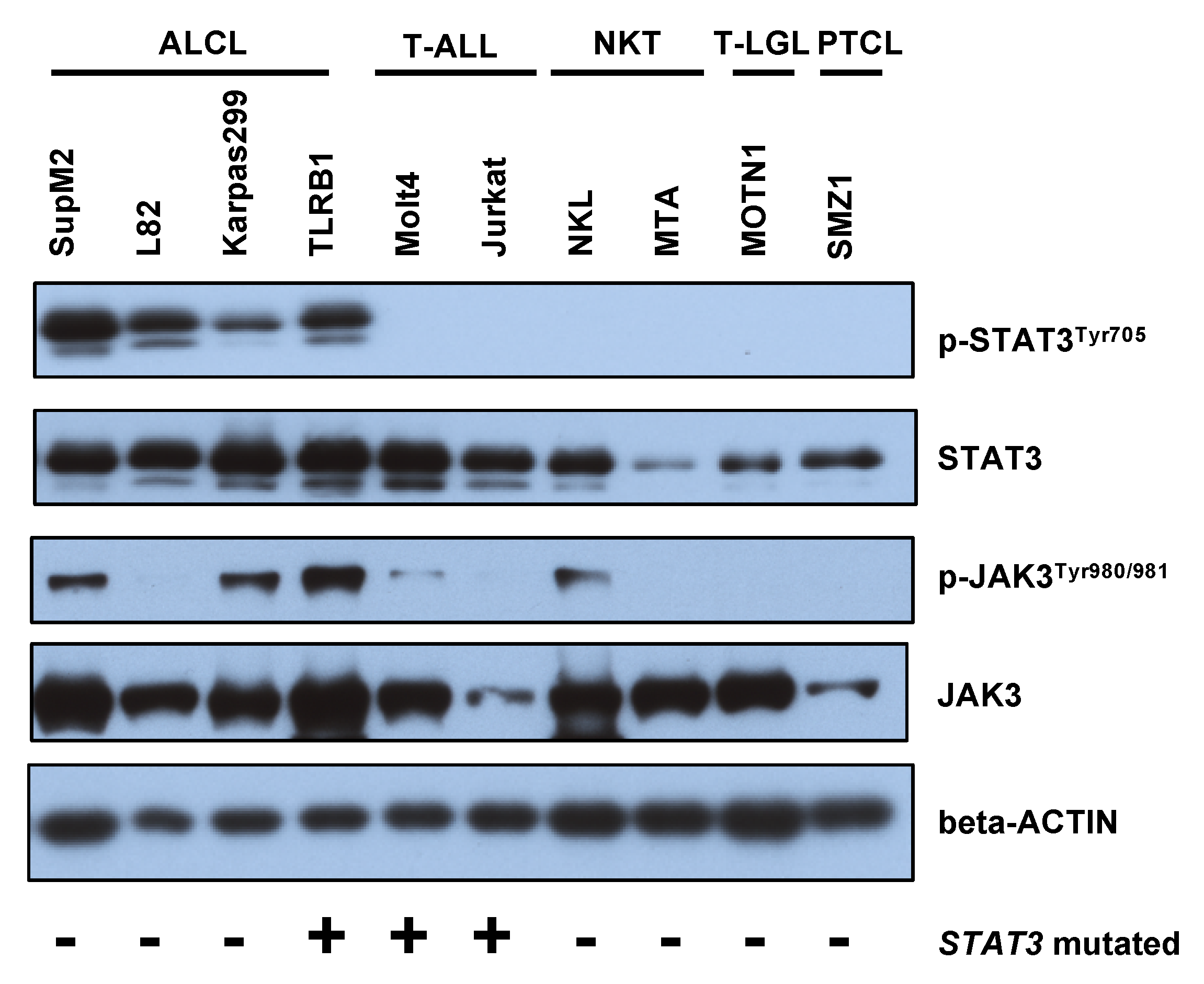
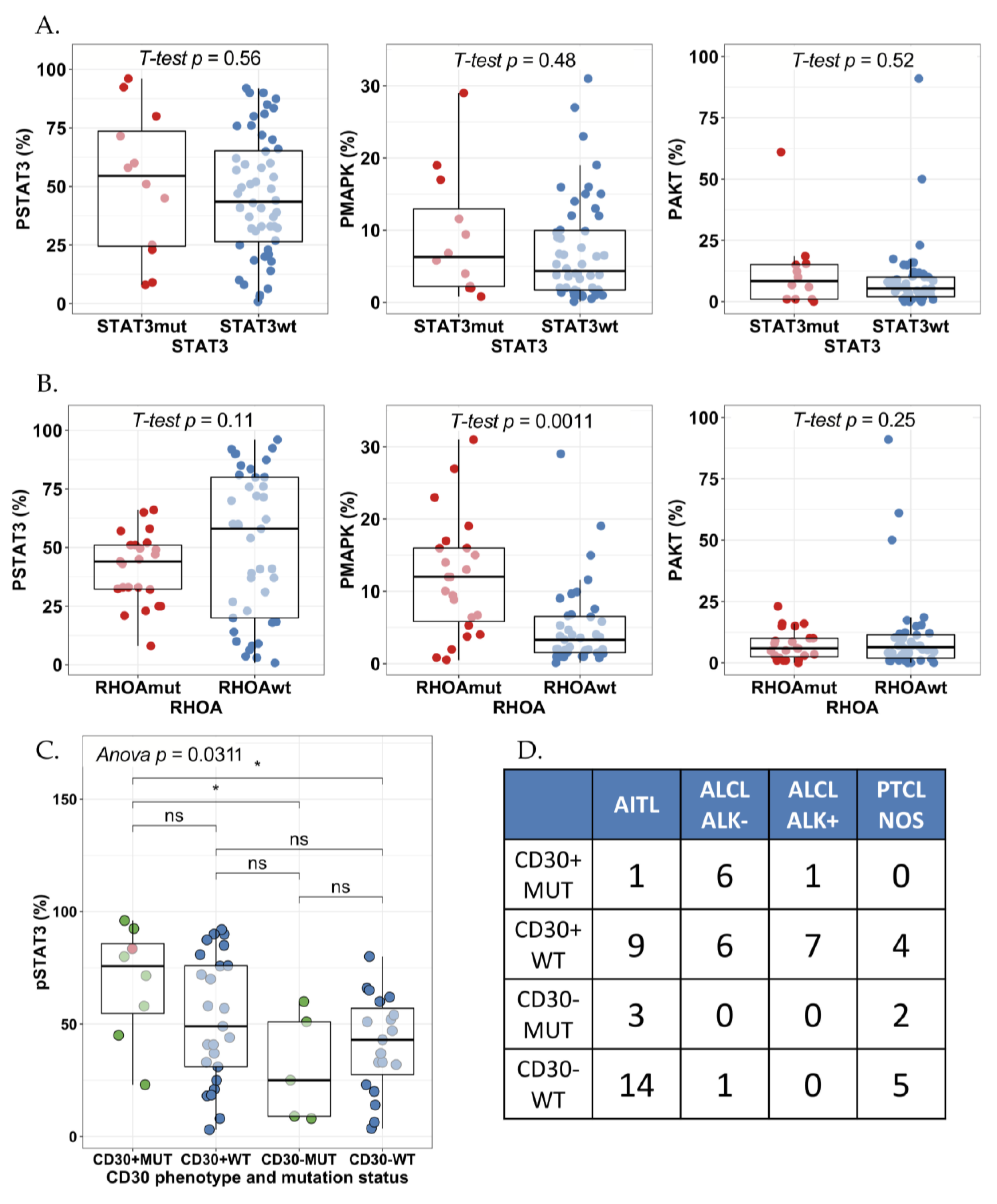
2.3. STAT3 Phosphorylation Is Associated with JAK1/STAT3 Mutation Status in CD30+ ALK− ALCLs
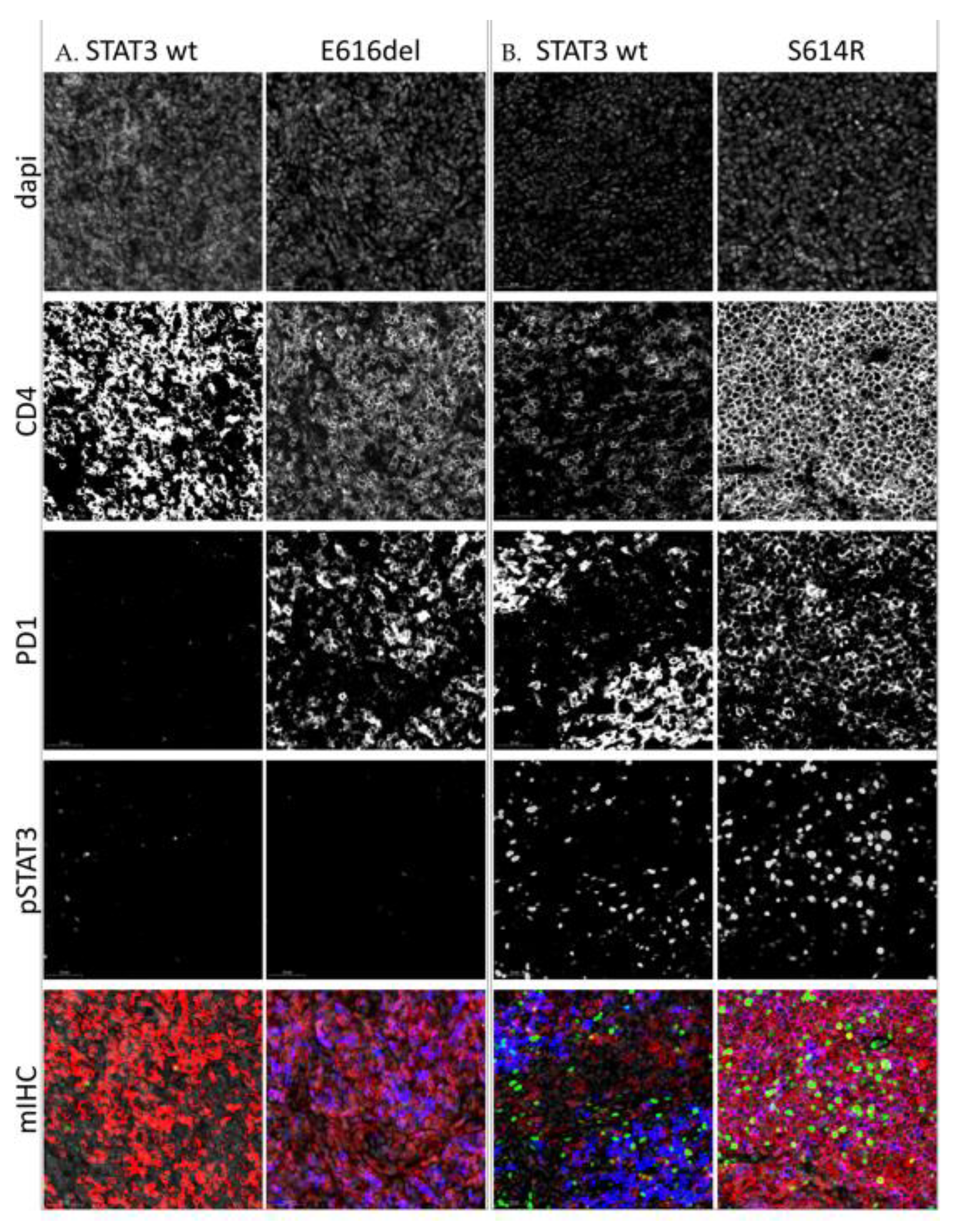
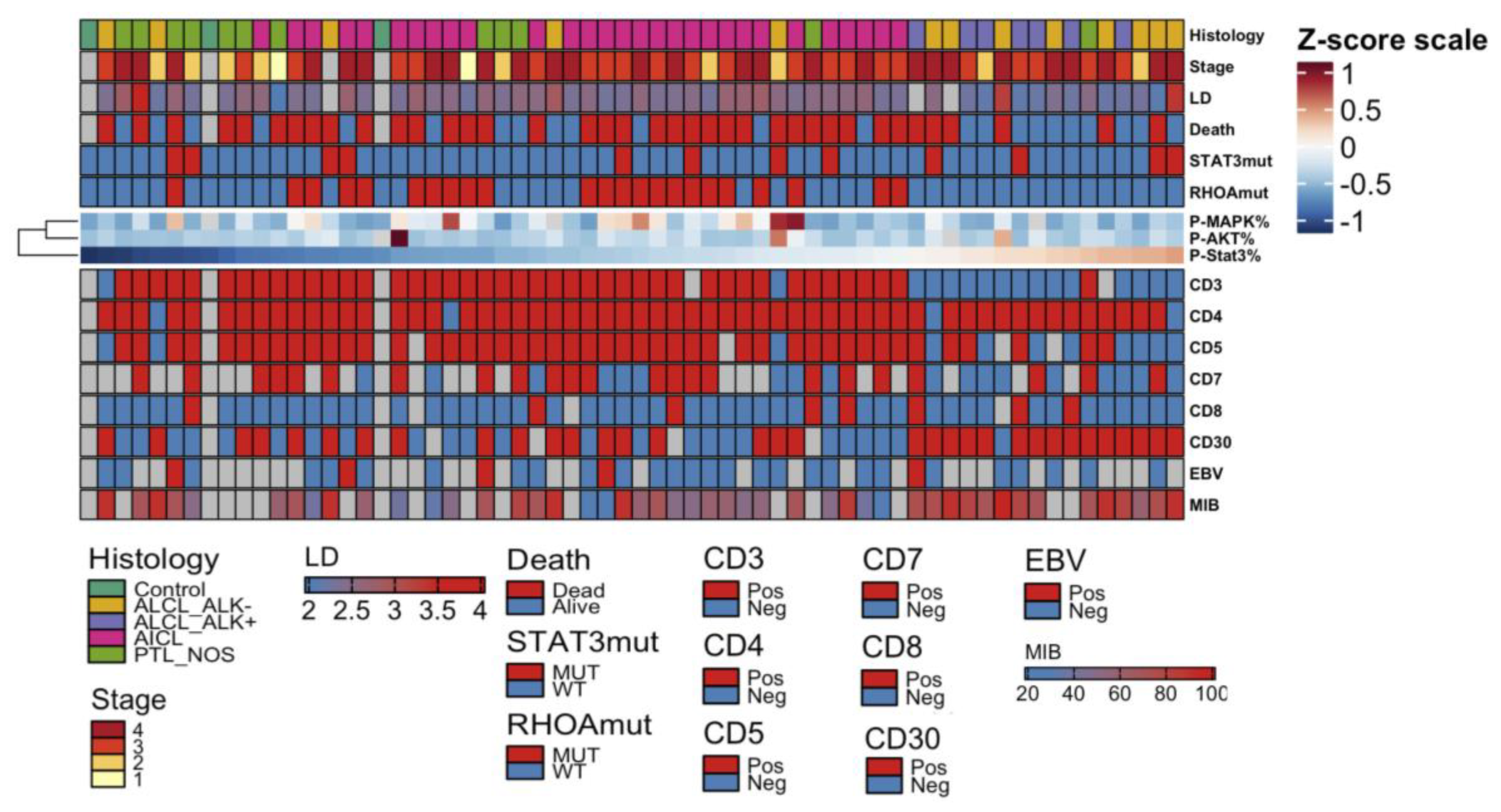
2.4. High pY-STAT3 Expression Is Associated with CD3− CD5− CD7− CD30+ Immunophenotype Common to ALCL
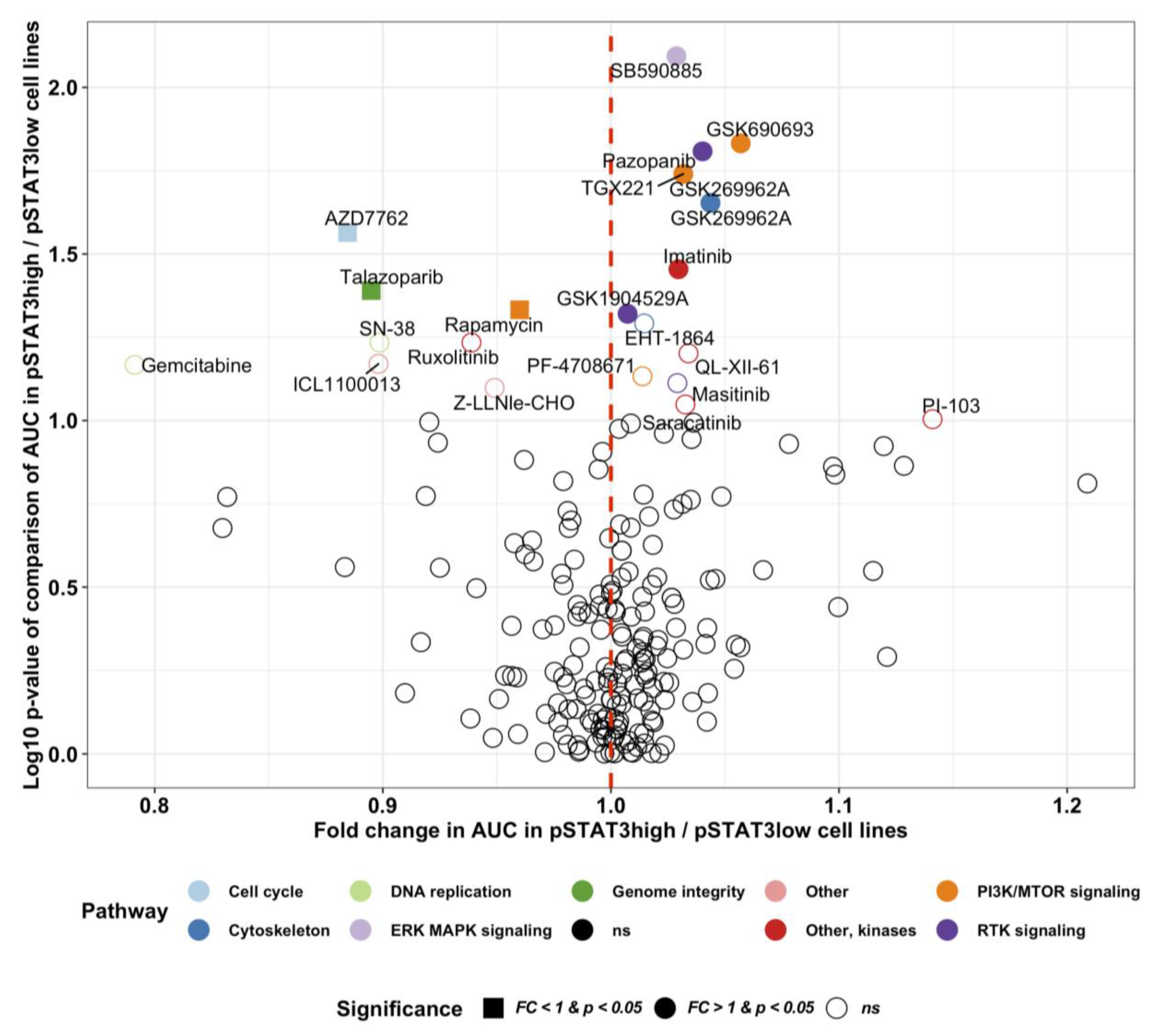
2.5. Novel Potential Inhibitors of STAT3 Activation
3. Discussion
4. Materials and Methods
4.1. Patients and Cell Lines
4.2. Cell Lines and Cell Culture Conditions
4.3. Sample Preparation
4.4. DNA Extraction
4.5. Targeted Deep Amplicon Sequencing
4.6. Immunohistochemistry
4.6.1. Single Color Immunohistochemistry
4.6.2. Multiplex Immunohistochemistry
4.7. Western Blot
4.8. CCLE and DepMap Data
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Project, T.N.-H.L.C. A Clinical Evaluation of the International Lymphoma Study Group Classification of Non-Hodgkin’s Lymphoma. Blood 1997, 89, 3909–3918. [Google Scholar]
- Ellin, F.; Landström, J.; Jerkeman, M.; Relander, T. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: A study from the Swedish Lymphoma Registry. Blood 2014, 124, 1570–1577. [Google Scholar] [CrossRef] [Green Version]
- International T-Cell Lymphoma Project; Vose, J.; Armitage, J.; Weisenburger, D. International Peripheral T-Cell and Natural Killer/T-Cell Lymphoma Study: Pathology Findings and Clinical Outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- D’Amore, F.; Relander, T.; Lauritzsen, G.F.; Jantunen, E.; Hagberg, H.; Anderson, H.; Holte, H.; Osterborg, A.; Merup, M.; Brown, P.; et al. Up-Front Autologous Stem-Cell Transplantation in Peripheral T-Cell Lymphoma: NLG-T-01. J. Clin. Oncol. 2012, 30, 3093–3099. [Google Scholar] [CrossRef]
- Mak, V.; Hamm, J.; Chhanabhai, M.; Shenkier, T.; Klasa, R.; Sehn, L.H.; Villa, D.; Gascoyne, R.D.; Connors, J.M.; Savage, K.J. Survival of Patients With Peripheral T-Cell Lymphoma After First Relapse or Progression: Spectrum of Disease and Rare Long-Term Survivors. J. Clin. Oncol. 2013, 31, 1970–1976. [Google Scholar] [CrossRef]
- Iqbal, J.; Wilcox, R.; Naushad, H.; Rohr, J.; Heavican, T.B.; Wang, C.; Bouska, A.; Fu, K.; Chan, W.C.; Vose, J.M.; et al. Genomic signatures in T-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev. 2016, 30, 89–100. [Google Scholar] [CrossRef]
- Lemonnier, F.; Couronné, L.; Parrens, M.; Jaïs, J.-P.; Travert, M.; Lamant, L.; Tournillac, O.; Rousset, T.; Fabiani, B.; Cairns, R.A.; et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012, 120, 1466–1469. [Google Scholar] [CrossRef] [Green Version]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.S.; Van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef]
- Iqbal, J.; Weisenburger, D.D.; Greiner, T.C.; Vose, J.M.; McKeithan, T.; Küçük, C.; Geng, H.; Deffenbacher, K.; Smith, L.; Dybkær, K.; et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010, 115, 1026–1036. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [Green Version]
- Pencik, J.; Pham, H.T.T.; Schmoellerl, J.; Javaheri, T.; Schlederer, M.; Culig, Z.; Merkel, O.; Moriggl, R.; Grebien, F.; Kenner, L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine 2016, 87, 26–36. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2012, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Migone, T.; Lin, J.; Cereseto, A.; Mulloy, J.; O’Shea, J.; Franchini, G.; Leonard, W. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 1995, 269, 79–81. [Google Scholar] [CrossRef]
- Heinrich, T.; Rengstl, B.; Muik, A.; Petkova, M.; Schmid, F.; Wistinghausen, R.; Warner, K.; Crispatzu, G.; Hansmann, M.-L.; Herling, M.; et al. Mature T-cell Lymphomagenesis Induced by Retroviral Insertional Activation of Janus Kinase 1. Mol. Ther. 2013, 21, 1160–1168. [Google Scholar] [CrossRef] [Green Version]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia 2014, 28, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Couronné, L.; Scourzic, L.; Pilati, C.; Della Valle, V.; Duffourd, Y.; Solary, E.; Vainchenker, W.; Merlio, J.-P.; Beylot-Barry, M.; Damm, F.; et al. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. Haematological 2013, 98, 1748–1752. [Google Scholar] [CrossRef] [Green Version]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Rajala, H.; Gómez-Seguí, I.; Olson, T.; McGraw, K.; Przychodzen, B.; Kulasekararaj, A.; Afable, M.; et al. STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood 2013, 122, 2453–2459. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.A.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.; Crispatzu, G.; Oberbeck, S.; Mayer, P.; Pützer, S.; Von Jan, J.; Vasyutina, E.; Warner, K.; Weit, N.; Pflug, N.; et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Sung, M.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, W.T.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA–VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2017, 32, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Han, J.J.; O’Byrne, M.; Stenson, M.J.; Maurer, M.; Wellik, L.E.; Feldman, A.L.; McPhail, E.D.; Witzig, T.E.; Gupta, M. Prognostic and therapeutic significance of phosphorylated STAT3 and protein tyrosine phosphatase-6 in peripheral-T cell lymphoma. Blood Cancer J. 2018, 8, 110. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine receptor signaling is required for the survival of ALK−anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar] [CrossRef] [Green Version]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Martinez, B.; Zehnder, J.L.; A Arber, D. STAT3 mutations are frequent in CD30+ T-cell lymphomas and T-cell large granular lymphocytic leukemia. Leukemia 2013, 27, 2244–2247. [Google Scholar] [CrossRef] [Green Version]
- Shair, K.H.Y.; Bendt, K.M.; Edwards, R.H.; Bedford, E.C.; Nielsen, J.N.; Raab-Traub, N. EBV Latent Membrane Protein 1 Activates Akt, NFκB, and Stat3 in B Cell Lymphomas. PLOS Pathog. 2007, 3, e166. [Google Scholar] [CrossRef] [Green Version]
- Montes-Mojarro, I.; Steinhilber, J.; Bonzheim, I.; Quintanilla-Martinez, L.; Fend, F. The Pathological Spectrum of Systemic Anaplastic Large Cell Lymphoma (ALCL). Cancers 2018, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, J.O. The aggressive peripheral T-cell lymphomas: 2017. Am. J. Hematol. 2017, 92, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, J.H.; Horwitz, S.; Teruya-Feldstein, J.; A Lunning, M.; Viale, A.; Huberman, K.; Socci, N.D.; Lailler, N.; Heguy, A.; Dolgalev, I.; et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia 2014, 29, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manso, R.; Sánchez-Beato, M.; González-Rincón, J.; Gómez, S.; Rojo, F.; Mollejo, M.; García-Cosio, M.; Menárguez, J.; Piris, M.A.; Rodríguez-Pinilla, S.M. Mutations in the JAK/STAT pathway genes and activation of the pathway, a relevant finding in nodal Peripheral T-cell lymphoma. Br. J. Haematol. 2017, 183, 497–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.-I.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Song, T.L.; Nairismägi, M.-L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.-M.; Pang, W.-L.; Kizhakeyil, A.; Wijaya, G.-C.; Huang, D.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef] [Green Version]
- Gäbler, K.; Behrmann, I.; Haan, C. JAK2 mutants (e.g., JAK2V617F) and their importance as drug targets in myeloproliferative neoplasms. JAK-STAT 2013, 2, e25025. [Google Scholar] [CrossRef]
- Fukuda, A.; Wang, S.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Alexander, W.; Hilton, D.S. The Role of Suppressors of Cytokine Signaling (SOCS) Proteins in Regulation of the Immune Response. Annu. Rev. Immunol. 2004, 22, 503–529. [Google Scholar] [CrossRef]
- Amador, C.; Greiner, T.; Heavican, T.B.; Smith, L.M.; Galvis, K.T.; Lone, W.G.; Bouska, A.; D’Amore, F.; Pedersen, M.B.; Pileri, S.A.; et al. Reproducing the Molecular Subclassification of Peripheral T-cell Lymphoma-NOS by Immunohistochemistry. Blood 2019, 134, 2159–2170. [Google Scholar] [CrossRef]
- Heavican, T.B.; Bouska, A.; Yu, J.; Lone, W.; Amador, C.; Gong, Q.; Zhang, W.; Li, Y.; Dave, B.J.; Nairismägi, M.-L.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzani, P.L.; Baliva, G.; Magagnoli, M.; Bendandi, M.; Modugno, G.; Gherlinzoni, F.; Orcioni, G.F.; Ascani, S.; Simoni, R.; Pileri, S.A.; et al. Gemcitabine Treatment in Pretreated Cutaneous T-Cell Lymphoma: Experience in 44 Patients. J. Clin. Oncol. 2000, 18, 2603–2606. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Dodero, A.; Chiappella, A.; Monaco, F.; Degl’Innocenti, D.; Salvi, F.; Vitolo, U.; Argnani, L.; Corradini, P.; Zinzani, P.L.; et al. A phase II study on the role of gemcitabine plus romidepsin (GEMRO regimen) in the treatment of relapsed/refractory peripheral T-cell lymphoma patients. J. Hematol. Oncol. 2016, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkenau, H.-T.; Chong, G.; Cunningham, D.; Watkins, D.; Sirohi, B.; Chau, I.; Wotherspoon, A.; Norman, A.; Horwich, A.; Matutes, E. Gemcitabine, cisplatin and methylprednisolone for the treatment of patients with peripheral T-cell lymphoma: The Royal Marsden Hospital experience. Haematology 2007, 92, 271–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, D.; Unger, J.M.; Spier, C.M.; Persky, D.O.; Young, F.; Leblanc, M.; Fisher, R.I.; Miller, T.P. Phase 2 trial of combined cisplatin, etoposide, gemcitabine, and methylprednisolone (PEGS) in peripheral T-cell non-Hodgkin lymphoma: Southwest Oncology Group Study S0350. Cancer 2012, 119, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, H.L.M.; Eldfors, S.; Kuusanmäki, H.; Van Adrichem, A.J.; Olson, T.; Lagström, S.; I Andersson, E.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [Green Version]
- Team, R. RStudio: Integrated Development for R; RStudio: Boston, MA, USA, 2015. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTCL Subtype | n | STAT3 Mutation Frequency | JAK1/3 Mutation Frequency | RHOA Mutation Frequency | Co-Occuring Mutations |
|---|---|---|---|---|---|
| AITL | 30 | 13% | 0% | 70% | 10% |
| NOS | 12 | 17% | 0% | 17% | 8% |
| ALK+ ALCL | 8 | 13% | 0% | 0% | None |
| ALK− ALCL | 13 | 38% | 15% | 0% | 8% |
| All | 63 | 19% | 3% | 37% | 8% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, E.I.; Brück, O.; Braun, T.; Mannisto, S.; Saikko, L.; Lagström, S.; Ellonen, P.; Leppä, S.; Herling, M.; Kovanen, P.E.; et al. STAT3 Mutation Is Associated with STAT3 Activation in CD30+ ALK− ALCL. Cancers 2020, 12, 702. https://doi.org/10.3390/cancers12030702
Andersson EI, Brück O, Braun T, Mannisto S, Saikko L, Lagström S, Ellonen P, Leppä S, Herling M, Kovanen PE, et al. STAT3 Mutation Is Associated with STAT3 Activation in CD30+ ALK− ALCL. Cancers. 2020; 12(3):702. https://doi.org/10.3390/cancers12030702
Chicago/Turabian StyleAndersson, Emma I., Oscar Brück, Till Braun, Susanna Mannisto, Leena Saikko, Sonja Lagström, Pekka Ellonen, Sirpa Leppä, Marco Herling, Panu E. Kovanen, and et al. 2020. "STAT3 Mutation Is Associated with STAT3 Activation in CD30+ ALK− ALCL" Cancers 12, no. 3: 702. https://doi.org/10.3390/cancers12030702