
Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease
by
 , , and
, , and
Yajie Tong
1,† ,
,
Shuqing Zhang
2,†, 
Suzette Riddle
3,
Lubo Zhang
4,
Rui Song
4,* and
Dongmei Yue
1,* 1
Department of Pediatrics, Shengjing Hospital of China Medical University, Shenyang 110004, China
2
School of Pharmacy, China Medical University, Shenyang 110122, China
3
Cardiovascular Pulmonary Research Laboratories, Departments of Pediatrics and Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA
4
Lawrence D. Longo, MD Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomedicines 2021, 9(8), 944; https://doi.org/10.3390/biomedicines9080944
Submission received: 24 June 2021
/
Revised: 29 July 2021
/
Accepted: 30 July 2021
/
Published: 2 August 2021
(This article belongs to the Special Issue MicroRNAs, tRNA Fragments, and Circular RNAs: Pivotal Regulators of Gene Expression and Their Roles in Human Diseases)
Abstract
:Clinically, intrauterine hypoxia is the foremost cause of perinatal morbidity and developmental plasticity in the fetus and newborn infant. Under hypoxia, deviations occur in the lung cell epigenome. Epigenetic mechanisms (e.g., DNA methylation, histone modification, and miRNA expression) control phenotypic programming and are associated with physiological responses and the risk of developmental disorders, such as bronchopulmonary dysplasia. This developmental disorder is the most frequent chronic pulmonary complication in preterm labor. The pathogenesis of this disease involves many factors, including aberrant oxygen conditions and mechanical ventilation-mediated lung injury, infection/inflammation, and epigenetic/genetic risk factors. This review is focused on various aspects related to intrauterine hypoxia and epigenetic programming in lung development and disease, summarizes our current knowledge of hypoxia-induced epigenetic programming and discusses potential therapeutic interventions for lung disease.
1. Introduction
Intrauterine hypoxia, also known as fetal hypoxia, refers to a scenario where the fetus is deprived of an adequate oxygen supply due to environmental, maternal, placental, and fetal factors [1,2,3,4,5,6]. Excessive hypoxia can prevent the fetus from completing its genetically determined growth potential, leading to approximately 10% of babies stunted in the womb and delivered prematurely. The intrauterine hypoxia is involved in perinatal “plasticity,” affecting lung development, injury, and repair processes [6]. Bronchopulmonary dysplasia (BPD) is a multifactorial neonatal chronic lung disease, which is influenced by oxygen and/or intrauterine environment, genetics, immaturity, infection, poor nutrition, and mechanical ventilation. These risk factors are inter-related and interact during fetal development. For instance, prenatal hypoxia followed with postnatal hyperoxia rather than normoxia can exaggerate retardation of morphological lung development leading to BPD-like alterations [7,8]. In addition, excess chronic hypoxia followed by relative hyperoxic exposure, mechanical and infectious stimuli incite oxidative stress, contributing to BPD pathogenesis [9]. ROS can lead to cell apoptosis and dysfunction in alveolar cells, bronchial epithelial cells, alveolar macrophages, and endothelial cells by directly disrupting proteins, carbohydrates, lipids, DNA, and RNA [10]. Additionally, ROS can also trigger epigenetic modifications altering related gene expression patterns [11]. Both direct and epigenetic effects of ROS can cause impaired alveolar and capillary development, boosted vascular permeability and inflammatory responses, and promote the development of BPD [10,12].
This review focuses on the damage of excessive chronic intrauterine hypoxia to lung development and its related effects on the pathogenesis of BPD.
BPD is a chronic lung injury syndrome involving lung developmental plasticity, damage, and repair, which can limit the remodeling ability of surviving infants’ lungs until adulthood [13]. BPD is a breathing disorder in which the infants’ lungs become irritated and develop abnormally [14,15]. Although intensive treatment and support have improved the infants’ life prospects, morbidities related to severe BPD persist. The vulnerability of BPD has also evolved. “Old BPD,” defined in the 1960s, is characterized by severe lung injury. In premature infants showing reduced vascular and alveolar development with improved survival, “new BPD” was identified [16,17,18]. There is preclinical and clinical evidence that the new form of BPD is caused by early developmental arrest and impaired lung development rather than acute lung injury. This highlights the need for a better understanding of the molecular pathways that guide normal lung development and discover of the mechanisms and applications of lung regeneration [19,20,21,22].
Notably, oxygen-dependent epigenetic programming and regulation are the primary focus of intensive studies today. Epigenetic programming and modification cause inheritable and stable gene expression patterns without coding sequence alterations. Epigenetic mechanisms including DNA methylation, histone modifications, and miRNA expression play a significant role during hypoxia regulating different lineage-specific expression profiles and the transcriptional program for gene expression [5,6,23,24]. These are also the essential mechanisms for developmental programming. Similar studies have emphasized that epigenetic modifications and programming in response to environmental stimuli are critical in achieving appropriate gene expression patterns, especially in specific lung tissues in relation to environmental signals [25,26]. This review focuses on aspects associated with chronic intrauterine hypoxia and epigenetic programming in bronchopulmonary dysplasia. Taking into account the common growth and development disorders in BPD infants, any small growth amelioration may support pulmonary rehabilitation.
2. Epigenetics and Lung Development
According to Yang and Schwartz [27], epigenetic mechanisms can control gene expression in chronic lung conditions such as idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease. Although few studies have explored this hypothesis, the expectation is that research on epigenetics and lung development will become prominent in the near future. Epigenetics is considered the study of heritable changes in gene expression triggered by mechanisms that are separate from the expressed DNA sequence [28,29]. There are environmentally induced epigenetic alterations to DNA that can disrupt cells during development, leading to gene expression changes. Changes in the transcriptomes and epigenetics of lung cells occur at developmentally sensitive time points (Figure 1) (Table 1). Such changes in the epigenome of the cells may change the structure and function of the lungs. Although epigenome changes may result in subsequent changes in the epigenetic platform [30], leading to the deviation from normal lung development during lung repair and after injury, epigenetic alterations during lung development enable the epigenome to guide gene expression response to future damage and guide lung repair.
2.1. DNA Methylation
DNA methylation is a reversible DNA modification resulting from the addition of a methyl group to cytosines in nucleic acids [25,45,46]. DNA methylation is carried out by maintaining DNA methyltransferase (DNMT) 1 and the de novo methyltransferases DNMT3A and DNMT3B. Active DNA demethylation is initiated by ten-eleven translocation (TET) enzymes (TET1, TET2, and TET3) [47,48,49]. Fetal lungs begin to form four weeks after conception and continue to develop after birth. With the capability to develop into every cell type in the body, the pluripotent embryonic cells differentiate into specific cell lineages [46]. As cells develop into specialized types, they undergo epigenetic changes on histones and DNA, resulting in specific cell lineages. Therefore, DNA methylation is crucial and plays a vital role in cellular differentiation.
In human embryonic lung cells, the methylation of CpG islands was frequently found in the proximal promoter regions of TP53BP2 (tumor protein p53 binding protein 2) and Apaf-1 (apoptotic protease activating factor-1) [31]. Notably, the inhibition of methylation markedly upregulated Apaf-1 expression in human embryonic lung cells. Apaf-1 may play an essential role in DNA damage-induced apoptosis. Moreover, the CpG island-related proximal promoter regions of Apaf-1 and TP53BP2 are the transcription factor binding sites. Therefore, the methylation of Apaf-1 and TP53BP2 may inhibit embryonic morphogenesis and play a vital role in early lung development [31]. At the pseudoglandular/canalicular stage of lung development, CpG island methylation in the VEGF-A promoter of primary fetal distal lung epithelial cells was demonstrated to play a crucial role in the vascular growth of the cardiopulmonary system [35]. DNA methylation-regulated genes were verified during normal alveolar septation [43,50]. These genes are involved in inflammation (e.g., B-cell CLL/lymphoma 6, chitinase-like 3, chemokine [C-C motif] receptor 6, Ccl5, signal transducer and activator of transcription 4, CD209a antigen, histocompatibility 2, O region β locus, CD19 antigen, killer cell lectin-like receptor subfamily B member 1B, B-cell linker, SH2 domain-containing 1A, protein kinase C theta, and β-2 microglobulin), antioxidant defense (e.g., extracellular superoxide dismutase 3, and peroxiredoxin 6), extracellular matrix formation (Collagen alpha-3 (VI), Collagen alpha-1 (XXVII), elastin, tenascin C), and lung cancer (ribonucleotide reductases M 1, and 2, and S-phase kinase-associated protein 2) [43]. A more recent study identified numerous differentially methylated CpGs at birth that reflect fetal lung developmental processes [51]. These findings may contribute to our understanding of how the DNA methylation mechanism is related to normal and altered lung development.
2.2. Histone Acetylation
Histone modifications also play a critical role in the epigenetics of lung development and complements regulation by DNA methylation. Gene expression patterns can be controlled by a complex combination of histone acetylation and deacetylation [30,52]. For instance, Joss-Moore et al. [53] determined that the regulation of gene expression depends on the fine regulation of histone modifications. Rapid local changes in histone conformation take place during the acetylation of nucleosomes. While the mechanisms remain unelucidated, rapid deacetylation and acetylation could facilitate the transit of polymerases in nucleosomes [30]. Histone acetyltransferases (HATs) mediate the acetylation of histone tails, thereby promoting gene transcription, while histone deacetylases (HDACs) remove acetyl groups leading to gene silencing [54,55]. HAT1 is essential for the acetylation of newly synthesized histones H3 and H4. In Hat1-/- mice embryonic fibroblasts are sensitive to DNA disruptors and exhibit high levels of genomic instability. In addition, HAT-1 deficiency can cause lung development defects, leading to neonatal mortality [32]. HDAC1/2 deficiency in proximal lung endoderm progenitors directly increased Bmp4 expression, subsequently decreased Sox2 expression, and inhibited the development of multiple proximal cells, contributing to defects in the proximal airways and lung morphogenesis [36].
2.3. MicroRNAs
MicroRNAs (miRNAs or miRs) are a class of small (18–25 nucleotides in length) noncoding RNAs. They usually inhibit gene expression by promoting mRNA degradation or disrupting its translation. Argonaute 1–4 and Dicer, two essential miRNA processing proteins, are expressed in the developing lung epithelium and mesoderm [56,57]. Notably, the loss of Dicer in lung epithelial cells can lead to severe defects in branching and epithelial structure, leading to perinatal death [58].
At the embryonic stage, miR-142-3p was found to be specifically upregulated in the embryonic lung mesenchyme. Furthermore, inhibition of miR-142-3p led to ectopic expression and differentiation of parabronchial smooth muscle cell progenitors in the mouse embryonic lung [33]. Similarly, miR-326 was found to play an essential role in the expansion of the distal epithelium and disruption of regular branching patterns and mesenchymal integrity in the embryonic lung [34].
At the pseudoglandular stage, miR-17 and its paralogs, miR-20a and miR-106b were highly expressed in the lung. The downregulation of miR-17, miR-20a, and miR-106b caused significant branching defects in embryonic lung epithelial explants [37]. The miR302-367 cluster was mainly expressed in the embryonic lung epithelium at the embryonic and pseudoglandular stages. This cluster promoted lung epithelial proliferation and suppressed subsequent differentiation through direct inhibition of the expression of tumor suppressors Rbl2 and Cdkn1a [38]. In addition, miR-449a was found to be upregulated at the pseudoglandular, canalicular and saccular stages, and decreased dramatically at birth. MiR-449a increased the Mycn and Sox9 mRNA levels, and the Ki-67 and SOX9 protein levels to stimulate distal epithelial progenitor proliferation and mucociliary differentiation, supporting an essential role of miR-449a in the mid-stages of lung development [39].
MiR-26a is highly expressed in the rat fetal lung, especially at the saccular stage [59]. The knockout of miR-26a-1/2 in the mouse promoted the formation of dilated lumens and aerated regions at the beginning of the canalicular stage, and maturation of the alveolar structure at the canalicular and saccular stages of lung development [41]. MiR-127 was also highly expressed in the late stage of fetal lung development and regulated terminal bud count and bud sizes in the developing lung [40]. MiR-17-92 can be downregulated by histone acetylation, promoting alveolar type 1 cell spreading and lung sacculation [42]. In the pseudoglandular and saccular stages of rat lung development, let-7a, miR-93, miR-125b-5p, miR-146b, miR-296, miR-1949, and miR-3560 were identified to be consistently expressed in the rat embryonic lungs; while miR-1949, miR-125b-5p, miR-296, and miR-93 were downregulated; and let-7a, miR-146b, and miR-3560 were upregulated [59]. A recent miRNA profiling of human and mouse postnatal alveolar tissue found fifty up- or downregulated miRNAs at the alveolar stage. Significantly, the inhibition of miR-539 and miR-590 markedly reduced alveolar development, with a reduction in radial alveolar count and lung compliance [44]. The above studies support the crucial role of miRNA in lung development and suggest the crosstalk among the epigenetic mechanisms, requiring further investigation.
Over time, epigenetics has become a critical aspect in determining lung development for fetuses and infants by considering the external environment and factors around the pregnant mother. From these studies on epigenetics and lung development of fetuses and infants growing up, some limitations or challenges arise, especially in understanding the role of epigenetic regulation in gene expression under different lung conditions.
3. Hypoxia and Vulnerability of Neonatal Chronic Lung Disease
Intrauterine hypoxia has significant impacts on fetal development, including breathing disorders. While high altitude, maternal smoking, chorioamnionitis, hypertension/preeclampsia, and intrapartum and post-placental complications all induce intrauterine hypoxia, there are limited reports on the production of bronchopulmonary dysplasia in such hypoxic conditions. In addition to being attributed to hypoxic injury, some of these risk factors (such as chorioamnionitis and preeclampsia) are complex pathological processes. Several clinical trials reported an association between higher altitude hypoxia and an increased prevalence of BPD [60,61]. Long-term hypoxia at high altitudes in pregnancy boosted ROS synthesis in ovine uterine arteries [62,63], providing additional evidence that hypoxia-induced ROS can perform a critical mechanistic part in the formation of irregular vasculature and BPD.
Unlike other organs, the lung completes a fraction of its development immediately before and after birth. During the final lung development stage (alveolarization), the secondary septation process breaks down alveolar ducts to alveolar sacs, including the expansion of capillary beds through angiogenesis to increase the lung’s surface area to facilitate gas exchange [17,64]. However, the growth completion in the postnatal stage means that the lung becomes highly vulnerable to external factors or environmental pressures, which interfere with the developmental program [53], this is particularly evident in the preterm birth setting, where alveolarization disruption results in BPD, which is commonly known to be the most common maturity complication [17,65,66]. The evolution of the lung’s vascular system resembles that of the airways, including peribronchial arteries rising and separating concurrently with the airways. Once splitting is done, angiogenesis and vasculogenesis join to create the alveolar-capillary bed inside the fetal lung mesenchyme. The canalicular and saccular levels of lung growth are when the alveolar capillary bed is formed [67]. Throughout capillary creation, epithelial growth factors such as VEGF, which is regulated by hypoxia/HIF-1α and other signaling, recruit forming capillaries towards the epithelial basement membrane, facilitating epithelial–endothelial interactions and the creation of alveolar septa in the late alveolar developmental phase [68]. Latest research indicates that interruption of normal lung vascular development can be a critical factor in the pathogenesis of BPD [69,70,71]. While lung vascular formation has been studied extensively in alveolar stage animal models, all hypoxia and hyperoxia result in emphysema with decreased alveolar numbers and capillary density in the alveolar stage [72,73]. It is unknown if these models accurately replicate BPD pathogenesis because newborns during the late saccular and alveolar developmental stages frequently experience BPD. The essential mechanisms underlying BPD, including possible vasculature dysregulation in the developing lung, most likely begin during the canalicular and saccular phases of lung growth. Thus, it is assumed that persistent hypoxia has an adverse effect on the pulmonary blood vessels, leading to BPD, which mainly occurs in the tubule/sac phase and may also be at the alveolar level. As is often the case, infants battling BPD require respiratory support in the early stages of infant life failure. Mechanical ventilation and high concentration oxygen supply can cause hyperoxia injury, which enhances hypoxia-induced lung developmental disorder. A considerable population would experience long-term deficits in pulmonary function, including persistent airway obstruction and delayed growth of the distal lungs.
To further understand the vulnerability to BPD, it is crucial to determine how the lung develops. As mentioned above, there are various stages of lung development. The alveolar stage begins before birth and extends up to some years after birth [17,64,74]. Chronic hypoxia impairs alveolarization and lung development [7,75,76]. Pulmonary angiogenesis and secondary septation markedly increase the lung’s surface area for gas exchange [17,27,64,77,78]. The early lung development stages involving the development of pulmonary vasculature appear to take place mainly through vasculogenesis [64,67]. Under intermittent hypoxia (12% O2), angiogenic gene and protein expression were downregulated, and thus, angiogenesis and lung development were perturbed in newborn mice [79,80]. At the same time, the secondary septation process is an integrated, complex series of events that involve paracrine signals between different cell types within the lung, and comprises epithelial cells, fibroblasts, endothelial cells, and inflammatory cells [81,82,83]. Taken together, these stages and the possible impact of the disease on the lungs illustrate their vulnerability under intrauterine hypoxia (Figure 2).
4. Epigenetic Programming and Neonatal Chronic Lung Disease
Epigenetics influences cell-specific developmental gene transcription and gene silencing. There is a timely requirement for gene transcription regulation during normal development. In this case, only the genes that are specific to a particular cell type and developmental stage are transcriptionally active [84,85]. The ability to conduct gene transcriptional modulation provides “plasticity” during development [86,87]. Dysregulation of chromatin remodeling pathways, which include DNA methylation, miRNA regulation, and histone acetylation, has also been identified in recent studies as part of epigenetic programming in response to hypoxia or hypoxia/hyperoxia in neonatal or infant lungs, different experimental models, and BPD patients [88,89,90,91,92,93] (Figure 2) (Table 2).
To determine the impact of epigenetic programming on lung development and BPD, epigenetic modifications assist in directing the associated factors and transcriptional machinery to the correct location within genes. One of the primary and better understood epigenetic programming features is DNA methylation. Here, the emphasis is on the alveolar septation process in neonatal lungs, accompanied by changed DNA methylation profiles coinciding with distinct gene expression changes. DNA methylation profiling identified 149 differentially methylated genes in lung tissue samples from preterm infants with BPD [43]. Out of 149 genes with altered methylation status, 23 genes had opposite patterns of methylation and expression. Among them, the methylation of zinc-finger protein 438 was decreased and associated with an increased expression in BPD, while the other genes displayed increased methylation corresponding with decreased expression in BPD [43]. Further analysis demonstrated that pathways associated with genes differentially methylated and expressed in BPD included ErbB signaling, neuregulin signaling, RhoA signaling, VEGF signaling, cardiomyocyte differentiation via BMP receptors, axonal guidance signaling, and glutathione-mediated detoxification [43]. Some of those pathways such as RhoA signaling, axonal guidance signaling and glutathione-mediated detoxification have been implicated in lung development and BPD pathogenesis [94,95,96].

{kind=link}
{kind=link}
{kind=link}
Table 2.
MiRNAs in hypoxia and BPD.
| miRNA | Regulation | Species and Samples | Targets | Disease/Condition | References |
|---|---|---|---|---|---|
| miR-17-92 | Downregulated | Human infant lungs | ? | Extremely and very preterm, BPD | [97] |
| miR-103a-3p and miR-185-5p miR-200a-3p | Downregulated Upregulated | Umbilical cord blood-derived exosomes from human infants | PI3K/Akt and angiogenesis-related signaling pathways | Very preterm, BPD | [98] |
| miR-15a | Upregulated | Chicken lung | Bcl2 | Hypoxia | [99] |
| miR-210 and miR-374a | Upregulated | Plasma of newborn piglets | ? | Hypoxia | [100] |
| miR-34a | Downregulated | Mouse lungs | ? | Postnatal hypoxia-induced BPD | [101] |
Other than DNA methylation, histone marker modifications, especially on the outer promoter regions, are important for the epigenetic programming of gene expression [102,103,104] and play a crucial role in the developmental origin of lung diseases [105,106,107,108,109]. DNA methylation of the promoter of miR-17-92 downregulated this miRNA cluster expression and mediated the molecular pathogenesis of bronchopulmonary dysplasia [97]. Indeed, miRNAs are also essential in mediating developmental lung disorders [110,111,112,113,114,115]. In the chicken lung, hypoxia stress stimulates miR-15a expression to direct target Bcl2 and inhibit its antiapoptotic activity at particular stages, which may be a novel therapeutic target for developmental lung disorders by hypoxia insults [99]. Saugstad et al. demonstrated that global hypoxia-ischemia in newborn piglets increased circulating miR-210 and miR-374a expression [100]. These two miRNAs were involved in the pulmonary vasculature and pulmonary hypertension and intrauterine growth restriction [116,117], which are closely associated with BPD. A more recent study found that umbilical cord blood-derived exosomal miRNAs from very preterm human infants with BPD were 90 downregulated (e.g., miR-103a-3p and miR-185-5p) and 328 upregulated (e.g., miR-200a-3p), which were associated with PI3K/Akt and angiogenesis-related signaling pathways [98]. Further, overexpression of miR-103a-3p and miR-185-5p augmented endothelial cell proliferation, migration and tube formation, while overexpression of miR-200a-3p impeded these angiogenic responses [98]. Additionally, miR-34a expression was found to be decreased in the postnatal hypoxia-induced mouse model of BPD/lung injury [101]. Although miR-34a was demonstrated to be upregulated in hyperoxia-induced mice BPD model and human BPD samples [101], it remains unclear the role of miR-34a and the downstream molecular mechanism in hypoxia-induced BPD. In addition, human BPD may be caused by both prenatal hypoxia and postnatal hyperoxia, so it deserves further investigation of the mechanistic role of miR-34a in prenatal hypoxia-induced BPD.
Recent studies in certain animal models, including intrauterine hypoxia, unveiled significant changes in gene expression patterns in the placenta and fetal organs/tissues (such as the heart, brain, cerebral artery, liver, pulmonary artery, and lung). It has been demonstrated that both HIF-1α and hypoxia-derived reactive oxygen species (ROS) can significantly mediate hypoxia-induced epigenetic programming and developmental disorders [6,118,119,120,121,122]. Although hypoxia-modulated epigenetics have been investigated in the placenta, fetal brain, and fetal heart, it remains unclear in the fetal lung. Further research is needed to investigate the effect of intrauterine hypoxia on global DNA and gene-specific DNA methylation patterns and other epigenetic patterns in the developing lung and understand how fetal stressors affect epigenetic changes at specific sites of associated genes in intrauterine hypoxia-related developmental diseases.
5. Perspectives for Treatment and Prevention of Neonatal Chronic Lung Disease
Despite the high prevalence of BPD, no effective therapeutics were available to treat this disease in the past twenty years. Only vitamin A, caffeine, and corticosteroid are clinically used after birth for BPD intervention [123,124,125,126]. Recently, research on the epigenetic involvement in BPD has been steadily increasing. As discussed above, epigenetic modifications, such as DNA methylation, histone acetylation, and miRNAs alteration, play important roles in BPD. Notably, some studies revealed that DNMT and HDAC inhibitors, or correcting miRNA expression could improve pathophysiological dysfunction in BPD [127,128,129,130]. Despite recent advances in the epigenetic field, clinical epigenetic therapy for BPD is still a challenge for future research. The mechanistic role of epigenetics in the occurrence and development of BPD is not completely understood, especially in pathological processes provoked by intrauterine hypoxia in the different stages of lung development.
A preliminary human miRNA profiling study in blood from preterm infants identified that miR-133b and miR-7 were more highly expressed in subjects with BPD compared to those without BPD, whereas the expression of miR-152 and miR-30a-3p decreased in the subjects with BPD. Furthermore, the downregulation of miR-152 and miR-30a-3p and upregulation of miR-133b and miR-7 were found in the BPD group, compared to the non-BPD group in the older-age set [93]. Therefore, more studies examining hypoxia-related epigenetic pathway alterations in the development of BPD the lung tissue and blood of different age sets will be necessary. This could provide evidence regarding the mechanical roles of the epigenetic modifications in the development of BPD and contribute to developing epigenetic pharmacological therapies.
In the past 20 years, clinical trials have made significant progress in prenatal gene therapy and have provided hope for patients who suffered from genetic diseases. These diseases occur before or shortly after birth and may cause severe morbidity or death in the uterus or after birth. Moreover, there is no specific and efficient postpartum treatment method. Because gene transduction is easy to attain in fluid-filled fetal lungs, they are ideal for prenatal gene therapy [131]. A recent study demonstrated that the knockout of HDAC3 improved alveolarization and pulmonary angiogenesis in BPD [130]. The knowledge gained from studies in epigenetic modification-based postnatal gene therapy is insufficient, but it is a necessary starting point and a potential and promising strategy for treating and preventing BPD. In addition, evolving gene editing approaches, including the use of nucleases such as TALENs (transcription activator-like effector nucleases), zinc-finger nucleases, and CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR-associated 9) to instigate a sequence-specific change in the DNA [132,133,134], encouraged the development of gene expression regulation, epigenetic modification, functional gene screening, gene diagnosis, and therapeutic drug discovery [135,136,137,138,139,140]. For example, dCas9 can be fused with DNMTs or TETs to regulate DNA methylation, thereby regulating specific gene expression [141,142,143]. Similarly, dCas9 can be fused with HDACs or HATs to regulate chromatin structure, thereby regulating gene expression [144,145,146]. Moreover, miRNA expression can be regulated by targeting the terminal loop or 5′ region of pre-miRNA by CRISPR-Cas9 technology [147,148,149]. The utero gene editing has been investigated in lung diseases [150,151,152,153]. Therefore, recent proof-of-concept studies support the feasibility of prenatal gene editing with high specificity for the lung and provide evidence that prenatal gene editing is potent and promising for the management of the developmental origin of lung diseases (Figure 3).
Nutritional supplement therapy has also been developed as a potential intervention for BPD, and various randomized clinical trials conducted in recent years have supported this approach. It is fascinating that many biologically active dietary components (“epigenetics diet”) change the epigenome [154,155]. These epigenetics diets include polyphenols (epigallocatechin-3-gallate, resveratrol, genistein), vitamins (vitamins c and d, folate, choline), isothiocyanates, withaferin a, and selenium. Perinatal vitamin D deficiency and low vitamin D levels have played a crucial role in neonatal BPD [156,157]. Significantly, vitamin D can directly stimulate fetal pulmonary artery endothelial cell and alveolar epithelial type II cell growth and function to prevent BPD development in preterm infants [158]. Dose- and time-dependent, and sex- and genetic background-specific epigenetic diets and their epigenome modification roles should be further investigated in BPD development.
Because the pathogenesis of alveolarization, vascularization, and tissue repair in immature lungs that cause BPD is multifactorial, the combined treatment of epigenetics and specific gene targeting methods will provide a broader prospect for the prevention and treatment of BPD.
Rodent-based forms of BPD provide significant advances in respect of genetic tools accessibility. Despite this, prenatal hypoxia, especially in preterm infants, and hyperoxia risk factors are far less significant clinical frameworks in animal BPD models. In addition, There there are limited utility for examining breathing dynamics, gas exchange, and pulmonary hemodynamics [159]. There seems to be an urgent need to adapt current models to more accurately reflect the pathological mechanisms at work in affected fetus and newborn infant, and conduct more rigorous evaluations of possible eligible pharmacological and nonpharmacological therapies for the treatment of BPD.
6. Concluding Remarks
Epigenetics plays a significant role in lung development. Lung development and the response to a conditioning injury (i.e., intrauterine hypoxia) require coordinated and structured proliferation and migration. For instance, lung development includes phenotypic modulation and programming to develop cells with specialized functions.
A better understanding of environmental influences (e.g., intrauterine hypoxia) and “developmental plasticity” on neonatal and fetal lung development will provide insights to establish preventive strategies to reduce or prevent abnormal lung development and the developmental origins of lung diseases.
Author Contributions
Conceptualization, R.S. and D.Y.; writing—original draft preparation, Y.T., and S.Z.; writing—review and editing, S.R., L.Z., R.S. and D.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Department of Science and Technology of Liaoning Province (CN) (NO. 2020 JH2/10300128) and Natural Science Foundation of Liaoning Province (NO. 201602873).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Moore, L.G.; Charles, S.M.; Julian, C.G. Humans at high altitude: Hypoxia and fetal growth. Respir. Physiol. Neurobiol. 2011, 178, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amark, H.; Sirotkina, M.; Westgren, M.; Papadogiannakis, N.; Persson, M. Is obesity in pregnancy associated with signs of chronic fetal hypoxia? Acta Obstet. Gynecol. Scand. 2020, 99, 1649–1656. [Google Scholar] [CrossRef]
- Turner, J.M.; Mitchell, M.D.; Kumar, S.S. The physiology of intrapartum fetal compromise at term. Am. J. Obstet. Gynecol. 2020, 222, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Zhang, L. Epigenetic programming of hypoxic-ischemic encephalopathy in response to fetal hypoxia. Prog. Neurobiol. 2015, 124, 28–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducsay, C.A.; Goyal, R.; Pearce, W.J.; Wilson, S.; Hu, X.Q.; Zhang, L. Gestational Hypoxia and Developmental Plasticity. Physiol. Rev. 2018, 98, 1241–1334. [Google Scholar] [CrossRef] [PubMed]
- Gortner, L.; Monz, D.; Mildau, C.; Shen, J.; Kasoha, M.; Laschke, M.W.; Roolfs, T.; Schmiedl, A.; Meier, C.; Tutdibi, E. Bronchopulmonary dysplasia in a double-hit mouse model induced by intrauterine hypoxia and postnatal hyperoxia: Closer to clinical features? Ann. Anat. 2013, 195, 351–358. [Google Scholar] [CrossRef]
- Schmiedl, A.; Roolfs, T.; Tutdibi, E.; Gortner, L.; Monz, D. Influence of prenatal hypoxia and postnatal hyperoxia on morphologic lung maturation in mice. PLoS ONE 2017, 12, e0175804. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Abdul-Hafez, A.; Gewolb, I.H.; Uhal, B.D. Oxygen injury in neonates: Which is worse? hyperoxia, hypoxia, or alternating hyperoxia/hypoxia. J. Lung Pulm. Respir. Res. 2020, 7, 4–13. [Google Scholar]
- Valencia, A.M.; Abrantes, M.A.; Hasan, J.; Aranda, J.V.; Beharry, K.D. Reactive Oxygen Species, Biomarkers of Microvascular Maturation and Alveolarization, and Antioxidants in Oxidative Lung Injury. React. Oxyg. Species 2018, 6, 373–388. [Google Scholar] [CrossRef]
- Lorente-Pozo, S.; Parra-Llorca, A.; Lara-Canton, I.; Solaz, A.; Garcia-Jimenez, J.L.; Pallardo, F.V.; Vento, M. Oxygen in the neonatal period: Oxidative stress, oxygen load and epigenetic changes. Semin. Fetal Neonatal Med. 2020, 25, 101090. [Google Scholar] [CrossRef]
- Wang, J.; Dong, W. Oxidative stress and bronchopulmonary dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef]
- Narayanan, M.; Owers-Bradley, J.; Beardsmore, C.S.; Mada, M.; Ball, I.; Garipov, R.; Panesar, K.S.; Kuehni, C.E.; Spycher, B.D.; Williams, S.E.; et al. Alveolarization continues during childhood and adolescence: New evidence from helium-3 magnetic resonance. Am. J. Respir. Crit. Care Med. 2012, 185, 186–191. [Google Scholar] [CrossRef]
- Thekkeveedu, R.K.; Guaman, M.C.; Shivanna, B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir. Med. 2017, 132, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, A.; Gwini, S.M.; Menahem, S.; Allison, B.J.; Miller, S.L.; Polglase, G.R. Preterm growth restriction and bronchopulmonary dysplasia: The vascular hypothesis and related physiology. J. Physiol. 2019, 597, 1209–1220. [Google Scholar] [CrossRef]
- Kramer, B.W.; Lievense, S.; Been, J.V.; Zimmermann, L.J. From classic to new bronchopulmonary dysplasia. Ned. Tijdschr. Geneeskd. 2010, 154, A1024. [Google Scholar]
- Baker, C.D.; Alvira, C.M. Disrupted lung development and bronchopulmonary dysplasia: Opportunities for lung repair and regeneration. Curr. Opin. Pediatr. 2014, 26, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voynow, J.A. “New” bronchopulmonary dysplasia and chronic lung disease. Paediatr. Respir. Rev. 2017, 24, 17–18. [Google Scholar] [CrossRef]
- Silva, D.M.; Nardiello, C.; Pozarska, A.; Morty, R.E. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1239–L1272. [Google Scholar] [CrossRef]
- Collins, J.J.P.; Tibboel, D.; de Kleer, I.M.; Reiss, I.K.M.; Rottier, R.J. The Future of Bronchopulmonary Dysplasia: Emerging Pathophysiological Concepts and Potential New Avenues of Treatment. Front. Med. 2017, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Abman, S.H.; Bancalari, E.; Jobe, A. The Evolution of Bronchopulmonary Dysplasia after 50 Years. Am. J. Respir. Crit. Care Med. 2017, 195, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R. Signaling Pathways Involved in the Development of Bronchopulmonary Dysplasia and Pulmonary Hypertension. Children 2020, 7, 100. [Google Scholar] [CrossRef]
- Nanduri, J.; Makarenko, V.; Reddy, V.D.; Yuan, G.; Pawar, A.; Wang, N.; Khan, S.A.; Zhang, X.; Kinsman, B.; Peng, Y.J.; et al. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 2515–2520. [Google Scholar] [CrossRef] [Green Version]
- Palma-Gudiel, H.; Eixarch, E.; Crispi, F.; Moran, S.; Zannas, A.S.; Fananas, L. Prenatal adverse environment is associated with epigenetic age deceleration at birth and hypomethylation at the hypoxia-responsive EP300 gene. Clin. Epigenet. 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasha, A.B.; Chen, X.Q.; Zhou, G.P. Bronchopulmonary dysplasia: Pathogenesis and treatment. Exp. Ther. Med. 2018, 16, 4315–4321. [Google Scholar] [CrossRef] [Green Version]
- Rood, K.; Lopez, V.; La Frano, M.R.; Fiehn, O.; Zhang, L.; Blood, A.B.; Wilson, S.M. Gestational Hypoxia and Programing of Lung Metabolism. Front. Physiol. 2019, 10, 1453. [Google Scholar] [CrossRef]
- Yang, I.V.; Schwartz, D.A. Epigenetic control of gene expression in the lung. Am. J. Respir. Crit. Care Med. 2011, 183, 1295–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagood, J.S. Beyond the genome: Epigenetic mechanisms in lung remodeling. Physiology 2014, 29, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Aboud, N.M.A.; Shahid, H.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Hoareau-Aveilla, C.; Meggetto, F. Crosstalk between microRNA and DNA Methylation Offers Potential Biomarkers and Targeted Therapies in ALK-Positive Lymphomas. Cancers 2017, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, J.; Wang, X.; Liu, Y.; Gu, B.; Zhao, G.; Li, Y. Methylation of TP53BP2 and Apaf-1 genes in embryonic lung cells and their impact on gene expression. Ann. Transl. Med. 2018, 6, 459. [Google Scholar] [CrossRef]
- Nagarajan, P.; Ge, Z.; Sirbu, B.; Doughty, C.; Garcia, P.A.A.; Schlederer, M.; Annunziato, A.T.; Cortez, D.; Kenner, L.; Parthun, M.R. Histone acetyl transferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS Genet. 2013, 9, e1003518. [Google Scholar] [CrossRef] [Green Version]
- Carraro, G.; Shrestha, A.; Rostkovius, J.; Contreras, A.; Chao, C.M.; El Agha, E.; Mackenzie, B.; Dilai, S.; Guidolin, D.; Taketo, M.M.; et al. miR-142-3p balances proliferation and differentiation of mesenchymal cells during lung development. Development 2014, 141, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Cushing, L.; Ai, X.; Lu, J. miR-326 is downstream of Sonic hedgehog signaling and regulates the expression of Gli2 and smoothened. Am. J. Respir. Cell Mol. Biol. 2014, 51, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.J.; Land, S.C. Regulation of vascular signalling by nuclear Sprouty2 in fetal lung epithelial cells: Implications for co-ordinated airway and vascular branching in lung development. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2018, 224, 105–114. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, Y.; Morley, M.P.; Lu, M.M.; Demayo, F.J.; Olson, E.N.; Morrisey, E.E. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2-Bmp4/Rb1 regulatory pathway. Dev. Cell 2013, 24, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, G.; El-Hashash, A.; Guidolin, D.; Tiozzo, C.; Turcatel, G.; Young, B.M.; De Langhe, S.P.; Bellusci, S.; Shi, W.; Parnigotto, P.P.; et al. miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev. Biol. 2009, 333, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhang, Y.; Hurd, L.; Hannenhalli, S.; Liu, F.; Lu, M.M.; Morrisey, E.E. Regulation of lung endoderm progenitor cell behavior by miR302/367. Development 2011, 138, 1235–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, E.L.; Choy, K.W.; Donahoe, P.K.; Tracy, A.A.; Hila, R.; Loscertales, M.; Longoni, M. MiR-449a Affects Epithelial Proliferation during the Pseudoglandular and Canalicular Phases of Avian and Mammal Lung Development. PLoS ONE 2016, 11, e0149425. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, M.; Wang, Y.; Zhang, H.; Weng, T.; Baviskar, P.; Guo, Y.; Gou, D.; Liu, L. MicroRNA-127 modulates fetal lung development. Physiol. Genom. 2009, 37, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.F.; Kan, Q.; Yang, Y.; Zhang, Y.H.; Shen, J.X.; Zhang, C.; Zhou, X.Y. Knockout of microRNA26a promotes lung development and pulmonary surfactant synthesis. Mol. Med. Rep. 2018, 17, 5988–5995. [Google Scholar] [CrossRef]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Cuna, A.; Halloran, B.; Faye-Petersen, O.; Kelly, D.; Crossman, D.K.; Cui, X.; Pandit, K.; Kaminski, N.; Bhattacharya, S.; Ahmad, A.; et al. Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am. J. Respir. Cell Mol. Biol. 2015, 53, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Ahangari, F.; Espinoza, C.R.; Chhabra, D.; Nicola, T.; Yan, X.; Lal, C.V.; Hagood, J.S.; Kaminski, N.; Bar-Joseph, Z.; et al. Integrating multiomics longitudinal data to reconstruct networks underlying lung development. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L556–L568. [Google Scholar] [CrossRef]
- Kumar, S.; Chinnusamy, V.; Mohapatra, T. Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond. Front. Genet. 2018, 9, 640. [Google Scholar] [CrossRef] [Green Version]
- Durham, A.L.; Adcock, I.M. Basic science: Epigenetic programming and the respiratory system. Breathe 2013, 9, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Fehl, J.; Pozarska, A.; Nardiello, C.; Rath, P.; Solaligue, D.E.S.; Vadasz, I.; Mayer, K.; Herold, S.; Seeger, W.; Morty, R.E. Control Interventions Can Impact Alveolarization and the Transcriptome in Developing Mouse Lungs. Anat. Rec. 2019, 302, 346–363. [Google Scholar] [CrossRef] [Green Version]
- Merid, S.K.; Novoloaca, A.; Sharp, G.C.; Kupers, L.K.; Kho, A.T.; Roy, R.; Gao, L.; Annesi-Maesano, I.; Jain, P.; Plusquin, M.; et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med. 2020, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bemer, M. Unraveling the Complex Epigenetic Mechanisms that Regulate Gene Activity. Methods Mol. Biol. 2018, 1675, 205–231. [Google Scholar] [CrossRef]
- Joss-Moore, L.A.; Albertine, K.H.; Lane, R.H. Epigenetics and the developmental origins of lung disease. Mol. Genet. Metab. 2011, 104, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.N.; Khan, A.U. Role of histone acetylation in cell physiology and diseases: An update. Clin. Chim. Acta 2010, 411, 1401–1411. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Alisch, R.S.; Jin, P.; Epstein, M.; Caspary, T.; Warren, S.T. Argonaute2 is essential for mammalian gastrulation and proper mesoderm formation. PLoS Genet. 2007, 3, e227. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Fazi, F.; Ciaudo, C. Argonaute Proteins: From Structure to Function in Development and Pathological Cell Fate Determination. Front. Cell Dev. Biol. 2019, 7, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.S.; Zhang, Z.; McManus, M.T.; Harfe, B.D.; Sun, X. Dicer function is essential for lung epithelium morphogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 2208–2213. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kai, G.; Pu, X.D.; Qing, K.; Guo, X.R.; Zhou, X.Y. Expression profile of microRNAs in fetal lung development of Sprague-Dawley rats. Int. J. Mol. Med. 2012, 29, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Ye, X.Y.; Singhal, N.; De La Rue, S.; Lodha, A.; Shah, P.S. Higher altitude and risk of bronchopulmonary dysplasia among preterm infants. Am. J. Perinatol. 2013, 30, 601–606. [Google Scholar] [CrossRef]
- Alshehri, M.A. Are preterm infants at high altitude at greater risk for the development of bronchopulmonary dysplasia? J. Trop. Pediatr. 2014, 60, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.Q.; Huang, X.; Xiao, D.; Zhang, L. Direct effect of chronic hypoxia in suppressing large conductance Ca(2+)-activated K(+) channel activity in ovine uterine arteries via increasing oxidative stress. J. Physiol. 2016, 594, 343–356. [Google Scholar] [CrossRef]
- Xiao, D.; Hu, X.Q.; Huang, X.; Zhou, J.; Wilson, S.M.; Yang, S.; Zhang, L. Chronic hypoxia during gestation enhances uterine arterial myogenic tone via heightened oxidative stress. PLoS ONE 2013, 8, e73731. [Google Scholar] [CrossRef] [Green Version]
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444. [Google Scholar] [CrossRef] [Green Version]
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef]
- Tracy, M.K.; Berkelhamer, S.K. Bronchopulmonary Dysplasia and Pulmonary Outcomes of Prematurity. Pediatr. Ann. 2019, 48, e148–e153. [Google Scholar] [CrossRef]
- Hislop, A. Developmental biology of the pulmonary circulation. Paediatr. Respir. Rev. 2005, 6, 35–43. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Vandivier, R.W.; Tuder, R.M. Vascular endothelial growth factor in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L209–L221. [Google Scholar] [CrossRef]
- Alvira, C.M. Aberrant Pulmonary Vascular Growth and Remodeling in Bronchopulmonary Dysplasia. Front. Med. 2016, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Hocq, C.; Vanhoutte, L.; Guilloteau, A.; Massolo, A.C.; Van Grambezen, B.; Carkeek, K.; Piersigilli, F.; Danhaive, O.; from the European Society for Pediatric Research. Early diagnosis and targeted approaches to pulmonary vascular disease in bronchopulmonary dysplasia. Pediatr. Res. 2021. [Google Scholar] [CrossRef]
- Mandell, E.W.; Abman, S.H. Fetal Vascular Origins of Bronchopulmonary Dysplasia. J. Pediatr. 2017, 185, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Remesal, A.; Pedraz, C.; San Feliciano, L.; Ludena, D. Pulmonary expression of vascular endothelial growth factor (VEGF) and alveolar septation in a newborn rat model exposed to acute hypoxia and recovered under conditions of air or hyperoxia. Histol. Histopathol. 2009, 24, 325–330. [Google Scholar] [CrossRef]
- Remesal, A.; San Feliciano, L.; Isidoro-Garcia, M.; Ludena, D. Effects of antenatal betamethasone and dexamethasone on the lung expression of vascular endothelial growth factor and alveolarization in newborn rats exposed to acute hypoxia and recovered in normoxia or hyperoxia. Neonatology 2010, 98, 313–320. [Google Scholar] [CrossRef]
- Gallacher, D.J.; Hart, K.; Kotecha, S. Common respiratory conditions of the newborn. Breathe 2016, 12, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Filby, C.E.; Hooper, S.B.; Wallace, M.J. Partial pulmonary embolization disrupts alveolarization in fetal sheep. Respir. Res. 2010, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Kang, N.Y.; Ivanovska, J.; Tamir-Hostovsky, L.; Belik, J.; Gauda, E.B. Chronic Intermittent Hypoxia in Premature Infants: The Link Between Low Fat Stores, Adiponectin Receptor Signaling and Lung Injury. Adv. Exp. Med. Biol. 2018, 1071, 151–157. [Google Scholar] [CrossRef]
- Schittny, J.C.; Mund, S.I.; Stampanoni, M. Evidence and structural mechanism for late lung alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L246–L254. [Google Scholar] [CrossRef] [Green Version]
- Mund, S.I.; Stampanoni, M.; Schittny, J.C. Developmental alveolarization of the mouse lung. Dev. Dyn. 2008, 237, 2108–2116. [Google Scholar] [CrossRef] [Green Version]
- Elberson, V.D.; Nielsen, L.C.; Wang, H.; Kumar, H.S. Effects of intermittent hypoxia and hyperoxia on angiogenesis and lung development in newborn mice. J. Neonatal Perinat. Med. 2015, 8, 313–322. [Google Scholar] [CrossRef]
- Chao, C.M.; Moiseenko, A.; Zimmer, K.P.; Bellusci, S. Alveologenesis: Key cellular players and fibroblast growth factor 10 signaling. Mol. Cell. Pediatr. 2016, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Castillo, J.A.; Perez, D.B.; Ntokou, A.; Seeger, W.; Morty, R.E.; Ahlbrecht, K. Understanding alveolarization to induce lung regeneration. Respir. Res. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammoto, A.; Mammoto, T. Vascular Niche in Lung Alveolar Development, Homeostasis, and Regeneration. Front. Bioeng. Biotechnol. 2019, 7, 318. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, Y.; Taylor, D.; Ovchinnikov, D.A.; Wolvetang, E.J.; de Torrente, L.; Mar, J.C. Variability of Gene Expression Identifies Transcriptional Regulators of Early Human Embryonic Development. PLoS Genet. 2015, 11, e1005428. [Google Scholar] [CrossRef]
- Dumeige, L.; Nehlich, M.; Viengchareun, S.; Perrot, J.; Pussard, E.; Lombes, M.; Martinerie, L. Preterm birth is associated with epigenetic programming of transgenerational hypertension in mice. Exp. Mol. Med. 2020, 52, 152–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, E.R.; Britt, R.D., Jr.; Trinidad, M.C.; Faksh, A.; Martin, R.J.; MacFarlane, P.M.; Pabelick, C.M.; Prakash, Y.S. Perinatal oxygen in the developing lung. Can. J. Physiol. Pharmacol. 2015, 93, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Lafuente, E.; Beldade, P. Genomics of Developmental Plasticity in Animals. Front. Genet. 2019, 10, 720. [Google Scholar] [CrossRef]
- Meiners, S.; Hilgendorff, A. Early injury of the neonatal lung contributes to premature lung aging: A hypothesis. Mol. Cell. Pediatr. 2016, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- McEvoy, C.T.; Spindel, E.R. Pulmonary Effects of Maternal Smoking on the Fetus and Child: Effects on Lung Development, Respiratory Morbidities, and Life Long Lung Health. Paediatr. Respir. Rev. 2017, 21, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Lignelli, E.; Palumbo, F.; Myti, D.; Morty, R.E. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L832–L887. [Google Scholar] [CrossRef]
- Sahoo, D.; Zaramela, L.S.; Hernandez, G.E.; Mai, U.; Taheri, S.; Dang, D.; Stouch, A.N.; Medal, R.M.; McCoy, A.M.; Aschner, J.L.; et al. Transcriptional profiling of lung macrophages identifies a predictive signature for inflammatory lung disease in preterm infants. Commun. Biol. 2020, 3, 259. [Google Scholar] [CrossRef]
- Lal, C.V.; Olave, N.; Travers, C.; Rezonzew, G.; Dolma, K.; Simpson, A.; Halloran, B.; Aghai, Z.; Das, P.; Sharma, N.; et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. J. Clin. Investig. Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Chen, W.J.; Hsieh, W.S.; Tsao, P.N.; Yu, S.L.; Lai, C.Y.; Lee, W.C.; Jeng, S.F. MicroRNA expression aberration associated with bronchopulmonary dysplasia in preterm infants: A preliminary study. Respir. Care 2013, 58, 1527–1535. [Google Scholar] [CrossRef]
- Wong, M.J.; Kantores, C.; Ivanovska, J.; Jain, A.; Jankov, R.P. Simvastatin prevents and reverses chronic pulmonary hypertension in newborn rats via pleiotropic inhibition of RhoA signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L985–L999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadivel, A.; Alphonse, R.S.; Collins, J.J.; van Haaften, T.; O’Reilly, M.; Eaton, F.; Thebaud, B. The axonal guidance cue semaphorin 3C contributes to alveolar growth and repair. PLoS ONE 2013, 8, e67225. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Bhattacharya, S.; Sridhar, A.; Iqbal, A.M.; Mariani, T.J. Recurrent copy number variants associated with bronchopulmonary dysplasia. Pediatr. Res. 2016, 79, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Rogers, L.K.; Robbins, M.; Dakhlallah, D.; Yang, Z.; Lee, L.J.; Mikhail, M.; Nuovo, G.; Pryhuber, G.S.; McGwin, G.; Marsh, C.B.; et al. Attenuation of miR-17 approximately 92 Cluster in Bronchopulmonary Dysplasia. Ann. Am. Thorac. Soc. 2015, 12, 1506–1513. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.Q.; Yan, Q.; Chen, Z.G.; Jia, C.H.; Li, X.H.; Liang, Z.Y.; Gu, J.; Wei, H.L.; Lian, C.Y.; Zheng, J.; et al. Umbilical Cord Blood-Derived Exosomes From Very Preterm Infants With Bronchopulmonary Dysplasia Impaired Endothelial Angiogenesis: Roles of Exosomal MicroRNAs. Front. Cell Dev. Biol. 2021, 9, 637248. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Hu, X.; Wu, C.; Li, N. Hypoxia-induced miR-15a promotes mesenchymal ablation and adaptation to hypoxia during lung development in chicken. PLoS ONE 2014, 9, e98868. [Google Scholar] [CrossRef]
- Garberg, H.T.; Huun, M.U.; Baumbusch, L.O.; Asegg-Atneosen, M.; Solberg, R.; Saugstad, O.D. Temporal Profile of Circulating microRNAs after Global Hypoxia-Ischemia in Newborn Piglets. Neonatology 2017, 111, 133–139. [Google Scholar] [CrossRef]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, V.E.; Howe, L.J. Histone acetylation: Where to go and how to get there. Epigenetics 2009, 4, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Engelen, E.; Brandsma, J.H.; Moen, M.J.; Signorile, L.; Dekkers, D.H.; Demmers, J.; Kockx, C.E.; Ozgur, Z.; van IJcken, W.F.J.; van den Berg, D.L.C.; et al. Proteins that bind regulatory regions identified by histone modification chromatin immunoprecipitations and mass spectrometry. Nat. Commun. 2015, 6, 7155. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Adcock, I.M.; Ito, K. Histone acetylation and deacetylation: Importance in inflammatory lung diseases. Eur. Respir. J. 2005, 25, 552–563. [Google Scholar] [CrossRef]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.F.; Ma, X.L.; Shen, Z.; Wu, X.L.; Cheng, F.; Du, L.Z. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 2010, 28, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Masjedi, M.R.; Barnes, P.J.; Adcock, I.M. Epigenetics and chromatin remodeling play a role in lung disease. Tanaffos 2011, 10, 7–16. [Google Scholar]
- Yao, Y.; Liu, Q.; Adrianto, I.; Wu, X.; Glassbrook, J.; Khalasawi, N.; Yin, C.; Yi, Q.; Dong, Z.; Geissmann, F.; et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat. Commun. 2020, 11, 3822. [Google Scholar] [CrossRef]
- Sessa, R.; Hata, A. Role of microRNAs in lung development and pulmonary diseases. Pulm. Circ. 2013, 3, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.; Rahman, M.; Nana-Sinkam, S.P. MicroRNAs in respiratory disease. A clinician’s overview. Ann. Am. Thorac. Soc. 2014, 11, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Nardiello, C.; Morty, R.E. MicroRNA in late lung development and bronchopulmonary dysplasia: The need to demonstrate causality. Mol. Cell. Pediatr. 2016, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Olave, N.; Lal, C.V.; Halloran, B.; Pandit, K.; Cuna, A.C.; Faye-Petersen, O.M.; Kelly, D.R.; Nicola, T.; Benos, P.V.; Kaminski, N.; et al. Regulation of alveolar septation by microRNA-489. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L476–L487. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Rivero, M.; Zhang, R.; Heilmann-Heimbach, S.; Mueller, A.; Bagci, S.; Dresbach, T.; Schroder, L.; Holdenrieder, S.; Reutter, H.M.; Kipfmueller, F. Circulating microRNAs are associated with Pulmonary Hypertension and Development of Chronic Lung Disease in Congenital Diaphragmatic Hernia. Sci. Rep. 2018, 8, 10735. [Google Scholar] [CrossRef]
- Dutta, R.K.; Chinnapaiyan, S.; Unwalla, H. Aberrant MicroRNAomics in Pulmonary Complications: Implications in Lung Health and Diseases. Mol. Ther. Nucleic Acids 2019, 18, 413–431. [Google Scholar] [CrossRef]
- White, K.; Lu, Y.; Annis, S.; Hale, A.E.; Chau, B.N.; Dahlman, J.E.; Hemann, C.; Opotowsky, A.R.; Vargas, S.O.; Rosas, I.; et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol. Med. 2015, 7, 695–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; MacIntyre, D.A.; Binkhamis, R.; Cook, J.; Sykes, L.; Bennett, P.R.; Terzidou, V. Maternal plasma miRNAs as potential biomarkers for detecting risk of small-for-gestational-age births. EBioMedicine 2020, 62, 103145. [Google Scholar] [CrossRef]
- Patterson, A.J.; Xiao, D.; Xiong, F.; Dixon, B.; Zhang, L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCepsilon gene in foetal rat hearts. Cardiovasc. Res. 2012, 93, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustelo, M.; Barkhuizen, M.; van den Hove, D.L.A.; Steinbusch, H.W.M.; Bruno, M.A.; Loidl, C.F.; Gavilanes, A.W.D. Clinical Implications of Epigenetic Dysregulation in Perinatal Hypoxic-Ischemic Brain Damage. Front. Neurol. 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Yu, R.M.K.; Wu, R.S.S.; Kong, R.Y.C. Overexpression and Knockdown of Hypoxia-Inducible Factor 1 Disrupt the Expression of Steroidogenic Enzyme Genes and Early Embryonic Development in Zebrafish. Gene Regul. Syst. Biol. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Pamenter, M.E.; Hall, J.E.; Tanabe, Y.; Simonson, T.S. Cross-Species Insights Into Genomic Adaptations to Hypoxia. Front. Genet. 2020, 11, 743. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef]
- Tyson, J.E.; Wright, L.L.; Oh, W.; Kennedy, K.A.; Mele, L.; Ehrenkranz, R.A.; Stoll, B.J.; Lemons, J.A.; Stevenson, D.K.; Bauer, C.R.; et al. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N. Engl. J. Med. 1999, 340, 1962–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londhe, V.A.; Nolen, T.L.; Das, A.; Higgins, R.D.; Tyson, J.E.; Oh, W.; Devaskar, S.U. Vitamin A supplementation in extremely low-birth-weight infants: Subgroup analysis in small-for-gestational-age infants. Am. J. Perinatol. 2013, 30, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Pakvasa, M.A.; Saroha, V.; Patel, R.M. Optimizing Caffeine Use and Risk of Bronchopulmonary Dysplasia in Preterm Infants: A Systematic Review, Meta-analysis, and Application of Grading of Recommendations Assessment, Development, and Evaluation Methodology. Clin. Perinatol. 2018, 45, 273–291. [Google Scholar] [CrossRef]
- Baud, O.; Watterberg, K.L. Prophylactic postnatal corticosteroids: Early hydrocortisone. Semin. Fetal Neonatal Med. 2019, 24, 202–206. [Google Scholar] [CrossRef]
- Cetinkaya, M.; Cansev, M.; Cekmez, F.; Tayman, C.; Canpolat, F.E.; Kafa, I.M.; Yaylagul, E.O.; Kramer, B.W.; Sarici, S.U. Protective Effects of Valproic Acid, a Histone Deacetylase Inhibitor, against Hyperoxic Lung Injury in a Neonatal Rat Model. PLoS ONE 2015, 10, e0126028. [Google Scholar] [CrossRef] [PubMed]
- Menden, H.; Xia, S.; Mabry, S.M.; Noel-MacDonnell, J.; Rajasingh, J.; Ye, S.Q.; Sampath, V. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L332–L346. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, L.; Yu, J.; Fu, H.; Zhou, D.; Liu, H. Inhibition of microRNA-29a alleviates hyperoxia-induced bronchopulmonary dysplasia in neonatal mice via upregulation of GAB1. Mol. Med. 2019, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hong, H.; Li, X.-X.; Li, J.; Zhang, Z.-Q. The involvement of HDAC3-mediated inhibition of microRNA cluster 17-92 in hyperoxia-mediated impairment of lung development in neonatal rats. bioRxiv 2020, 1–26. [Google Scholar] [CrossRef] [Green Version]
- David, A.L.; Waddington, S.N. Candidate diseases for prenatal gene therapy. Methods Mol. Biol. 2012, 891, 9–39. [Google Scholar] [CrossRef]
- Cathomen, T.; Joung, J.K. Zinc-finger nucleases: The next generation emerges. Mol. Ther. 2008, 16, 1200–1207. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Scott, A. How CRISPR is transforming drug discovery. Nature 2018, 555, S10–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Tong, R.; Li, M.; Liu, Y.; Xue, J.; Lu, Y. Advancements and Obstacles of CRISPR-Cas9 Technology in Translational Research. Mol. Ther. Methods Clin. Dev. 2019, 13, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Schacker, M.; Seimetz, D. From fiction to science: Clinical potentials and regulatory considerations of gene editing. Clin. Transl. Med. 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Lu, A.; Wang, J.; Sun, W.; Huang, W.; Cai, Z.; Zhao, G.; Wang, J. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov. 2019, 5, 22. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [Green Version]
- Sapozhnikov, D.M.; Szyf, M. Unraveling the functional role of DNA methylation using targeted DNA demethylation by steric blockage of DNA methyltransferase with CRISPR/dCas9. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Lin, Y.T.; Gallegos, D.A.; Hazlett, M.F.; Gomez-Schiavon, M.; Yang, M.G.; Kalmeta, B.; Zhou, A.S.; Holtzman, L.; Gersbach, C.A.; et al. Enhancer Histone Acetylation Modulates Transcriptional Bursting Dynamics of Neuronal Activity-Inducible Genes. Cell Rep. 2019, 26, 1174–1188. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhou, X.; Wei, M.; Gao, X.; Zhao, L.; Shi, R.; Sun, W.; Duan, Y.; Yang, G.; Yuan, L. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019, 19, 19–28. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, K.; Cheng, Y.; Zhong, Z.; Tian, L.; Tang, X.; Tang, A.; Zheng, X.; Zhang, T.; Qi, Y.; et al. CRISPR-Cas9 Based Genome Editing Reveals New Insights into MicroRNA Function and Regulation in Rice. Front. Plant Sci. 2017, 8, 1598. [Google Scholar] [CrossRef] [Green Version]
- Basso, M.F.; Ferreira, P.C.G.; Kobayashi, A.K.; Harmon, F.G.; Nepomuceno, A.L.; Molinari, H.B.C.; Grossi-de-Sa, M.F. MicroRNAs and new biotechnological tools for its modulation and improving stress tolerance in plants. Plant Biotechnol. J. 2019, 17, 1482–1500. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; Lopez-Giraldez, F.; Coskun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef] [Green Version]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alapati, D.; Morrisey, E.E. Gene Editing and Genetic Lung Disease. Basic Research Meets Therapeutic Application. Am. J. Respir. Cell Mol. Biol. 2017, 56, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Hodges, C.A.; Conlon, R.A. Delivering on the promise of gene editing for cystic fibrosis. Genes Dis. 2019, 6, 97–108. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, S.H.; Lee, M.S.; Kim, M.S. Epigenetic modification by dietary factors: Implications in metabolic syndrome. Mol. Aspects Med. 2017, 54, 58–70. [Google Scholar] [CrossRef]
- Li, S.; Chen, M.; Li, Y.; Tollefsbol, T.O. Prenatal epigenetics diets play protective roles against environmental pollution. Clin. Epigenet. 2019, 11, 82. [Google Scholar] [CrossRef]
- Mao, X.; Qiu, J.; Zhao, L.; Xu, J.; Yin, J.; Yang, Y.; Zhang, M.; Cheng, R. Vitamin D and IL-10 Deficiency in Preterm Neonates With Bronchopulmonary Dysplasia. Front. Pediatr. 2018, 6, 246. [Google Scholar] [CrossRef]
- Park, H.W.; Lim, G.; Park, Y.M.; Chang, M.; Son, J.S.; Lee, R. Association between vitamin D level and bronchopulmonary dysplasia: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0235332. [Google Scholar] [CrossRef]
- Mandell, E.; Seedorf, G.; Gien, J.; Abman, S.H. Vitamin D treatment improves survival and infant lung structure after intra-amniotic endotoxin exposure in rats: Potential role for the prevention of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L420–L428. [Google Scholar] [CrossRef] [Green Version]
- Morty, R.E. Using Experimental Models to Identify Pathogenic Pathways and Putative Disease Management Targets in Bronchopulmonary Dysplasia. Neonatology 2020, 117, 233–239. [Google Scholar] [CrossRef]
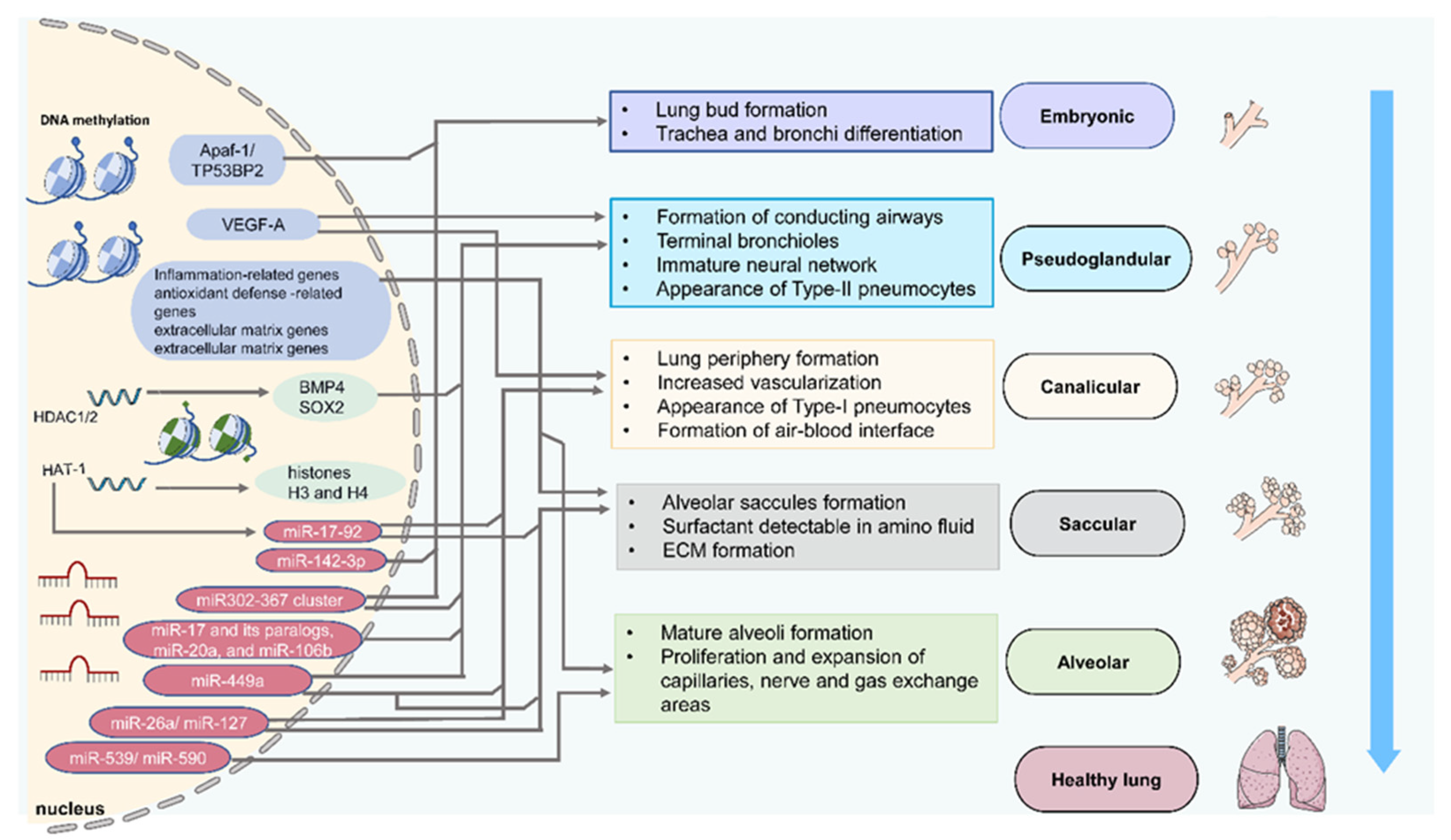
Figure 1.
Epigenetic regulation of lung development. Schematic depicting of epigenetic components such as DNA methylation, histone acetylation, and miRNA expression to modify targeted genes and determine phenotypes at different lung development stages. These epigenetic components may have a crosstalk effect.
Figure 1.
Epigenetic regulation of lung development. Schematic depicting of epigenetic components such as DNA methylation, histone acetylation, and miRNA expression to modify targeted genes and determine phenotypes at different lung development stages. These epigenetic components may have a crosstalk effect.

Figure 2.
Intrauterine hypoxia increases the susceptibility to BPD. Intrauterine hypoxia results in different pathophysiological phenotypes of the alveoli (for example, defects in alveolarization, gas exchange, and blood vessel growth), airway smooth muscle (for example, reduction of airway smooth muscle cells proliferation and increased airway smooth muscle thickness and inflammation) and airway epithelium (for example, decreased Na+ transport and increased mucus secretion). These structural or functional defects mediated by hypoxia signals lead to infant BPD susceptibility.
Figure 2.
Intrauterine hypoxia increases the susceptibility to BPD. Intrauterine hypoxia results in different pathophysiological phenotypes of the alveoli (for example, defects in alveolarization, gas exchange, and blood vessel growth), airway smooth muscle (for example, reduction of airway smooth muscle cells proliferation and increased airway smooth muscle thickness and inflammation) and airway epithelium (for example, decreased Na+ transport and increased mucus secretion). These structural or functional defects mediated by hypoxia signals lead to infant BPD susceptibility.

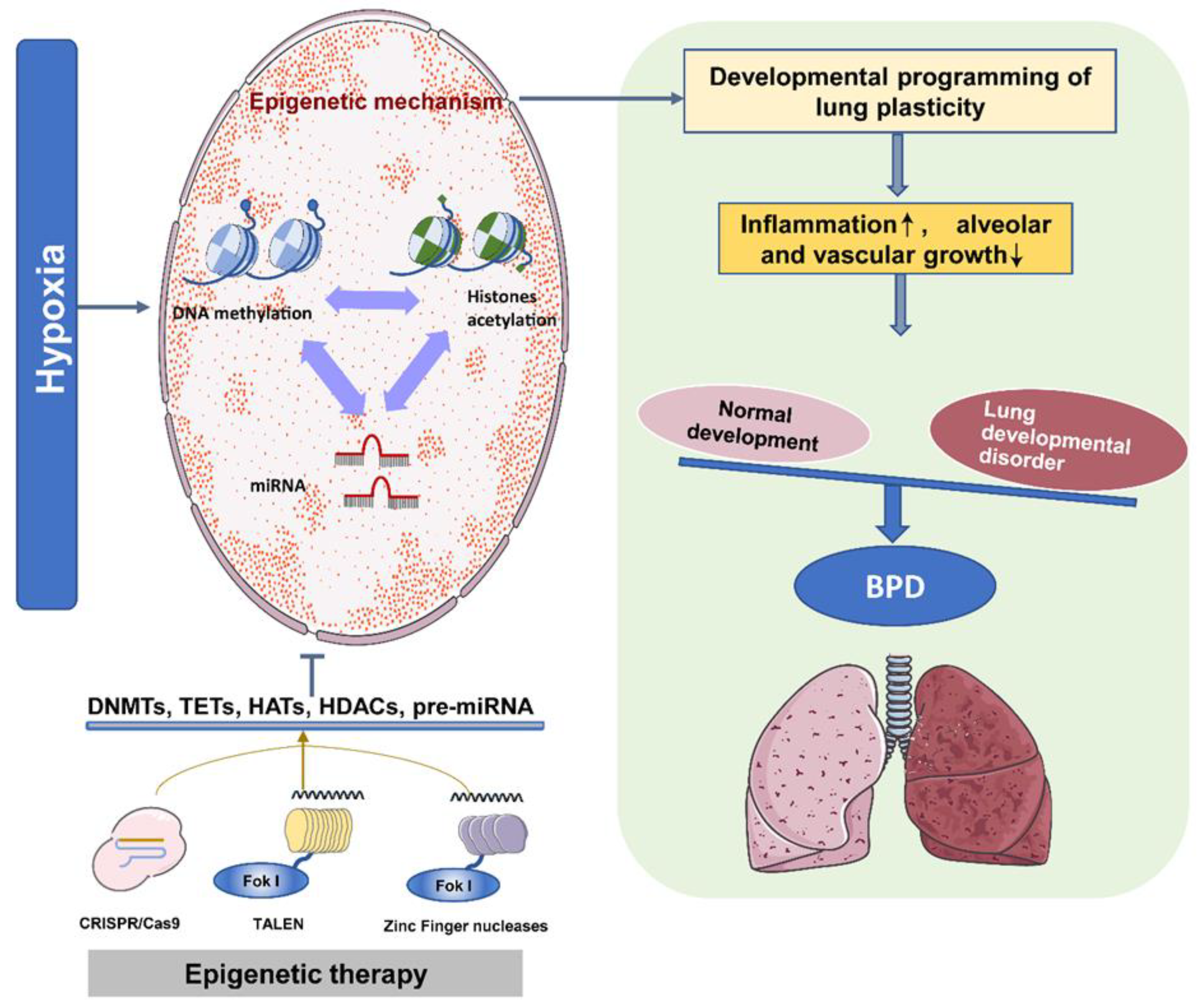
Figure 3.
The epigenetic program controls the development of BPD. Under intrauterine hypoxia, different epigenetic mechanisms coordinate the development of lung plasticity. Therefore, inflammation is induced during lung development, and the growth of alveoli and blood vessels is inhibited. Eventually, normal and abnormal lung development are unbalanced, leading to the development of BPD. Epigenetic therapy based on DNMT inhibitors, HDAC inhibitors, and miR modulators can improve lung development and reduce the pathogenesis of neonatal chronic lung disease.
Figure 3.
The epigenetic program controls the development of BPD. Under intrauterine hypoxia, different epigenetic mechanisms coordinate the development of lung plasticity. Therefore, inflammation is induced during lung development, and the growth of alveoli and blood vessels is inhibited. Eventually, normal and abnormal lung development are unbalanced, leading to the development of BPD. Epigenetic therapy based on DNMT inhibitors, HDAC inhibitors, and miR modulators can improve lung development and reduce the pathogenesis of neonatal chronic lung disease.

Table 1.
Lung development and epigenetic regulatory components.
| Stage | Species and Samples | Epigenetics | Targeted Genes | Characteristic Events | References |
|---|---|---|---|---|---|
| Embryonic | Human embryonic lung cells | DNA Methylation | TP53BP2 and Apaf-1 | Cell proliferation | [31] |
| Mouse embryonic fibroblasts | HAT-1 | Histones H3 and H4 | Embryonic lung development | [32] | |
| Mouse lung primordia | miR-142-3p | WNT signaling | Lung mesenchymal cells proliferation and differentiation | [33] | |
| Mouse embryonic lung explants | miR-326 | Smo and Gli2 | Lung mesenchymal cells proliferation and differentiation | [34] | |
| Pseudoglandular | Rat fetal distal lung epithelial cells | DNA Methylation | VEGF-A | Airway and vascular branching | [35] |
| Mouse proximal lung endoderm progenitors | HDAC1/2 | BMP4/SOX2 | Branching morphogenesis | [36] | |
| Mouse embryonic lung epithelial explants | miR-17 and its paralogs, miR-20a, and miR-106b | Stat3 and Mapk14 | Epithelial bud morphogenesis | [37] | |
| Early lung endoderm | miR302–367 cluster | Rbl2 and Cdkn1a | Lung epithelial proliferation | [38] | |
| Human, murine, and avian fetal lungs | miR-449a | Mycn and Sox9 | Epithelial proliferation and mucociliary differentiation | [39] | |
| Rat fetal lungs | miR-127 | Lung branching | [40] | ||
| Canalicular | Rat fetal distal lung epithelial cells | DNA Methylation | VEGF-A | Airway and vascular branching | [35] |
| Human, murine, and avian fetal lungs | miR-449a | Mycn and Sox9 | Epithelial proliferation and mucociliary differentiation | [39] | |
| Mouse fetal lungs | miR-26a | SFTPA1, SFTPB, SFTPC | Formation of dilated lumens and aerated regions, maturation of the alveolar structure | [41] | |
| Saccular | Human, murine, and avian fetal lungs | miR-449a | Mycn and Sox9 | Epithelial proliferation and mucociliary differentiation | [39] |
| miR-26a | SFTPA1, SFTPB, SFTPC | Maturation of the alveolar structure | [41] | ||
| Mouse fetal lungs | HDAC3/miR-17-92 | TGF-β | Alveolar type 1 cell spreading and lung sacculation | [42] | |
| Alveolar | Mouse newborn lung | DNA Methylation | ? | Alveolar septation | [43] |
| Mouse postnatal and early child lung | miR-539 and miR-590 | Alveolar development | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tong, Y.; Zhang, S.; Riddle, S.; Zhang, L.; Song, R.; Yue, D. Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease. Biomedicines 2021, 9, 944. https://doi.org/10.3390/biomedicines9080944
AMA Style
Tong Y, Zhang S, Riddle S, Zhang L, Song R, Yue D. Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease. Biomedicines. 2021; 9(8):944. https://doi.org/10.3390/biomedicines9080944
Chicago/Turabian StyleTong, Yajie, Shuqing Zhang, Suzette Riddle, Lubo Zhang, Rui Song, and Dongmei Yue. 2021. "Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease" Biomedicines 9, no. 8: 944. https://doi.org/10.3390/biomedicines9080944
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.