
Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases
by
 , and
, and
Simone Patergnani
1,† ,
,
Esmaa Bouhamida
1,†, 
Sara Leo
1,
Paolo Pinton
1,2 and
Alessandro Rimessi
1,2,*
1
Department of Medical Sciences and Laboratory for Technologies of Advanced Therapies (LTTA), University of Ferrara, 44121 Ferrara, Italy
2
Center of Research for Innovative Therapies in Cystic Fibrosis, University of Ferrara, 44121 Ferrara, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomedicines 2021, 9(2), 216; https://doi.org/10.3390/biomedicines9020216
Submission received: 27 January 2021
/
Revised: 16 February 2021
/
Accepted: 18 February 2021
/
Published: 20 February 2021
(This article belongs to the Special Issue Oxidative Stress and Inflammation: From Mechanisms to Therapeutic Approaches 2.0)
Abstract
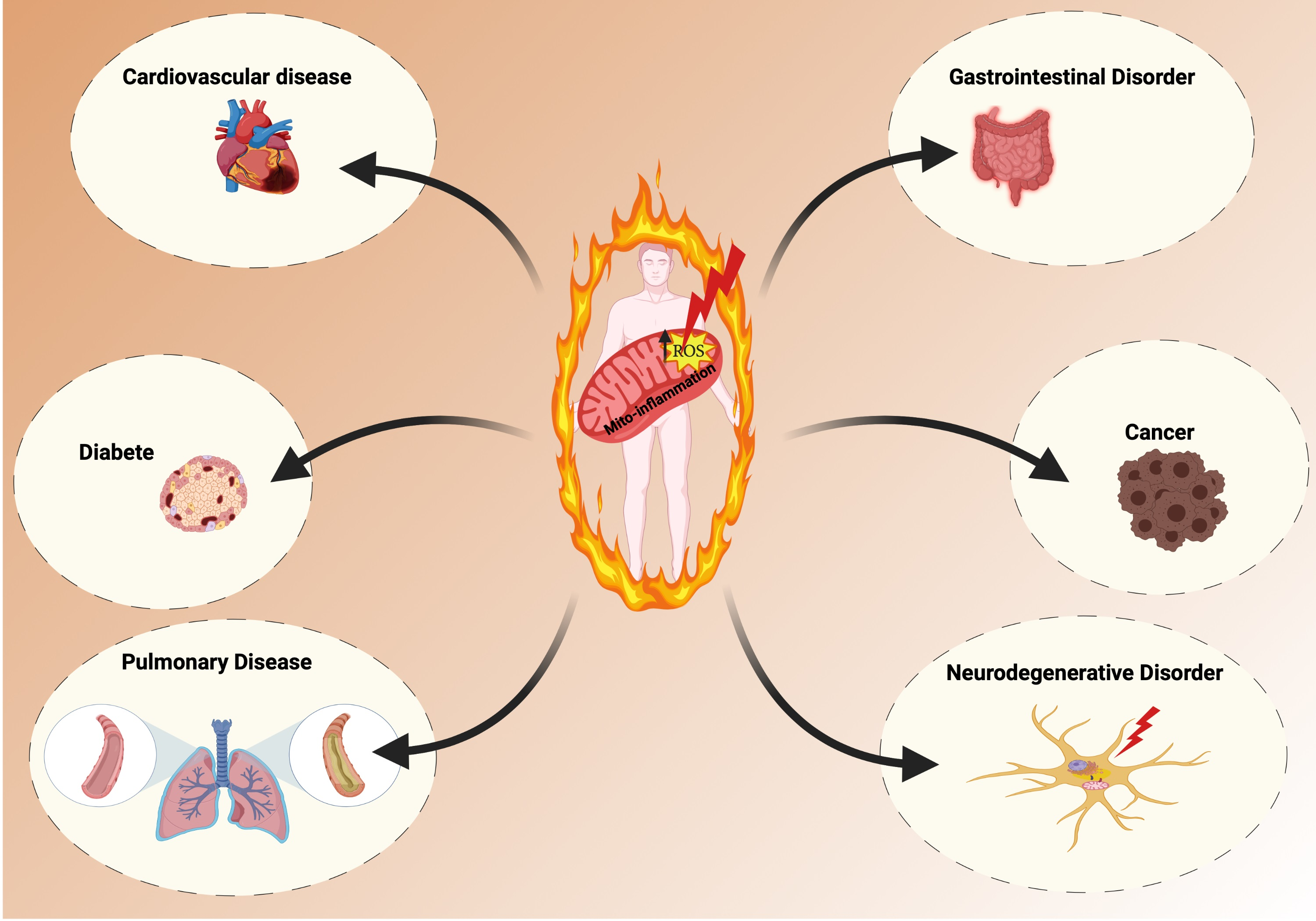
:A decline in mitochondrial redox homeostasis has been associated with the development of a wide range of inflammatory-related diseases. Continue discoveries demonstrate that mitochondria are pivotal elements to trigger inflammation and stimulate innate immune signaling cascades to intensify the inflammatory response at front of different stimuli. Here, we review the evidence that an exacerbation in the levels of mitochondrial-derived reactive oxygen species (ROS) contribute to mito-inflammation, a new concept that identifies the compartmentalization of the inflammatory process, in which the mitochondrion acts as central regulator, checkpoint, and arbitrator. In particular, we discuss how ROS contribute to specific aspects of mito-inflammation in different inflammatory-related diseases, such as neurodegenerative disorders, cancer, pulmonary diseases, diabetes, and cardiovascular diseases. Taken together, these observations indicate that mitochondrial ROS influence and regulate a number of key aspects of mito-inflammation and that strategies directed to reduce or neutralize mitochondrial ROS levels might have broad beneficial effects on inflammatory-related diseases.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Free radicals and other reactive oxygen species (ROS) are frequently associated with being harmful. However, several physiological functions (differentiation, cellular signaling, phosphorylation/dephosphorylation events and apoptosis) are dependent on the presence of reactive species inside the cells [1]. To regulate these cellular functions, the ROS production must be kept low. When it increases, ROS become dangerous, determining undesirable effects on cellular structures, including intracellular organelles, and participating in the onset and/or progression of several human disorders, such as neurodegenerative disorders, cancer, pulmonary, and cardiovascular diseases [2,3,4,5]. ROS can be produced at cytosolic levels by the NADPH oxidase (NOX) and the nitric oxide (NO) synthase enzyme. The well-characterized source of cytosolic ROS is NOX2, which produces anion superoxide (O2·−) through NAPDH electron exchange [6]. Alternatively, significant amounts of ROS are generated in the peroxisomes compartment by oxidases and NO synthases that produce hydrogen peroxide (H2O2) and NO, respectively [7]. The endoplasmic reticulum (ER) represents a place where high amounts of ROS are produced. In this compartment, ROS result in being the byproduct of oxidases and oxygenase involved during the protein folding process, such as ER oxidoreductin 1 [8]. Moreover, ER present iron deposits that contribute to form the reactive species hydroxyl radical (·OH), but undoubtedly, mitochondria represent the primary source of ROS (mitochondrial ROS, mtROS) for the cell [9].
Mitochondria are essential semi-autonomous cellular organelles with a double membrane composed by an inner (IMM) and an outer membrane (OMM). OMM separates the mitochondria from cytoplasm and it may be freely traversed by many proteins (5000 daltons or less), ions, such as Calcium (Ca2+), and metabolites, while the larger molecules are imported by specific translocase [10,11,12]. IMM is highly specialized and permits the passage of small molecules, including oxygen, carbon dioxide, and water. The passage throughout IMM of any other macromolecules is permitted by the action of dedicated translocase, like TIM (translocase of the inner membrane). Within the organelle, OMM and IMM allow the formation of two different compartments: the intermembrane space (IMS) and the matrix [13]. IMS plays a crucial role in regulating different cellular activities, such as mitochondrial respiration, proteins transport, lipid homeostasis and metal ion exchange; while the matrix contains the mitochondrial DNA (mtDNA), mitochondrial ribosomes and multiple diverse metabolic pathways [tricarboxylic acid (TCA) cycle, β-oxidation, and heme synthesis], that modulate the assembly and genes expression, as well as, Ca2+ uptake which is critically important to cellular function [14,15,16] mtDNA is a closed circle double-stranded DNA, without histones and of effective repair mechanism. mtDNA encodes for 37 genes, including 13 components of the mitochondrial electron transport chain (mETC) [17]. Mitochondrial matrix is also the site of a series of enzyme that convert pyruvate and fatty acid in acetyl-CoA. Once produced, acetyl-CoA enters in TCA cycle which produces NADH and FADH2 that are used by mETC [18].
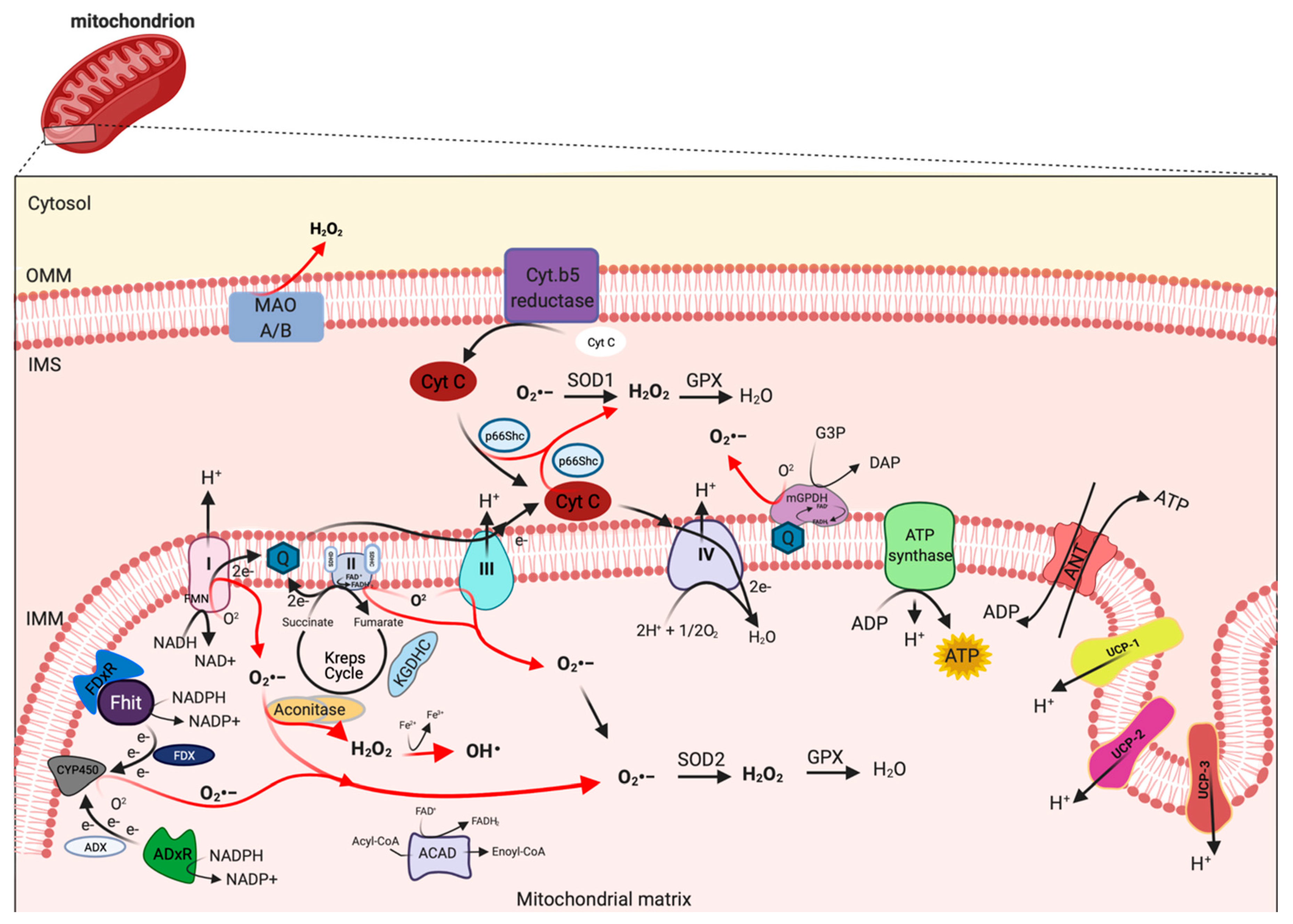
mETC is composed by four complexes (I-IV) that are embedded in the IMM and are responsible to create the electrochemical gradient required to generate ATP. Notably, complex-I (NADH-CoQ reductase) and complex III (cytochrome c reductase) represent the primary sites of production of mitochondrial O2·− (Figure 1). This byproduct of the mitochondrial respiration is also considered the most quantitatively important mtROS source in higher organisms [2]. However, mitochondria are not only an ROS-producing compartment but, in turn, are their targets. In particular, mitochondrial phospholipids and DNA are susceptible to mtROS-induced oxidative damage (Figure 2). Mitochondrial phospholipids loss their permeability and fluidity compromising the functioning of all factor associated to the membrane, in particular mETC members and ion channels. mtDNA loses its integrity, acquiring mutations and reducing in the number of mtDNA copies. In turn, mtDNA mutations can determine further alteration in mitochondrial functioning and signaling that may be determinant for various human diseases [19]. To avoid accumulating oxidative damages, mitochondria have evolved different mechanisms to reduce the mtROS-induced oxidative damage or eliminate injured mitochondria. The mitochondrial antioxidant systems and the mitochondrial stress responses (including mitophagy, mitochondrial unfolded protein response (mtUPR), and apoptosis) represent some classical examples [2]. In addition, it is widely accepted that alterations in mitochondrial functions (like in Ca2+ dynamics and/or in lipid transfer from ER to mitochondria) modulate the mtROS production [2]. Thus, increased levels of mitochondrial Ca2+ activate ROS-generating enzyme and the formation of radicals.
Clearly, mitochondria and mtROS production are primary signal hub for the cells. It is not surprising that diverse studies highlight their involvement in pathogenesis of various diseases, including neurodegenerative disorders, cancer, viral and bacterial infection, cardiovascular diseases, metabolic syndromes, and autoimmune disorders [20]. In particular, it has been proposed that mitochondrial dysfunction and excessive mtROS levels sustain inflammation in these pathological conditions. In these contexts, mtROS-induced inflammation acts as a feedback system creating a stressful environment, where the exacerbation of inflammation provokes tissue damage and becomes a chronic event [20]. Considering the importance of oxidative stress in inflammation, it is easy to speculate that therapeutic manipulations aimed to prevent oxidative damage, targeting the mitochondrial (dys)function and generating an antioxidant power, may represent an opportunity to disrupt the reciprocal relation between mtROS and “mito-inflammation”. A new concept that identifies the compartmentalization of the inflammatory process in which the mitochondrion acts as central regulator, checkpoint, and arbitrator.
The present review will summarize the current literature surrounding the role of mitochondria in ROS production and signaling in both physiology and pathophysiology. In particular, it will be discussed how mitochondrial oxidative stress regulates diverse human diseases throughout the modulation of “mito-inflammation”, with a focus on the therapeutic mitochondria-targeted strategies for avoid mtROS production and reduce the oxidative stress, in order to counteract, or better, prevent the inflammatory state at the base of pathological conditions.
2. Mitochondria: A ROS Production Machinery
Mitochondria are widely recognized as a source of ROS production within most mammalian cells in both physiological and pathological conditions. The generation of mtROS from mitochondria was first discovered during the early 1970s [21,22]. According to estimates, 1–2% of total cellular oxygen consumption is going to ROS production [23].
mtROS are produced as byproduct of bioenergetic metabolism during the process of oxidative phosphorylation (OXPHOS) and formed by one-electron transfers, generating O2•− that can be converted to H2O2 by superoxide dismutase (SOD) enzymes [24]. Multiple sites of mtROS production have long been identified in the organelle and mtROS can take place both in the mitochondrial matrix by the core metabolic machinery present in the IMM and in the intermembrane space [25]. Generation of mtROS mainly takes place at the ETC, located on the IMM, during the process of OXPHOS [26], which includes the major sites of the respiratory chain Complex I (NADH dehydrogenase (ubiquinone), 45 protein subunits), Complex III (ubiquinol-cytochrome c reductase, 10 proteins subunits), and also the dihydrolipoamide dehydrogenase enzyme [27,28]. Complex I generates O2•− by reducing flavin mononucleotide (FMN) site on complex I and reversing electron transfer from the coenzyme Q (CoQ) pool back to complex I [2]. Basically, complex II (succinate dehydrogenase) was not considered a source of ROS per se, instead its contribution to ROS generation is linked to reverse electron transfer, in which electrons are transferred from succinate to ubiquinone through complex II and then back to complex I [29]. Complex II consists of four subunits. Two subunits are located on the matrix side of the IMM: SDHA (succinate dehydrogenase), the flavoprotein subunit covalently bound a FAD cofactor, which removes electrons from succinate; SDHB, the iron-sulfur protein subunit, contains the binding site of the substrate succinate, three clusters (2Fe-2S), (4Fe-4S), and (3Fe-4S) that mediate electron transfer to the ubiquinone molecule; and SDHC and SDHD the two anchor subunits to the IMM, the assembly factors participate in the biogenesis of complex II [30,31,32]. Brand’s group demonstrated that complex II can produce superoxide through flavin adenine nucleotide (FAD) [33]. Indeed, it has been suggested that other less well-described sites may also participates in ROS production including, the electron transferring flavoprotein/ETF:Q oxidoreductase (ETF/ETF:QOR) system of fatty acid β-oxidation [34], dihydroorotate dehydrogenase [35], and proline dehydrogenase [36]. These different mtROS’s sites have distinct signaling roles and subsequently the primary production sites change under different physiological conditions [37]. Indeed, the production of radicals from the mitochondrial respiratory chain is conditioned by multiple factors, including mitochondrial membrane potential, metabolic state of mitochondria and oxygen levels [38].
Other mitochondrial proteins may participate to increment the mtROS production. In particular, the mitochondrial enzyme dehydrogenase of phosphate dehydrogenase (glycerol phosphate dehydrogenase, mGPDH), located at the outer surface of the IMM, is a flavoprotein containing FAD and serves as an electron shuttle linking the cytosolic NADH/ NAD recycling to the mETC implicated in lipid metabolism capable of producing cytosolic NAD+ from the NADH formed in glycolysis [39]. However, it is unequally expressed in numerous mouse tissues mediating the generation of H2O2 [40]. Therefore, other potential sources of mtROS are still more poorly explored, such as acyl-CoA dehydrogenases (ACAD), which are flavoproteins and have increasingly been recognized as oxidant sources in mitochondria, involved in lipid catabolism [41]. Both of these proteins are implicated in mtROS production in tissues during the oxidation lipid-derived substrates [42]. The mitochondrial enzyme adrenodoxin reductase (ADxR)-adrenodoxin (ADX)-cytochrome P450scc (CYP450) system (cholesterol side chain cleavage) is also involved in superoxide generation and is coupled with NADPH in the mitochondrial matrix [43]. The onco-suppressor Fhit protein, imported into mitochondria, interacts with ferredoxin reductase (FDxR), which transfers electrons from NADPH to CYP450 via ferredoxin (FDX), increasing the intracellular superoxide production [44,45]. The redox enzyme, p66shc, regulates the oxidative stress acting a different level. This adaptor protein may induce ROS generation by: (i) Translocating into mitochondria, after PKC-dependent phosphorylation, to transfer electrons from reduced cytochrome C to oxygen [46,47], (ii) activating the Rac-1-dependent plasma membrane NADPH oxidase [48], and (iii) downregulating the expression of antioxidant enzymes, such as glutathione peroxidase-1 and manganese superoxide dismutase (Mn-SOD) [49,50]. The enzymes monoamine oxidase (MAO-A and MAO-B) are located in the OMM and expressed in various mammalian tissues catalyze the oxidation of biogenic amines accompanied by the release of H2O2 [51]. Mitochondrial aconitase is an enzyme positioned in the matrix, the enzyme contains an iron-sulfur cluster that can be oxidized by O2•− or H2O2 generating •OH [52].
Uncoupling proteins (UCPs) are IMM proteins that belong to a family of mitochondrial transporters regulating the proton transport across the IMM. In particular, UCPs determine an inducible proton leak by causing mild-uncoupling events. During mild-uncoupling process, the mitochondrial membrane potential (ΔΨ) and the reduction events in the ETC diminished, thereby resulting in decreases in ROS production from the ETC [53].
UCPs exists in three forms: UCP 1-3. UCP1 is primary found in mitochondria from brown adipose tissue, where it regulates the thermogenesis in response to cold exposure and modulates the ROS production. Consistently, UCP1 knockdown mice are characterized by low levels of ΔΨ, reduced O2 consumption rate, and increased ROS production [54]. In addition, it has been recently demonstrated that UCP1 regulates the ROS production also in kidney. Deletion of UCP1 results in increased oxidative stress in kidney and exacerbates an ischemic condition or a nephrotoxic drug-induced acute kidney injury. Contrarily, viral-based expression of UCP1 suppresses the mtROS production and alleviates the induced kidney injury [55]. UCP2 and UCP3 mRNA expression are ubiquitous and their proteins are present in different tissues, in particular skeletal muscle, central nervous system, pancreas, and spleen. UCP2 and UCP3 protect mitochondrial proteins from endogenous ROS (such as O2•−), and UCP3 and UCP2 knockout models present increased ROS levels. In addition, it has been demonstrated that ROS, 4-hydroxy-2-nonenal (4-HNE), and lipid peroxidation determine activation of UCPs [56,57]. These findings suggest that UCPs exist in a feed-back mechanism, where increased ROS production activates a protective mechanism (uncoupling events) necessary to reduce the ROS formation and the consequent ROS damage. UCP2 has also an important role during insulin secretion in pancreatic β cells. In fact, UCP2 contributes to regulate the intracellular ROS levels of β cells, thereby controlling the excessive glucose-stimulated insulin secretion (GSIS) [58]. UCP2 represent an essential metabolite transporter that regulates the mitochondrial export into the cytosol of metabolites: malate, oxaloacetate and aspartate. Because of this transport, UCP2 prevents the oxidation of glucose and sustains the glutaminolysis [59]. Another protein that preserves an efficient electron transport and control the ROS production is hexokinase (HK) 2. HK2 is recruited to the OMM where it binds itself to the ADP/ATP exchange complex formed by VDAC and ANT. The role of HK2 is to provide continuously ADP in order to generate the ADP/ATP recycling mechanism essential to preserve the optimal respiration rate, thereby preventing the dangerous electron leak producing ROS [60]. In line with this evidence, HK2 knockdown and the dissociation of HK2 from mitochondria increase ROS production and activate the mitochondrial permeability transition (MPT) both in vivo and in vitro [61]. Accordingly, HK2 overexpression decreases the mtROS levels [61].
Sirtuins (SIRTs) are nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylases that play pivotal role in diverse cellular functions, including protein acetylation and deacetylation, metabolism, mitochondrial functioning, and cell survival. In addition, all seven SIRTs isoforms have been associated to process related to antioxidant and redox signaling. In particular, the functions of SIRT1 (the most well-studied member) have been associated to mediate protection from ROS. At demonstration, small molecule activators of SIRT1, such as SRT1720, determine activation of SOD2 levels, accompanied by reduction in the formation of 4-HNE and in protein carbonylation levels [62]. SIRT1 can be also directly activated by oxidative stress and is required for DNA repairing mechanisms following H2O2-induced damage [63]. Similarly, the antioxidant compound resveratrol activates SIRT1 to protect cells from H2O2-induced cell death [64]. Another important SIRTs modulating ROS levels is SIRT3. Downregulation of SIRT3 is associated with increases of ROS production and activation of the protumorigenic transcription factor HIF-1α [65]. Furthermore, SIRT3 may activate the human 8-oxoguanine-DNA glycosylase 1 (OGG1), which is an enzyme crucial for the repairing of the mtDNA [66].
However, the levels of mtROS are conditioned by the specific action of mitochondrial antioxidant systems, recruited to detox the radicals produced. SODs are the antioxidant enzymes that convert O2•− to H2O2 (Figure 1). Two isoforms control the level of O2•−: SOD1/copper-zinc superoxide dismutase (Cu, Zn-SOD) and SOD2/ Mn-SOD isoform. SOD1 is widely distributed throughout the cell, cytoplasm, nucleus, and intermembrane space of mitochondria (IMS), whereas SOD2 is expressed only in the mitochondrial matrix [67] (Figure 1). The rapid conversion into H2O2 is counteract by catalase and glutathione peroxidase (GPX), which neutralize H2O2 in H2O and oxygen [68,69] (Figure 1). Catalase is mainly located to cytosol, indicating that the scavenge capability in mitochondria it is leaved to GPX [70]. So far, in mammals, there have been eight GPX isoforms identified, where GPX1-4 and GPX6 are selenoproteins with a selenocysteine as catalytic moiety and only GPX1 and GPX4 are expressed to mitochondria. The GPX-dependent scavenge capability is associated to use glutathione (GSH) as cofactor and electron source to neutralize H2O2.
3. Mitochondrial Oxidative Stress and Mito-Inflammation in Inflammatory-Related Diseases
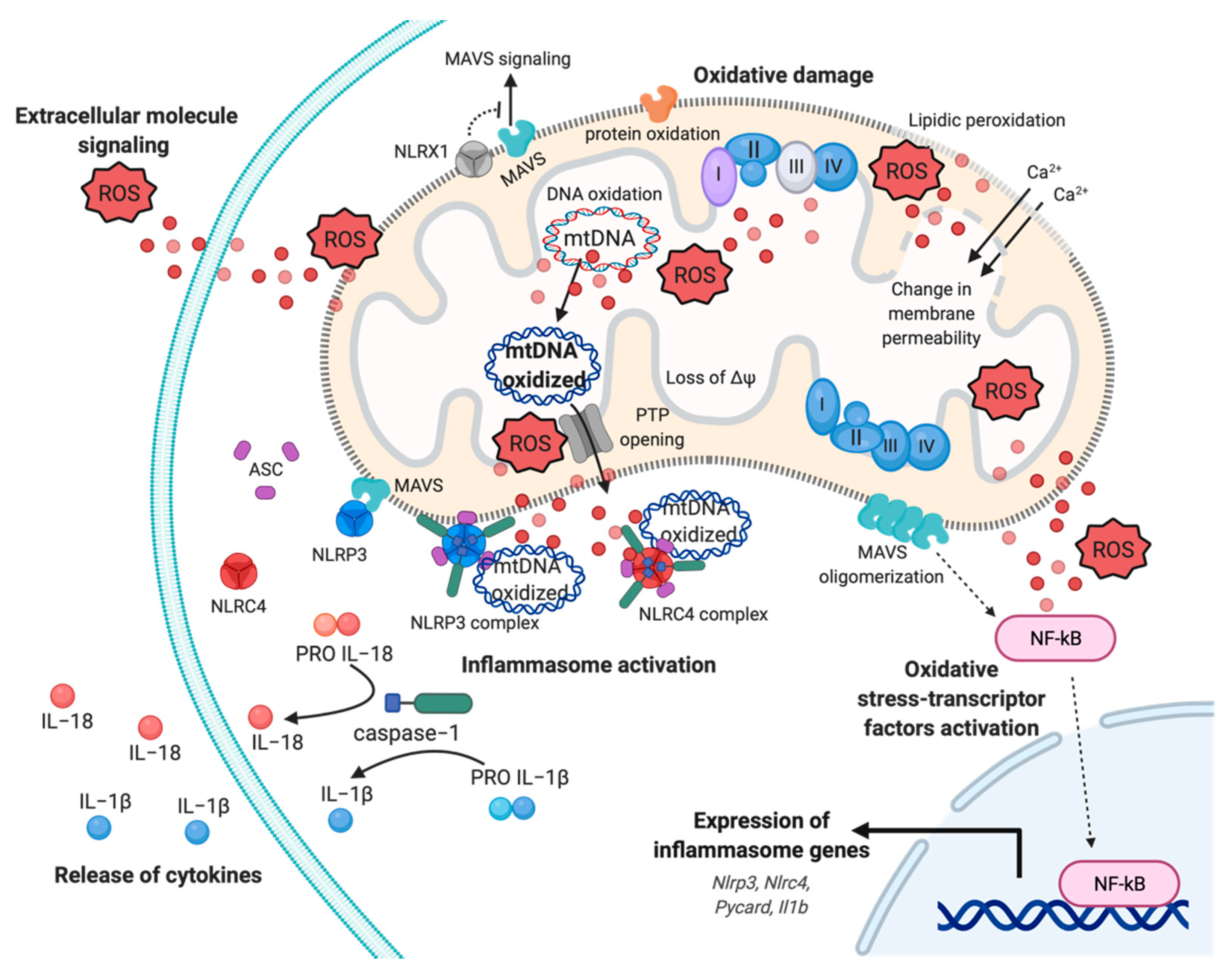
Mitochondrion is historically defined as energy powerhouse and arbiter of cellular destiny. Now, evidence assign to this organelle the supplementary role of “hub” in inflammation, becoming a druggable target to modulate the amplitude of inflammatory response and, eventually, its exacerbation. A new concept is arising around mitochondria and inflammation: The mito-inflammation, a mitochondria-related compartmentalized inflammatory response, where the organelle acts both downstream in intracellular signaling pathways triggered by exogenous pathogen-associated molecular patterns (PAMPs), and as font of mitochondrial damage-associated molecular patterns (mtDAMPs). During mito-inflammation, mtROS, mtDNA, ATP, cardiolipin, and mitochondrial Ca2+ are reversed in the cytosol or in the extracellular milieu to induce the expression and release of numerous pro-inflammatory mediators [71,72,73]. Among mtDAMPs, mtROS may directly act into the organelle, promoting oxidative damage to intra-organelle molecules and mtDNA, or freely move through the OMM (Figure 2). Once in the cytosol, mtROS may trigger the activation of pro-inflammatory signaling pathways, in particular, induce the activation of redox-sensitive transcription factors, such as nuclear factor kappa B (NF-kB), HIF and AP-1, which contribute to production of pro-inflammatory cytokines, including IL−1 and IL−8 (Figure 2) [74,75,76,77,78].
Oxidative damage to mtDNA and mitochondrial proteins alter the OXPHOS activity with several implications on ΔΨ and structure, producing in turn additional mtROS. Oxidized mtDNA is released from matrix mediating impaired mitophagy, Ca2+- and oxidative-dependent mitochondrial PTP opening, and OMM permeabilization [72,79]. In the cytosol, or in the extracellular milieu, mtROS and oxidized mtDNA may also be sensed by germline pattern recognition receptors (PPRs), localized at the plasma membrane and cytosol in immune and tissue resident cells [80]. These receptors are divided in four main sub-families on the basis of their location, function, and specific ligand: Membrane bound Toll-like receptors (TLRs), C-type lectin receptors (CLRs), the cytosolic NOD (nucleotide-binding oligomerization domain)-like receptors (NLRs), and RIG (retinoic acid- inducible gene)-l-like receptors (RLRs) [81].
In macrophages, binding of TLR-1, TLR-2, and TLR-4 induce the tumor necrosis factor receptor-associated factor 6 (TRAF-6) to move to mitochondria to induce antibacterial activity through mtROS [82]. It engages and ubiquitinates the ECSIT (evolutionarily conserved signaling intermediate in Toll pathways), which assembles the membrane arm complex I, promoting the translocation of mitochondria to the phagosomal membrane and the ECSIT-dependent macrophage oxidative burst useful to kill the engulfed microbes [82,83]
While TLR receptor have a plasma membrane localization, RLRs have a cytosol distribution. Among them, RIG-l-like (RIG-I) and melanoma differentiation associated gene 5 (MDA5) receptors can respectively sense short and long viral dsRNA. Once these proteins have been bound by cytosolic viral RNA, they interact with the mitochondrial antiviral signaling protein (MAVS) at the OMM. This coupling activates and promotes MAVS oligomerization leading to the activation of transcription factors IRF3, IRF7, and NF-kB, which induce the synthesis of interferon type-I and of antiviral molecules (Figure 2) [84]. MAVS interacts with NLR family member X1 (NLRX1), a regulator of mitochondrial antiviral immunity that localizes to OMM to block the MAVS-mediated interferon promoter and antiviral activity [85]. The strong connection between mtROS and MAVS has been enforced by evidences that linked an increase in mtROS production to an amplification of RLR-signaling and a mtROS-dependent modulation of the biophysical properties of the OMM required for MAVS oligomerization [86,87,88].
Another class of cytosolic PPRs activated by PAMPs and/or mtDAMPs are NLR proteins, where most of them after activation, form a multi-protein complex termed “inflammasome”, which leads to caspase-1 maturation and secretion of IL−1 and IL−18 [89]. A defined role for mitochondria in NLR activation and function has been confirmed for NLRX1, NLR family pyrin domain containing 3 (NLRP3), and NLR family CARD domain containing 4 (NLRC4/IPAF) [90]. The activation of the well-characterized inflammasome NLRP3 is split in two phases: Priming and activation. In the priming step, the expression and post-translational modifications of different inflammasome components (including NLRP3, NLRC4, pro-IL−1, pro-IL−18) are induced in response to pro-inflammatory stimuli and consequent NF-kB activation (Figure 2) [91]. The activation is controlled by a wide range of signals and requires a physical interaction with mitochondria for the assembly of NLRP3-inflammasome and the subsequent auto-cleavage of pro caspase-1, responsible for the production of mature cytokines IL−1 and IL−18 (Figure 2) [92]. Mitochondrial localization, due by MAVS and mitochondrial-anchored protein ligase (MAPL) binding, has a central role in the modulation of inflammasome response not only because of its role of scaffold, but also because this interaction favors the activation of NLRP3 through the release of mtROS and mtDAMPS (e.g., mtDNA) [93,94].
Moreover, it has been shown how the priming and the activation phases of NLRP3 inflammasome is linked to the new synthesis of mtDNA for inflammasome activation, highlighting the role of mitochondria in both the steps required [95]. In addition, NLRC4/IPAF inflammasome may be activated by oxidized mtDNA. This inflammasome is known to be activated by many pathogens, such as Pseudomonas aeruginosa, and their products by activation of TLRs on the plasma membrane via the microbial type III secretion system (T3SS) [96,97]. Nevertheless, few years ago an alternative mechanism of activation promoted by direct binding of oxidized mtDNA has been identified in bone-marrow macrophages [98].
The contribution of mtROS in inflammation is strictly influenced by mitochondrial status. mtROS levels increase especially during mitochondria malfunctioning, that cover a wide range of abnormalities from accumulation of unfolded proteins or excessive Ca2+, to OXPHOS impairment. Besides their role of signaling molecules at low concentrations and of inflammatory response activators at moderate concentrations, when mtROS concentration becomes too high, they are also responsible of cellular injuries. In order to avoid cell damage, the production of mtROS is fine-tuned by both mitochondrial antioxidant systems and mitochondrial stress responses that trigger the restore of mitochondrial homeostasis. Functional fusion complementation, mitophagy, and mitochondrial unfolded protein response (UPRmt) intervene to recover and preserve the mitochondrial homeostasis to regulate metabolism and innate immune response and cell viability. Mitochondrial fusion compensation optimizes the functional efficiency of organelle under stressful conditions, allowing the exchange of materials among partially damaged mitochondria [99]. Mitophagy neutralizes excess of mtROS, oxidized mtDNA, and other mtDAMPs relevant for inflammation, removing dysfunctional mitochondria mediating lysosomal degradation [100]. Mitochondrial-targeted kinase PINK1 and E3 ubiquitin ligase Parkin are the principal actors in the mitophagic response [101]. Parkin is recruited to OMM by altered mitochondrial import of PINK1 in depolarized mitochondria, where catalyzes the ubiquitination of OMM proteins to sequester the organelle in autophagosome, while PINK1 contributes to strengthen the mitophagy, phosphorylating both parkin and ubiquitin; and recruiting the mitophagic receptors NDP-52 and optineurin to mitochondria [102,103]. Finally, the mitochondrial unfolded protein response (UPRmt) is a transcriptional response activated by dysfunctional mitochondria, where upon mitochondrial stress, Activated Transcription Factor 5 (ATF5) fails to be imported into mitochondria and moves to nucleus to induce the transcription of mitochondrial chaperones and proteases, ROS detoxification protein, and innate immune genes [104,105,106]. UPRmt induces also the synthesis and secretion of nuclear-encoding and mitochondrial derived mediators, called mitochondrial cytokines or mitokines, able to reshape cell metabolism and viability [98,107,108]. Although, the signaling role of these mitokines has not yet been fully established, they may be considered pivotal actors for the modulation of systemic response, such as metabolism and or inflammation, during a disease progression. Furthermore, the excess of mitochondrial Ca2+ levels is critical for oxidative stress and inflammation. Perturbation of mitochondrial Ca2+ signaling boosts the mtROS production with consequent repercussions on the mitochondrial stress responses, inflammasomes activations and release of proinflammatory mediators [109]. The mtROS production correlates with the mitochondrial metabolic rate, which in turn, determines the effects of mitochondrial Ca2+ signaling on mtROS levels [110]. Mitochondrial Ca2+ signaling may promote mtROS production: (i) directly, by stimulating mitochondrial resident ROS-generating enzymes, such as -ketoglutarate and glycerol 3-phosphate dehydrogenase [111,112]; (ii) indirectly, by the Ca2+-dependent activation of nitric oxide synthase, which mediating nitric oxide blocks the mitochondrial complex IV; and (iii) or via reverse electron transport induced by Ca2+-dependent mitochondrial membrane depolarization [113,114]. In turn, mtROS contribute to perturb the Ca2+ signaling affecting the activity of receptors, Ca2+-effectors, and molecules involved in Ca2+ signaling pathways, reshaping intracellular, and compartmentalized Ca2+ signals [110]. An example is given by mitochondrial Ca2+ uniporter (MCU), which post-translational modification on cysteine at position 97, mediated by oxidative stress, induces clustering and persistent channel activation, leading to increased mitochondrial Ca2+-uptake [115].
The loss of mitochondrial homeostasis and the excessive accumulation of mtROS have been demonstrated to be linked to development of various pathologies, such cancer, diabetes, neurodegeneration, and cardiovascular diseases. This review is aimed at discussing some pathological conditions associated with an inflammatory state, which is strictly conditioned by the abnormal presence of these two factors, where mitochondrial dysfunction and mtROS become the principal actors in these pathological sceneries.
3.1. Cancer
The mtROS are involved in the different phases of tumorigenesis. In the initiation and promotion stage, mtROS induce mtDNA oxidation and damage, promoting mutations and structural alterations to mtDNA with consequent effects on gene expression and mitochondrial signaling. All these conditions, in turn, favor the cell proliferation and promote the apoptotic evasion in the progression stage. Wang et al., examining human lung, bladder and head and neck cancers, showed that the mutation in mtDNA were about 20-200 times more frequent respect to nuclear DNA [116]. The mtROS are responsible not only for the genetic instability but also for influencing the inflammatory microenvironments of tumors and to induce the activation of oxidative-sensitive transcription factors, such as HIF, which modulate the energy status in cancer cells [117,118]. Cancer cells adopt a different metabolism to produce energy, where the use of aerobic glycolysis results is preferred with respect to mitochondrial oxidative phosphorylation. This metabolic switch is known as the “Warburg effect” and is characterized by the fact that cancer cells use glucose and excrete lactate. This phenomenon was unveiled in the 1920s by Warburg and Cori. Subsequently, they also observed that mitochondria of cancer cells were dysfunctional. Several other investigations confirmed the Warburg effect throughout the years. Fantin et al. in 2006 demonstrated that it was possible to prevent the tumor progression by blocking the conversion of pyruvate into lactate by inhibiting lactate dehydrogenase [119]. In the same year, another work demonstrated that by activating the mitochondrial metabolism, in particular OXPHOS, the cancer growth arrested [120]. Therefore, it is well assumed that cancer cells avidly consume glucose for their maintenance. Consistent with this, the Warburg effect has been observed in many cancer types. In colorectal carcinoma, a strong correlation was observed between the malignant potential of the tumor and the levels of the expression of the glucose transporter (GLUT) proteins, responsible for the cellular glucose uptake [121]. Increased levels of GLUT have been also evidenced in human breast cancer [122]. Another example may be found in lung cancer, where a great dependency on glycolysis and a parallel impairment in mitochondrial respiration was found [123]. Finally, it has been demonstrated that aerobic glycolysis may be not only a consequence of carcinogenesis, but also favors the transformation process [124].
However, last decades have been characterized by very great improvements in the field of the scientific research. These advances have permitted a deeper and more detailed analysis of the continuous metabolic changes that happen in cancer cells, and in particular, it has been demonstrated that the Warburg effect is not consistent in all cancer types. In fact, in a range of tumor types, the mitochondria remain functional [125,126]. This happens in ovarian cancer, where it has not only been found to have a high dependence on mitochondria of cancer cells, but also that anti proliferative agents reduce the tumor growth by suppressing the mitochondrial metabolism [127]. By analyzing the metabolism in non-small cell lung cancer (NSCLC) patients in vivo, a great metabolic heterogeneity has been observed between different tumor regions: Some regions were characterized by high glycolytic rates, while others presented elevated levels of complete glucose oxidation [128]. High mitochondrial functioning has been found in liver cancers and in metastatic brain tumors [125,126,129]. Overall, these findings suggest that during the tumorigenesis, cancer cells may undergo a metabolic reprogramming, in which the Warburg effect represents a key event. Although, changes in glucose metabolism during tumor initiation and growth affect cellular processes that may generate ROS [130].
Indeed, oncogenes and tumor suppressor genes in cancer cells promote mtROS production, including oncogenic H-Ras that induces mtROS formation for mitogenic signaling [131,132]. This increment in ROS levels is due to mitochondrial hyperactivity mediated by major changes in Ψ and Ca2+-affinity/accumulation. The oncogenic mitochondrial hyperactivity is not sustainable for all the stages of tumor process. mtROS accumulation may damage the organelle, exacerbating the mitochondrial oncogenic stress. Under these conditions, the cancer cells active the mitochondrial stress responses, such as mitophagy and UPRmt, to restore the mitochondrial integrity potentiating the cellular survivor and resistance to stress [133]. mtROS produced by cancer cells are signaling molecules released in tumor microenvironment to condition cancer-associated cells and tumor-infiltrating leukocytes [117]. mtROS affect the function of cancer-associated fibroblasts, inducing differentiation and metabolic reprogramming to sustain and augment the tumorigenesis [134,135,136]. Although T-cells use complex I and III of ETC to generate a low level of mtROS to induce T-cell activation, the excessive accumulation of ROS in the tumor microenvironment has an immunosuppressive effect on the tumor-infiltrating immune cells [134]. High levels of ROS block the proliferation of infiltrating T-cells and inhibit their anti-tumor function [137]. These effects are counteracted with mtROS scavengers, such as MitoQ and MitoTEMPO, which restore the T-cell activation [138]. The controversial role of ROS in tumor microenvironment is represented by the fact that ROS are also produced by tumor-associated macrophage and myeloid-derived suppressor cells. In the case of macrophages, the ROS are implicated in cell activation and in killing processes, while for the myeloid-derived suppressor cells, the ROS are implicated as immunosuppressive signals to regulate immune cell functions [139,140].
3.2. Pulmonary Diseases
The mtROS formation is excessive in Cystic Fibrosis (CF) cells [73]. This pathology is characterized by genetic defects of cystic fibrosis transmembrane conductance regulator (CFTR) gene and recurrent pulmonary infection, which cause chronic inflammation of airways and respiratory failure. The persistent bacterial infections, in particular of P. aeruginosa, induce mitochondrial Ca2+-overload and mtROS formation in human CF bronchial cells, which in turn promoted the activation of NLRP3 and NLRC4 inflammasome and consequent release of IL−1 and IL−18 [109]. This interplay between NLRP3 and NLRC4 inflammasome has also been observed in CFTR-null mice, in alveolar CF macrophage, and in CF neutrophils, due to mtROS and direct binding with oxidized mtDNA [98,141]. Limiting the abnormal mitochondrial Ca2+-uptake in CF and mediating pharmacological inhibition of MCU, with KB-R7943, attenuated the pathogen-dependent ROS production and mitochondrial dysfunction, preventing the activation of NLRP3 inflammasome and UPRmt in vitro and in vivo [142].
The high levels of mtROS promote additional mitochondrial impairments in a feed-back stimulatory manner that sustains the activation of oxidative-sensitive transcription factors, which exacerbate the chronic pulmonary inflammation [2,143]. In fact, NF-kB, HIF-1, and AP-1 are hyperactivated in CF, favoring an elevated production of cytokines and chemokines, such as IL−8, and priming the inflammasome NLRP3 and its members. Their activation is, due to intrinsic defects, associated with defective CFTR, such as the oxidative stress and abnormal intracellular Ca2+ signaling [144,145,146,147,148,149].
The oxidative-sensitive nuclear recruitment of NF-kB also has a pivotal role in the chronic obstructive pulmonary disease (COPD). Here, its involvement is a determinant in sustaining the chronic inflammatory response through the up-regulation of different pro-inflammatory and chemotactic genes, including IL−8 and TNF-α, both important to COPD pathogenesis [150].
3.3. Gastrointestinal Disorders
In the gastrointestinal (GI) tract the NOX and xanthine oxidase (XO) systems represent the primary ROS producers. Notably, XO, which is mainly expressed in the liver and in the intestinal mucosa, catalyzes the oxidation of hypoxanthine (HX) to xanthine and the conversion of xanthine to uric acid. During these two reactions O2·− is produced. Excessive ROS production in GI is linked to inflammation and may be the cause of severe cellular damages that can disrupt the tract barrier of GI, thus determining gut permeability and contributing to different GI disorders. Gastroesophageal reflux (GR) represents a very common disorder, and all elements associated with GR, such as bile salts and acids, have been found to be potent ROS inducers as well as a primary cause of loss of antioxidant defenses. As a result, the squamous epithelium of esophagus undergoes erosion and ulceration [151]. Accordingly, by injecting antioxidants in a rat duodenogastroesophageal reflux model, ROS amounts and esophagitis resulted reduced [152]. Gastritis represents the inflammation of the stomach mucosa. When gastritis is chronic, it may be a cause of peptic ulcer disease. Different conditions provoke gastritis, such as bacterial infection, stress, cigarette smoke, and excessive alcohol consumption. All of them contribute to accumulating ROS and activate inflammation, with a consequent infiltration of neutrophils and macrophages in the gastric mucosa that can exacerbate the oxidative stress [153]. Interestingly, it has been demonstrated that RNS results in being protective for gastric tract. Indeed, NO radicals stimulate mucus secretion and inhibit the expression of adhesion molecule in the gastric epithelium. In this manner, ROS-mediated ulceration is prevented as neutrophils cannot adhere to the gastric mucosa [154]. In human intestinal diseases, the intracellular ROS levels are increased, generating cell stress and a reduction in the diversity of microbial community in the gut [155]. An increase of 10-100 times in mucosal ROS concentration has been observed in ulcerative colitis, in gastroduodenal mucosal inflammation, Chron’s, and inflammatory bowel diseases [156,157,158]. The epithelium barrier works to protect itself and the gastrointestinal district by stressors, such as pathogens, secreting mucus and antimicrobial peptides and low levels of ROS [159]. In this, the mitochondrial signaling and concomitant metabolic changes contribute significantly to intestinal equilibrium. In bowel epithelium, the mitochondrial impairment induces a metabolic imbalance that causes reduction of stemness and generation of dysfunctional Paneth cells, which predicts the Chron’s disease recurrence [160]. Indeed, an excessive increase in ROS production in gut has detrimental effects on the gastrointestinal membranes with consequent repercussion on tissue permeability and microbial biodiversity. Many inflammatory-related gastrointestinal disorders are associated with dysbiosis, an imbalance of gut microbiome that is linked to the release of PAMPs that induce inflammation [161].
3.4. Cardiovascular Diseases
The importance of oxidative stress in cardiovascular diseases (CVDs) has been continuously demonstrated. Overproduction of ROS and other oxidative stress-related factors are indeed the primary cause of diverse CVDs, in particular during atherosclerosis and myocardiac infarction. Low-density lipoproteins (LDL) are major cholesterol carriers, and their levels play a crucial role in atherosclerosis. Increased oxidative stress determines conversion of LDL in oxidized LDL (ox-LDL), which represent pathogenic, immunogenic, and atherogenic particles. In this conformation, LDL can enter the monocyte-macrophage system present in the arterial wall and cause the atherosclerotic process [162,163]. In particular, ox-LDL may increase the expression of the intercellular adhesion molecule-1 (ICAM-1) and vascular-cell adhesion molecule-1 (VCAM-1), improving the adhesion of monocytes to the arterial endothelium [164]. Ox-LDL also determine the secretion of monocyte chemotactic protein-1 (MCP-1) and monocyte colony stimulating factor (mCSF) by stimulating smooth muscle cells (SMCs) and endothelial cells (ECs) migration and proliferation [165]. Increase in inflammatory levels during atherosclerosis may also be mediated by oxidative stress. High ROS levels, in particular H2O2, directly stimulate macrophages to express chemokines and inflammatory cytokines to boost the inflammatory response at the site of the atherosclerotic endothelial injury. The immune receptor NLRP3 is also involved in atherosclerosis. Consistently, NLRP3 components are present in SMCs and ECs [166]. Most importantly, high NLRP3 levels characterize patients with coronary atherosclerosis and correlate with the severity of disease and with concomitant. Interestingly, increased NLRP3 levels also correlate with the presence of concomitant risk factors in patients with coronary artery disease. In particular, it has demonstrated that the presence of oxLDL primes and activates NLRP3 components. Accordingly, by treating endothelial cells with an inhibitor of component of NLRP3 ASK1 (GS-4997) it is possible to attenuate the inflammatory process, the ROS production, and, most importantly, the effects of oxLDL on the cholesterol efflux [167]. Cigarette smoking represent one of the major risk factors for the development of atherosclerotic plaque. It has been demonstrated that nicotine improves the mtROS levels and activate a molecular pathway where NLRP3 and pyroptosis cooperate to promote monocyte recruitment, release of pro-inflammatory factors, and pyroptotic cell death of ECs [168]. In particular, OGG1 is responsible for removing 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-OH-dG), the most abundant form of oxidative DNA damage [169]. It has been demonstrated that OGG1 downregulation leads to increased mtDNA damage as well as activation of NLRP3 inflammasome in atherosclerotic lesion [170]. Consistent with this, increased mtDNA damage levels have been observed in human atherosclerotic plaques (Yu et al., 2013).
Ischemic cardiac injury happens during myocardial infarction and represents the result of an occlusion of a coronary vessel, in which cardiac cells suffer a hypoxic condition, ATP depletion, and mitochondrial impairments. It has been demonstrated that during the ischemic condition complexes I and III result compromised, favoring the excessive mtROS production. This situation provokes severe damage to different phospholipid components of mitochondrial membranes, such as cardiolipin, leading to impairments and destabilization of ETC members, depletion of ATP production, mitochondrial permeability transition, and finally, cell death. Accordingly, to these findings, for several years, a quick restore of the blood supply (reperfusion) was though essential to limit the cellular damage. Consistently, reperfusion restores oxygen delivery, ATP production, and mitochondrial Ca2+ levels. At the same time, the reintroduction of blood rich in oxygen is deleterious for hypoxic tissue, which triggers an uncontrolled mtROS production by complexes I and III, determining mPTP opening, mitochondrial membranes permeabilization, and cell death [171,172,173]. Recently, it has been proposed that during ischemia/reperfusion (IR), the ETC may be not the only source of mtROS, indicating that other mtROS producers are involved [174].
As reported above, MAO are proteins localized on the OMM and are responsible for the generation of H2O2 [175]. It has been demonstrated that during IR the MAO-A expression increases in cardiomyocyte and it is responsible for producing excessive H2O2 production, which provokes mitochondrial damage. Interestingly, it has been proposed that during IR, the excessive ROS production mediated by MAO-A is sufficient to block the activity of sphingosine kinase, degrades sphingosine-1-phosphate (S1P) and increases the ceramide levels [176]. Notably, S1P suppresses the ceramide-mediated cell death [177]. Consistent with these findings, inhibition of MAO-A reduces mitochondrial lipid peroxidation and limits the cardiac infarct size, representing a potential therapeutic intervention against cardiac IR [178].
Another important contributor for mtROS production is the protein p66Shc, which is expressed in cardiomyocyte and is responsible for regulating different cellular processes [46,179,180]. Genetic silencing of p66Shc reduces the mtROS production and limits the lipid peroxidation, determining cardioprotective effects during IR model.
Among the different mechanisms through which ROS provoke IR injury of the heart, ROS-mediated inflammasome activation has an important contributor. In fact, mediator of inflammasomes, such as IL−1 and ASC, have key roles during cardiac IR injury and their inhibition exerts protective role against myocardium in mice exposed to IR [181,182]. In addition, it has been suggested that the role of NLRP3 in cardiac IR may be related to the member of α-arrestin protein superfamily, Thioredoxin-interacting protein (TXNIP). In a recent work, it has been demonstrated that during myocardial IR the interaction between TXNIP and NLRP3 resulted in an increase, and that following intramyocardial injection of TXNIP siRNA, the NLRP3 activation as well as the infarct size was reduced [183]. Moreover, when mtROS scavengers were added, the association between TXNIP and NLRP3 and the inflammasome activation were abrogated, suggesting that mtROS are responsible for activating the TXNIP -mediated activation of NLRP3 [183].
3.5. Neurodegenerative Disorders
Alzheimer’s (AD), Parkinson’s (PD), and Huntington Disease (HD) are neurodegenerative disorders with significant motor and cognitive decline, and, to date, incurable diseases associated to aging [184].
The progression of neurodegenerative diseases is associated with neuroinflammation promoted by protein aggregation and/or neuronal damage via damage-associated molecular patterns (DAMPs) and consequent activation of PRRs, such as CR3 and TLR-4, with the pivotal contribution of mtROS [185,186]. The activation of these receptors induces neuroinflammation mediating the recruitment pro-inflammatory signaling transducers, such as NLRP3 and NF-kB, which, activated by oxidative stress, promote the synthesis and release of pro-inflammatory mediators [187,188,189]. The susceptibility of neural tissue to oxidative stress is not only due to elevated ROS production but also reduced regeneration capacity of neurons and their modest antioxidant defenses [190]. The immune responses in neurodegenerative diseases are regulated by microglia cells, which mediate both protective and deleterious responses [191].
In AD, the release of pro-inflammatory cytokines, IL−1, is mediated by assembled NLRP3 inflammasome in activated microglia in response to aggregation of amyloid- proteins and consequent plaque formation [192,193,194]. Indeed, amyloid- has been shown to target the mitochondria, affecting the mitochondrial enzymes, alcohol dehydrogenase and cytochrome C oxidase [195]. A direct link between mtROS and the pathology has been observed also in transgenic mouse model and in human brain tissue [196,197]. The neurotoxicity in AD is also favored by activation of NLRP3 inflammasome, which induces tau abnormality favoring tau phosphorylation and aggregation [198,199]. Its deactivation has protective effects in AD [200]. Mitochondrial dysfunction, mtROS formation, and consequent NLRP3 inflammasome activation are also associated with bipolar and intracerebral hemorrhage disorders [201,202].
The progression of pathology is also conditioned by insidious aspect of mitochondrial signaling under oxidative stress, such as the persistent activation of UPRmt, which may exacerbate the disruption of neurons in vivo or induce the release of neuroprotective mitokines in AD, such as humanin [202,203]. Humanin reduce the neuroinflammation and the oxidative stress-induced injury in AD, diminishing the level of protein aggregation and of plaque deposition [204,205].
The production of ROS and the activation of NLRP3 is sustained and elevated also in PD, where microglia cells, in response to Lewy bodies and -synuclein protein, also release cytokines IL−6, TNF, and prostaglandins [206,207,208]. In particular, the alterations in complex I of ETC are the primary source of mtROS formation in PD patients, causing nigrostriatal degeneration observed in PD [209,210,211].
In HD patients, it is the activity of complex II of ETC that is diminished [212], with significant repercussions on mitochondria and mtDNA integrity, the cause of massive inflammation that damages the striatum [213]. In this area, only the pro-inflammatory cytokines IL−1 and TNF were increased, while IL−6, IL−8, and MMP-9 were up-regulated in cortex and cerebellum, two districts that normally are spared in HD [214].
The direct involvement of mitochondria in AD and PD is confirmed by the compartmentalized ROS suppression mediating mitoTEMPO, where the treatment reduced the expression of pro-inflammatory mediators, IL−1, IL−6, TNF, iNOS, and COX-2 in murine microglia cells, limiting the nuclear translocation of NF-kB and MAPKs [215].
3.6. Diabetes
Diabetes is described as a multifactorial and complex metabolic syndrome characterized by deficit in metabolism of carbohydrates, fats, and proteins. Consistent with this, hyperglycemia represents the main pathological condition.
A hyperglycemic condition can determine increase the mitochondrial flux determining increase in oxidative stress [216]. This elevation may be due to an increase in production of ROS/RNS (such as ONOO−, OH, 8-OHdG, and H2O2) through the ETC as well as decreases of antioxidant defense systems, in particular, catalase (CAT), glutathione peroxidase (GSH), and SOD [217,218].
In addition to this, nutrient overload determines increases in insulin synthesis demand, which forces -cells to product insulin [219]. ER is the primary organelle involved in protein synthesis. An uncontrolled insulin demand promotes an increase in disulfide bond formation for correct protein folding at ER, a condition that increases the ROS formation [220]. As a consequence, protein folding fails and ER functions are impaired [221]. Mitochondrial oxidative stress and ER stress create a vicious cycle that increases oxidative stress and affects further the functioning of these determinant organelle. Considering that both ER and mitochondria are crucial for controlling the glucose levels in blood and in -cells, the hyperglycemic condition becomes chronic. When this situation occurs, it is highly harmful for both insulin secretion and survival of -cells and diabetes may develop.
In addition, the excess in ROS production may deregulate the expression of important key factors (such as MafA and PDX-1) necessary for the activity of different genes involved in insulin generation [222].
ROS-dependent hyperglycemic damage is also associated with other cellular processes, in particular inflammation, and one of the primary targets of hyperglycemic damage are the vascular endothelial cells (VES). Notably, VES are not able to regulate the intracellular glucose concentration and when blood presents excessive amounts of glucose, they cannot prevent the glucose entry. This condition, together with uncontrolled ROS levels, promote damage and vascular complications [223,224]. Consistently, it has been demonstrated that in human aortic endothelial cells, the inhibition of ROS production by uncoupling of ETC reduced the chemokine IL−8 expression induced by high level of glucose [225]. The hyperglycemia-induced monocyte-endothelial adhesion and successiv transmigration was also enhanced in human coronary artery, where the mtROS promoted the redox-sensitive NF-kB activation and the expression of adhesion molecules, such as ICAM-1 and VCAM-1 [226].
The activation of the nuclear transcription factor NF-kB in diabetes is the result of ROS generation promoted by the excessive glucose and by saturated fatty-acids. TLR-4 senses the excess of saturated fatty-acids favoring the oxidative-dependent up-regulation of inflammatory genes, which determine the markedly release of pro-inflammatory mediators involved in monocyte adhesion and chemotaxis, such as IL−8 [227,228].
Vascular tissue is not the only target for ROS-driven inflammation in diabetes. The presence of inflammatory partner has also been identified in pancreas. Indeed, activated macrophages are present in the pancreas of diabetic persons and are responsible for the damage and cell death of pancreatic islet through ROS [229]. In this context, it has been proved that by using ROS scavenger, it is possible to reduce the inflammatory events and increase the pancreatic cell survival [229].
4. Conclusions
In recent years, the mitochondria have been increasingly assuming a key role in the pathogenesis of human diseases. Always considered the cellular powerhouse, the mitochondrion also acts as a strategic hub in diverse cellular process, such as inflammation. The mitochondrion is involved in the regulation of inflammation through multiple mechanisms, not only by releasing mDAMPs but also by affecting the mitochondrial stress responses or influencing the inter-organelle communications between ER and/or nucleus. Their involvement in the inflammation has suggested a new concept, the mito-inflammation, a compartmentalized inflammatory response that may be targeted to cure a wide range of inflammatory-associated pathogenic conditions. Consistently, different studies show how mtROS are the principal actors, fuel that feeds and sustains this process. It has been established that mtROS are endogenously signaling molecules, involved in a complexity of interactions. At low levels, they are necessary to regulate cellular functions and stimulate the mitohormesis, while their excessive accumulation is damaging to cells. Understanding the mechanisms by which mtROS communicate will aid in the development of mitochondrial targeted therapies for human inflammatory-related diseases. The mitochondrial antioxidant is the compartmentalized antioxidant strategy finer to counteract the ROS production to mitochondria. To this, targeted antioxidants have been developed, such as MitoQ and SkQ1 that reduce the severity of many inflammatory-related pathologies [230]. The mitochondrially antioxidant strategy has demonstrated cardioprotective efficacy decreasing the mtROS accumulation in many cardiac diseases [231]. MitoTEMPO and MitoQ protect from reperfusion injury and heart failure [232,233,234,235], while the mitochondrial Szeto-Schiller-31 peptide prevents oxidative stress and mitochondrial dysfunction targeting cardiolipin [236]. mtROS formation may be inhibited using MAO inhibitors. Several MAO inhibitors are clinically available for the treatment of neurological disorders and are currently the better compartmentalized strategy to counteract the mtROS formation in clinical use [237].
Many questions regarding the molecular function of mtROS and of mito-inflammation remain to be addressed. These findings and those that will be obtained in the coming years will increasingly reinforce the contribution of mitochondria and the mitochondrial quality control machinery in the mito-inflammation regulation, warranting the efforts of researchers to develop new mitochondria-targeted therapies.
Author Contributions
All authors contributed substantially to discussions of the content. All authors contributed to writing the article and to reviewing and/or editing the manuscript before submission. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by grants from Italian Cystic Fibrosis Research Foundation FFC#12/2010 and #19/2014, Italian Association for Cancer Research (AIRC, IG-23670), Telethon (GGP11139B), local funds from the University of Ferrara, and the Italian Ministry of Education, University and Research (PRIN Grant 2017E5L5P3) to PP. SP is supported by Fondazione Umberto Veronesi. AR is supported by Italian Cystic Fibrosis Research Foundation grant FFC #20/2015, local founds from University of Ferrara, FIR-2017, the Italian Ministry of Health (GR-2016-02364602) and the Italian Ministry of Education, University and Research (PRIN Grant 2017XA5J5N). PP is grateful to Camilla degli Scrovegni for continuous support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The Authors declare that there are no competing interests in relation to this work.
References
- Kaludercic, N.; Deshwal, S.; Di Lisa, F. Reactive oxygen species and redox compartmentalization. Front. Physiol. 2014, 5, 285. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81 Pt B, 281–293. [Google Scholar] [CrossRef]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Aguilera, A.; Rull, A.; Rodriguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menendez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediat. Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Toit, A. Transcription: A mitochondrial switch between transcription and replication. Nat. Rev. Mol. Cell Biol. 2015, 16, 128. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [Green Version]
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1090–1097. [Google Scholar] [CrossRef]
- Loschen, G.; Flohe, L.; Chance, B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett 1971, 18, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Brand, M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012, 287, 42921–42935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.A.; Krishnan, N.; Becker, D.F.; Tanner, J.J. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J. Biol. Chem. 2007, 282, 14316–14327. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Mracek, T.; Drahota, Z.; Houstek, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesina, P.; Kholova, D.; Bolehovska, R.; Cervinkova, Z.; Drahota, Z.; Houstek, J. Glycerophosphate-dependent hydrogen peroxide production by rat liver mitochondria. Physiol. Res. 2004, 53, 305–310. [Google Scholar]
- Cardoso, A.R.; Chausse, B.; da Cunha, F.M.; Luevano-Martinez, L.A.; Marazzi, T.B.; Pessoa, P.S.; Queliconi, B.B.; Kowaltowski, A.J. Mitochondrial compartmentalization of redox processes. Free Radic. Biol. Med. 2012, 52, 2201–2208. [Google Scholar] [CrossRef]
- Lambertucci, R.H.; Hirabara, S.M.; Silveira Ldos, R.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell. Physiol. 2008, 216, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Hanukoglu, I.; Rapoport, R.; Weiner, L.; Sklan, D. Electron leakage from the mitochondrial NADPH-adrenodoxin reductase-adrenodoxin-P450scc (cholesterol side chain cleavage) system. Arch. Biochem. Biophys. 1993, 305, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Marchi, S.; Fotino, C.; Romagnoli, A.; Huebner, K.; Croce, C.M.; Pinton, P.; Rizzuto, R. Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 12753–12758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapasso, F.; Pichiorri, F.; Gaspari, M.; Palumbo, T.; Aqeilan, R.I.; Gaudio, E.; Okumura, H.; Iuliano, R.; Di Leva, G.; Fabbri, M.; et al. Fhit interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis of cancer cells. J. Biol. Chem. 2008, 283, 13736–13744. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Rogers, B.; Kachi, S.; Hackett, S.F.; Sick, A.; Campochiaro, P.A. Reduction of p66Shc suppresses oxidative damage in retinal pigmented epithelial cells and retina. J. Cell. Physiol. 2006, 209, 996–1005. [Google Scholar] [CrossRef]
- Haga, S.; Terui, K.; Fukai, M.; Oikawa, Y.; Irani, K.; Furukawa, H.; Todo, S.; Ozaki, M. Preventing hypoxia/reoxygenation damage to hepatocytes by p66(shc) ablation: Up-regulation of anti-oxidant and anti-apoptotic proteins. J. Hepatol. 2008, 48, 422–432. [Google Scholar] [CrossRef]
- Kunduzova, O.R.; Bianchi, P.; Parini, A.; Cambon, C. Hydrogen peroxide production by monoamine oxidase during ischemia/reperfusion. Eur. J. Pharmacol. 2002, 448, 225–230. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Dlaskova, A.; Clarke, K.J.; Porter, R.K. The role of UCP 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim. Biophys. Acta 2010, 1797, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine 2019, 49, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Echtay, K.S.; Blaikie, F.H.; Asin-Cayuela, J.; Cocheme, H.M.; Green, K.; Buckingham, J.A.; Taylor, E.R.; Hurrell, F.; Hughes, G.; et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: Studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. J. Biol. Chem. 2003, 278, 48534–48545. [Google Scholar] [CrossRef] [Green Version]
- Robson-Doucette, C.A.; Sultan, S.; Allister, E.M.; Wikstrom, J.D.; Koshkin, V.; Bhattacharjee, A.; Prentice, K.J.; Sereda, S.B.; Shirihai, O.S.; Wheeler, M.B. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 2011, 60, 2710–2719. [Google Scholar] [CrossRef] [Green Version]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Wyatt, E.; Chawla, K.; Tran, M.; Ghanefar, M.; Laakso, M.; Epting, C.L.; Ardehali, H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol. Med. 2012, 4, 633–646. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A.; et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Abdelmohsen, K.; Pullmann, R., Jr.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015, 10, e0115341. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Panday, S.; Talreja, R.; Kavdia, M. The role of glutathione and glutathione peroxidase in regulating cellular level of reactive oxygen and nitrogen species. Microvasc. Res. 2020, 131, 104010. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Vitto, V.A.M.; Pinton, P.; Rimessi, A. Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front. Pharmacol. 2020, 11, 581114. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Malo, D.; Hekimi, S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/- mouse mutants. J. Immunol. 2010, 184, 582–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, H.; Parthasarathi, K.; Quadri, S.; Issekutz, A.C.; Bhattacharya, J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J. Clin. Investig. 2003, 111, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Escames, G.; Lopez, L.C.; Garcia, J.A.; Garcia-Corzo, L.; Ortiz, F.; Acuna-Castroviejo, D. Mitochondrial DNA and inflammatory diseases. Hum. Genet. 2012, 131, 161–173. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.B.; Bergstralh, D.T.; Duncan, J.A.; Lei, Y.; Morrison, T.E.; Zimmermann, A.G.; Accavitti-Loper, M.A.; Madden, V.J.; Sun, L.; Ye, Z.; et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Sun, X.; Nie, X.; Sun, L.; Tang, T.S.; Chen, D.; Sun, Q. COX5B regulates MAVS-mediated antiviral signaling through interaction with ATG5 and repressing ROS production. PLoS Pathog. 2012, 8, e1003086. [Google Scholar] [CrossRef]
- Nobre, L.; Wise, D.; Ron, D.; Volmer, R. Modulation of Innate Immune Signalling by Lipid-Mediated MAVS Transmembrane Domain Oligomerization. PLoS ONE 2015, 10, e0136883. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Girardin, S.E. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011, 21, 558–560. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Mijares, L.A.; Li, L.; Ogura, Y.; Kazmierczak, B.I.; Flavell, R.A. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 2007, 204, 3235–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolle, L.; Yu, F.S.; Kovach, M.A.; Ballinger, M.N.; Newstead, M.W.; Zeng, X.; Nunez, G.; Standiford, T.J. Redundant and cooperative interactions between TLR5 and NLRC4 in protective lung mucosal immunity against Pseudomonas aeruginosa. J. Innate Immun. 2015, 7, 177–186. [Google Scholar] [CrossRef]
- Jabir, M.S.; Hopkins, L.; Ritchie, N.D.; Ullah, I.; Bayes, H.K.; Li, D.; Tourlomousis, P.; Lupton, A.; Puleston, D.; Simon, A.K.; et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 2015, 11, 166–182. [Google Scholar] [CrossRef] [Green Version]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194. [Google Scholar]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Chen, Z.; Wu, Y.; Zhu, B.; Yang, L.; Yang, C. Metabolic remodeling induced by mitokines in heart failure. Aging 2019, 11, 7307–7327. [Google Scholar] [CrossRef]
- Yong, C.Q.Y.; Tang, B.L. A Mitochondrial Encoded Messenger at the Nucleus. Cells 2018, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid. Med. Cell. Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, B.; Mathur, A.; Duchen, M.R.; Erusalimsky, J.D.; Moncada, S. The effect of nitric oxide on cell respiration: A key to understanding its role in cell survival or death. Proc. Natl. Acad. Sci. USA 2000, 97, 14602–14607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasutto, L.; Azzolini, M.; Szabo, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta 2016, 1863, 2515–2530. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Choi, S.; Lee, J.; Huang, Y.T.; Chen, F.; Zhao, Y.; Lin, X.; Neuberg, D.; Kim, J.; Christiani, D.C. Mitochondrial Variations in Non-Small Cell Lung Cancer (NSCLC) Survival. Cancer Inform. 2015, 14 (Suppl. 1), CIN-S13976. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Thierbach, R.; Voigt, A.; Drewes, G.; Mietzner, B.; Steinberg, P.; Pfeiffer, A.F.; Ristow, M. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J. Biol. Chem. 2006, 281, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Sakashita, M.; Aoyama, N.; Minami, R.; Maekawa, S.; Kuroda, K.; Shirasaka, D.; Ichihara, T.; Kuroda, Y.; Maeda, S.; Kasuga, M. Glut1 expression in T1 and T2 stage colorectal carcinomas: Its relationship to clinicopathological features. Eur. J. Cancer 2001, 37, 204–209. [Google Scholar] [CrossRef]
- Grover-McKay, M.; Walsh, S.A.; Seftor, E.A.; Thomas, P.A.; Hendrix, M.J. Role for glucose transporter 1 protein in human breast cancer. Pathol. Oncol. Res. 1998, 4, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017, 24, 1694–1704. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C] glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, S.M.; Locasale, J.W. Revisiting the Warburg Effect: Some Tumors Hold Their Breath. Cell Metab. 2018, 28, 669–670. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014, 33, 2329–2340. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [PubMed] [Green Version]
- Cemerski, S.; Cantagrel, A.; Van Meerwijk, J.P.; Romagnoli, P. Reactive oxygen species differentially affect T cell receptor-signaling pathways. J. Biol. Chem. 2002, 277, 19585–19593. [Google Scholar] [CrossRef] [Green Version]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, e93411. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, R.G.; Napolioni, V.; Oikonomou, V.; De Luca, A.; Galosi, C.; Pariano, M.; Massi-Benedetti, C.; Borghi, M.; Puccetti, M.; Lucidi, V.; et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 2016, 7, 10791. [Google Scholar] [CrossRef]
- Rimessi, A.; Pozzato, C.; Carparelli, L.; Rossi, A.; Ranucci, S.; De Fino, I.; Cigana, C.; Talarico, A.; Wieckowski, M.R.; Ribeiro, C.M.P.; et al. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci. Adv. 2020, 6, eaax9093. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Legendre, C.; Mooij, M.J.; Adams, C.; O’Gara, F. Impaired expression of hypoxia-inducible factor-1alpha in cystic fibrosis airway epithelial cells—A role for HIF-1 in the pathophysiology of CF? J. Cyst. Fibros. 2011, 10, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, H.; Murphy, A.; Zou, F.; Gerard, C.; Klanderman, B.; Schuemann, B.; Lazarus, R.; Garcia, K.C.; Celedon, J.C.; Drumm, M.; et al. IL1B polymorphisms modulate cystic fibrosis lung disease. Pediatr. Pulmonol. 2009, 44, 580–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, N.; Gallup, M.; Sucher, A.; Maltseva, I.; McKemy, D.; Basbaum, C. AsialoGM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate Erk1/2. Am. J. Respir. Cell Mol. Biol. 2006, 34, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, D.P.; Chmiel, J.F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 2015, 50 (Suppl. 40), S39–S56. [Google Scholar] [CrossRef]
- Saadane, A.; Eastman, J.; Berger, M.; Bonfield, T.L. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J. Inflamm. 2011, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Tabary, O.; Boncoeur, E.; de Martin, R.; Pepperkok, R.; Clement, A.; Schultz, C.; Jacquot, J. Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cell Signal. 2006, 18, 652–660. [Google Scholar] [CrossRef]
- Bialas, A.J.; Sitarek, P.; Milkowska-Dymanowska, J.; Piotrowski, W.J.; Gorski, P. The Role of Mitochondria and Oxidative/Antioxidative Imbalance in Pathobiology of Chronic Obstructive Pulmonary Disease. Oxid. Med. Cell. Longev. 2016, 2016, 7808576. [Google Scholar]
- Wild, C.P.; Hardie, L.J. Reflux, Barrett’s oesophagus and adenocarcinoma: Burning questions. Nat. Rev. Cancer 2003, 3, 676–684. [Google Scholar] [CrossRef]
- Wetscher, G.J.; Perdikis, G.; Kretchmar, D.H.; Stinson, R.G.; Bagchi, D.; Redmond, E.J.; Adrian, T.E.; Hinder, R.A. Esophagitis in Sprague-Dawley rats is mediated by free radicals. Dig. Dis. Sci. 1995, 40, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, E.H.; Hahm, K.B. Oxidative stress in inflammation-based gastrointestinal tract diseases: Challenges and opportunities. J. Gastroenterol. Hepatol. 2012, 27, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, J.W.O.; Towarnicki, S.G. Mitochondria, the gut microbiome and ROS. Cell Signal. 2020, 75, 109737. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Dalmasso, G.; Wang, L.; Sitaraman, S.V.; Merlin, D.; Murthy, N. Orally delivered thioketal nanoparticles loaded with TNF-alpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010, 9, 923–928. [Google Scholar] [CrossRef]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial function—Gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef]
- Kountouras, J.; Chatzopoulos, D.; Zavos, C. Reactive oxygen metabolites and upper gastrointestinal diseases. Hepato-Gastroenterol. 2001, 48, 743–751. [Google Scholar]
- Brynildsen, M.P.; Winkler, J.A.; Spina, C.S.; MacDonald, I.C.; Collins, J.J. Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat. Biotechnol. 2013, 31, 160–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaloian, S.; Rath, E.; Hammoudi, N.; Gleisinger, E.; Blutke, A.; Giesbertz, P.; Berger, E.; Metwaly, A.; Waldschmitt, N.; Allez, M.; et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 2020, 69, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef]
- Zuliani, G.; Morieri, M.L.; Volpato, S.; Vigna, G.B.; Bosi, C.; Maggio, M.; Cherubini, A.; Bandinelli, S.; Guralnik, J.M.; Ferrucci, L. Determinants and clinical significance of plasma oxidized LDLs in older individuals. A 9 years follow-up study. Atherosclerosis 2013, 226, 201–207. [Google Scholar]
- Perrin-Cocon, L.; Diaz, O.; Andre, P.; Lotteau, V. Modified lipoproteins provide lipids that modulate dendritic cell immune function. Biochimie 2013, 95, 103–108. [Google Scholar] [CrossRef]
- Frostegard, J.; Haegerstrand, A.; Gidlund, M.; Nilsson, J. Biologically modified LDL increases the adhesive properties of endothelial cells. Atherosclerosis 1991, 90, 119–126. [Google Scholar] [CrossRef]
- Catapano, A.L.; Maggi, F.M.; Tragni, E. Low density lipoprotein oxidation, antioxidants, and atherosclerosis. Curr. Opin. Cardiol. 2000, 15, 355–363. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Hang, L.; Peng, Y.; Xiang, R.; Li, X.; Li, Z. Ox-LDL Causes Endothelial Cell Injury Through ASK1/NLRP3-Mediated Inflammasome Activation via Endoplasmic Reticulum Stress. Drug Des. Dev. Ther. 2020, 14, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.; Bischoff, C.; Piotrowski, J.; Bohr, V.A. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem. 1998, 273, 33811–33816. [Google Scholar] [CrossRef] [Green Version]
- Tumurkhuu, G.; Shimada, K.; Dagvadorj, J.; Crother, T.R.; Zhang, W.; Luthringer, D.; Gottlieb, R.A.; Chen, S.; Arditi, M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ. Res. 2016, 119, e76–e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine Oxidase Is Overactivated in Left and Right Ventricles from Ischemic Hearts: An Intriguing Therapeutic Target. Oxid. Med. Cell. Longev. 2016, 2016, 4375418. [Google Scholar] [CrossRef]
- Patergnani, S.; Marchi, S.; Rimessi, A.; Bonora, M.; Giorgi, C.; Mehta, K.D.; Pinton, P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy 2013, 9, 1367–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpi, A.; Menabo, R.; Kaludercic, N.; Pelicci, P.; Di Lisa, F.; Giorgio, M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim. Biophys. Acta 2009, 1787, 774–780. [Google Scholar] [CrossRef]
- Pomerantz, B.J.; Reznikov, L.L.; Harken, A.H.; Dinarello, C.A. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc. Natl. Acad. Sci. USA 2001, 98, 2871–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.N. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. Neurol. 2019, 18, 459–480. [Google Scholar]
- Hou, L.; Wang, K.; Zhang, C.; Sun, F.; Che, Y.; Zhao, X.; Zhang, D.; Li, H.; Wang, Q. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. 2018, 14, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Na, R.; Boldt, E.; Ran, Q. NLRP3 inflammasome activation by mitochondrial reactive oxygen species plays a key role in long-term cognitive impairment induced by paraquat exposure. Neurobiol. Aging 2015, 36, 2533–2543. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A. DAMP-Mediated Activation of NLRP3-Inflammasome in Brain Sterile Inflammation: The Fine Line between Healing and Neurodegeneration. Front. Immunol. 2014, 5, 99. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Mielke, M.L.; Gomez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef] [Green Version]
- Dumont, M.; Ho, D.J.; Calingasan, N.Y.; Xu, H.; Gibson, G.; Beal, M.F. Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic. Biol. Med. 2009, 47, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Lee, D.C.; Rizer, J.; Selenica, M.L.; Reid, P.; Kraft, C.; Johnson, A.; Blair, L.; Gordon, M.N.; Dickey, C.A.; Morgan, D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflamm. 2010, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Ma, Q.; Chen, S.; Hu, Q.; Feng, H.; Zhang, J.H.; Tang, J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann. Neurol. 2014, 75, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Andreazza, A.C.; Elmi, N.; Chen, W.; Young, L.T. Nod-like receptor pyrin containing 3 (NLRP3) in the post-mortem frontal cortex from patients with bipolar disorder: A potential mediator between mitochondria and immune-activation. J. Psychiatr. Res. 2016, 72, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, W.; Li, Z.; Hao, J.; Zhang, Z.; Liu, L.; Mao, N.; Miao, J.; Zhang, L. S14G-humanin improves cognitive deficits and reduces amyloid pathology in the middle-aged APPswe/PS1dE9 mice. Pharmacol. Biochem. Behav. 2012, 100, 361–369. [Google Scholar] [CrossRef]
- Yen, K.; Mehta, H.H.; Kim, S.J.; Lue, Y.; Hoang, J.; Guerrero, N.; Port, J.; Bi, Q.; Navarrete, G.; Brandhorst, S.; et al. The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging 2020, 12, 11185–11199. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Srivastava, S.Y.; Brickey, W.J.; Iocca, H.; Toews, A.; Morrison, J.P.; Chen, V.S.; Gris, D.; Matsushima, G.K.; Ting, J.P. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 2010, 30, 15811–15820. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Lim, H.W.; More, S.V.; Kim, B.W.; Koppula, S.; Kim, I.S.; Choi, D.K. The role of free radicals in the aging brain and Parkinson’s Disease: Convergence and parallelism. Int. J. Mol. Sci. 2012, 13, 10478–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.K.; Cheng, C.H.; Chen, S.D.; Liou, C.W.; Huang, C.R.; Chuang, Y.C. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet. Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Siddiqui, A.; Rivera-Sanchez, S.; Castro Mdel, R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Moller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef]
- Park, J.; Min, J.S.; Kim, B.; Chae, U.B.; Yun, J.W.; Choi, M.S.; Kong, I.K.; Chang, K.T.; Lee, D.S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef]
- Burgos-Moron, E.; Abad-Jimenez, Z.; Maranon, A.M.; Iannantuoni, F.; Escribano-Lopez, I.; Lopez-Domenech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Al-Aubaidy, H.A.; Jelinek, H.F. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur. J. Endocrinol. 2011, 164, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.N.; Naqvi, F.N.; Shafiq, F. Lipid peroxidation and serum antioxidant enzymes in patients with type 2 diabetes mellitus. Ann. NY Acad. Sci. 2006, 1084, 481–489. [Google Scholar] [CrossRef] [PubMed]
- West, I.C. Radicals and oxidative stress in diabetes. Diabet. Med. 2000, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Feige, M.J.; Hendershot, L.M. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 2011, 23, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zhang, Z.; Hu, Y.; Lu, Y.; Li, D.; Liu, J.; Liao, S.; Hu, M.; Wang, Y.; Zhang, D.; et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc. Natl. Acad. Sci. USA 2019, 116, 21732–21738. [Google Scholar] [CrossRef] [Green Version]
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem. 2005, 280, 11107–11113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Haffner, S.J.; Cassells, H. Hyperglycemia as a cardiovascular risk factor. Am. J. Med. 2003, 115 (Suppl. 8A), 6S–11S. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Yeh, M.; Danziger, E.C.; Hatley, M.E.; Riggan, A.E.; Leitinger, N.; Berliner, J.A.; Hedrick, C.C. Glucose regulates monocyte adhesion through endothelial production of interleukin-8. Circ. Res. 2003, 92, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Batkai, S.; Hasko, G.; Liaudet, L.; Drel, V.R.; Obrosova, I.G.; Pacher, P. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H610–H619. [Google Scholar] [CrossRef] [Green Version]
- Yeop Han, C.; Kargi, A.Y.; Omer, M.; Chan, C.K.; Wabitsch, M.; O’Brien, K.D.; Wight, T.N.; Chait, A. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: Dissociation of adipocyte hypertrophy from inflammation. Diabetes 2010, 59, 386–396. [Google Scholar] [PubMed] [Green Version]
- Cimini, F.A.; Barchetta, I.; Porzia, A.; Mainiero, F.; Costantino, C.; Bertoccini, L.; Ceccarelli, V.; Morini, S.; Baroni, M.G.; Lenzi, A.; et al. Circulating IL-8 levels are increased in patients with type 2 diabetes and associated with worse inflammatory and cardiometabolic profile. Acta Diabetol. 2017, 54, 961–967. [Google Scholar] [CrossRef]
- Choudhury, S.; Ghosh, S.; Gupta, P.; Mukherjee, S.; Chattopadhyay, S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-kappaB and SAPK/JNK pathway. Free Radic. Res. 2015, 49, 1371–1383. [Google Scholar] [CrossRef]
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Di Lisa, F.; Ferdinandy, P. Pharmacology of oxidative stress: Translational opportunities. Br. J. Pharmacol. 2017, 174, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mitochondrial sites of ROS production. Mitochondrial complex I and III of respiratory chain are the principal sites of O2•− production within a cell, which can be converted to H2O2 by superoxide dismutase (SOD1 and SOD2) enzymes. H2O2 in turn is rapid neutralized to H2O and oxygen by glutathione peroxidase (GPX). However, other mitochondrial proteins, localized from OMM to matrix, may also contribute to mtROS production, including monoamine oxidase A and B (MAO A/B), cytochrome (Cyt.) b5 reductase, mitochondrial glycerol-phosphate dehydrogenase (mGPDH), p66Shc, Fhit with ferredoxin reductase (FDxR), adrenodoxin reductase (ADxR)-adrenodoxin (ADX)-cytochrome P450scc (CYP450) system, α-ketoglutharate dehydrogenases (KGDHC), acyl-CoA dehydrogenases (ACAD) and aconitase. This figure has been created with “BioRender.com.”.
Figure 1.
Mitochondrial sites of ROS production. Mitochondrial complex I and III of respiratory chain are the principal sites of O2•− production within a cell, which can be converted to H2O2 by superoxide dismutase (SOD1 and SOD2) enzymes. H2O2 in turn is rapid neutralized to H2O and oxygen by glutathione peroxidase (GPX). However, other mitochondrial proteins, localized from OMM to matrix, may also contribute to mtROS production, including monoamine oxidase A and B (MAO A/B), cytochrome (Cyt.) b5 reductase, mitochondrial glycerol-phosphate dehydrogenase (mGPDH), p66Shc, Fhit with ferredoxin reductase (FDxR), adrenodoxin reductase (ADxR)-adrenodoxin (ADX)-cytochrome P450scc (CYP450) system, α-ketoglutharate dehydrogenases (KGDHC), acyl-CoA dehydrogenases (ACAD) and aconitase. This figure has been created with “BioRender.com.”.

Figure 2.
Mitochondrial ROS-induced inflammatory response. Representation of multifaceted aspects of mitochondrial ROS in inflammation. Increased mtROS cause oxidative damage to mitochondrial membrane, with events of lipidic peroxidation and changes in membrane permeability, molecules, proteins, and mtDNA, which contribute to mitochondrial dysfunction and exacerbation of mtROS production. The dissemination of mtROS actives the redox-sensitive transcription factor NF-kB, inducing the expression of inflammasome genes, such as Nlrp3, Nlrc4, and Il1b genes. In turn, the mtROS and mtDNA promote the cytokines release mediating the inflammasome NLRP3 and NLRC4 complex activation, through the recruitment of pro-caspase−1 and the processing of pro-IL−1 and pro-IL−18. Finally, mtROS are reversed to extracellular milieu to sustain and exacerbate the inflammatory responses, affecting proximal cells and conditioning the inflammatory microenvironment. Abbreviations: Reactive oxygen species, ROS; Interleukin−18, IL−18; Interleukin−1, IL−1; NLR Family CARD Domain Containing 4, NLRC4; ASC; NLR Family Pyrin Domain Containing 3, NLRP3; Mitochondrial antiviral-signaling protein, MAVS; mitochondrial deoxyribonucleic acid, mtDNA; NLR Family member X1 precursor, NLRX1; Calcium, Ca2+; Nuclear factor kappa-light-chain-enhancer of activated B cells, NF-kB; Mitochondrial membrane potential, ΔΨ. This figure has been created with “BioRender.com.”.
Figure 2.
Mitochondrial ROS-induced inflammatory response. Representation of multifaceted aspects of mitochondrial ROS in inflammation. Increased mtROS cause oxidative damage to mitochondrial membrane, with events of lipidic peroxidation and changes in membrane permeability, molecules, proteins, and mtDNA, which contribute to mitochondrial dysfunction and exacerbation of mtROS production. The dissemination of mtROS actives the redox-sensitive transcription factor NF-kB, inducing the expression of inflammasome genes, such as Nlrp3, Nlrc4, and Il1b genes. In turn, the mtROS and mtDNA promote the cytokines release mediating the inflammasome NLRP3 and NLRC4 complex activation, through the recruitment of pro-caspase−1 and the processing of pro-IL−1 and pro-IL−18. Finally, mtROS are reversed to extracellular milieu to sustain and exacerbate the inflammatory responses, affecting proximal cells and conditioning the inflammatory microenvironment. Abbreviations: Reactive oxygen species, ROS; Interleukin−18, IL−18; Interleukin−1, IL−1; NLR Family CARD Domain Containing 4, NLRC4; ASC; NLR Family Pyrin Domain Containing 3, NLRP3; Mitochondrial antiviral-signaling protein, MAVS; mitochondrial deoxyribonucleic acid, mtDNA; NLR Family member X1 precursor, NLRX1; Calcium, Ca2+; Nuclear factor kappa-light-chain-enhancer of activated B cells, NF-kB; Mitochondrial membrane potential, ΔΨ. This figure has been created with “BioRender.com.”.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. https://doi.org/10.3390/biomedicines9020216
AMA Style
Patergnani S, Bouhamida E, Leo S, Pinton P, Rimessi A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines. 2021; 9(2):216. https://doi.org/10.3390/biomedicines9020216
Chicago/Turabian StylePatergnani, Simone, Esmaa Bouhamida, Sara Leo, Paolo Pinton, and Alessandro Rimessi. 2021. "Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases" Biomedicines 9, no. 2: 216. https://doi.org/10.3390/biomedicines9020216
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.