


Pancreatic Cancer Research beyond DNA Mutations
 , , ,
, , ,  and
and 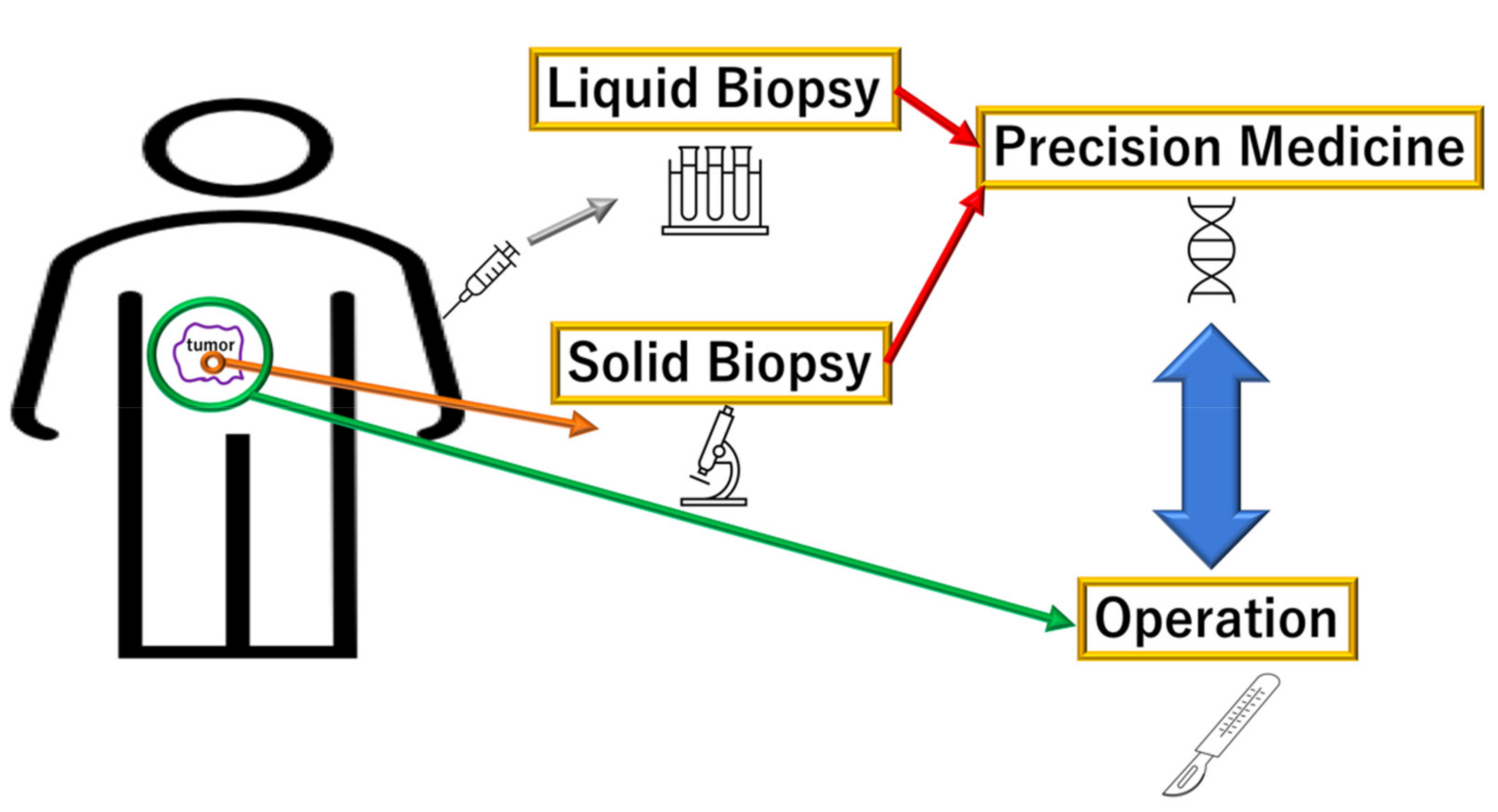{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Liquid Diagnosis of PDAC via DNA Mutations
3. Diagnosis through Methods other Than DNA Sequencing
3.1. Metagenomics
3.2. Polyamines
3.3. Driver Mutations-Dependent Effects in PDAC
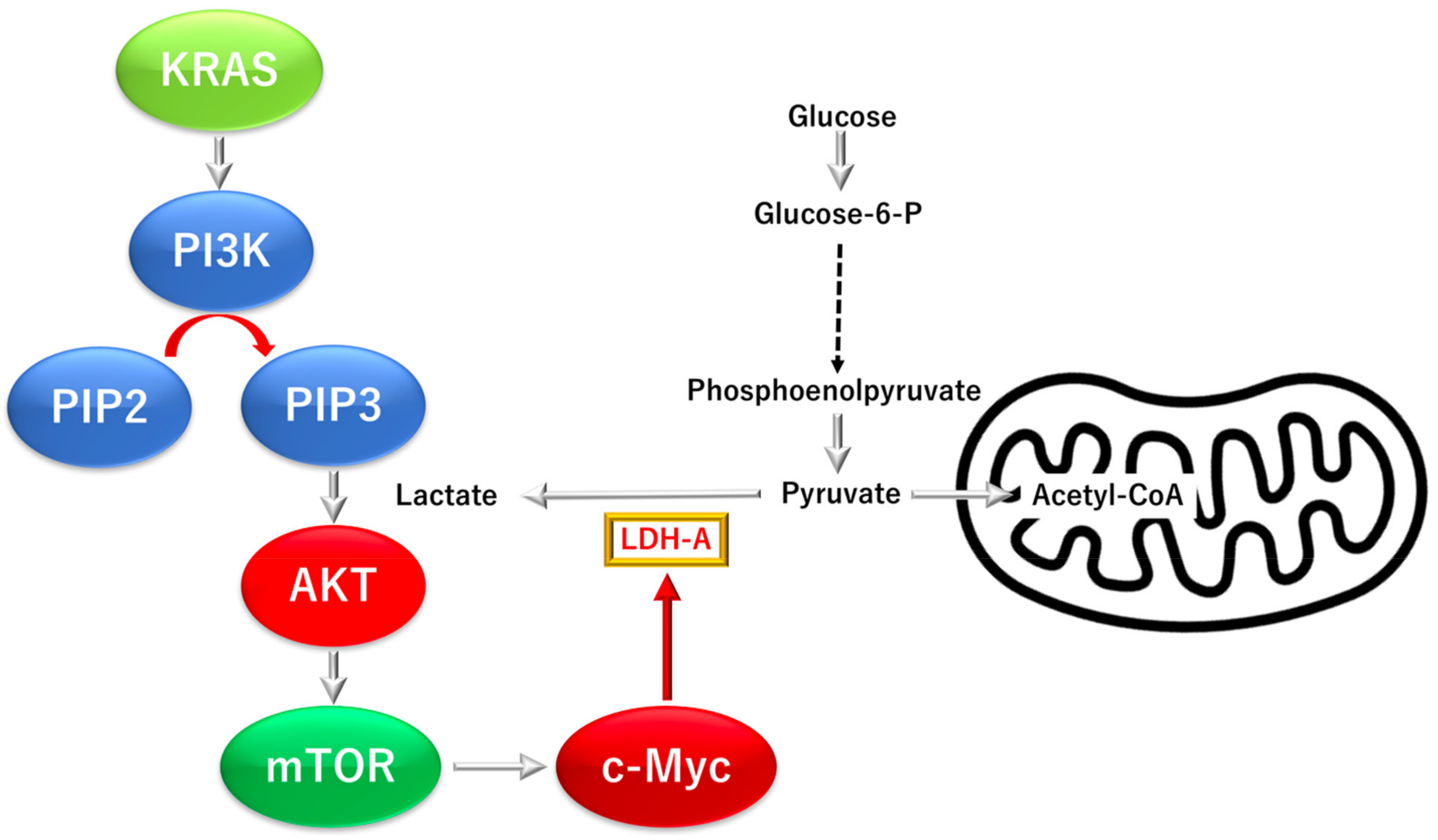
3.3.1. KRAS Proto-Oncogene GTPase (KRAS)-Dependent Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PI3K), a Mechanistic Target of Rapamycin Kinase Pathway
3.3.2. KRAS Proto-Oncogene and GTPase (KRAS)-Dependent Oncogene V-myc Avian Myelocy-Tomatosis Viral Oncogene Homolog (C-MYC) Pathway
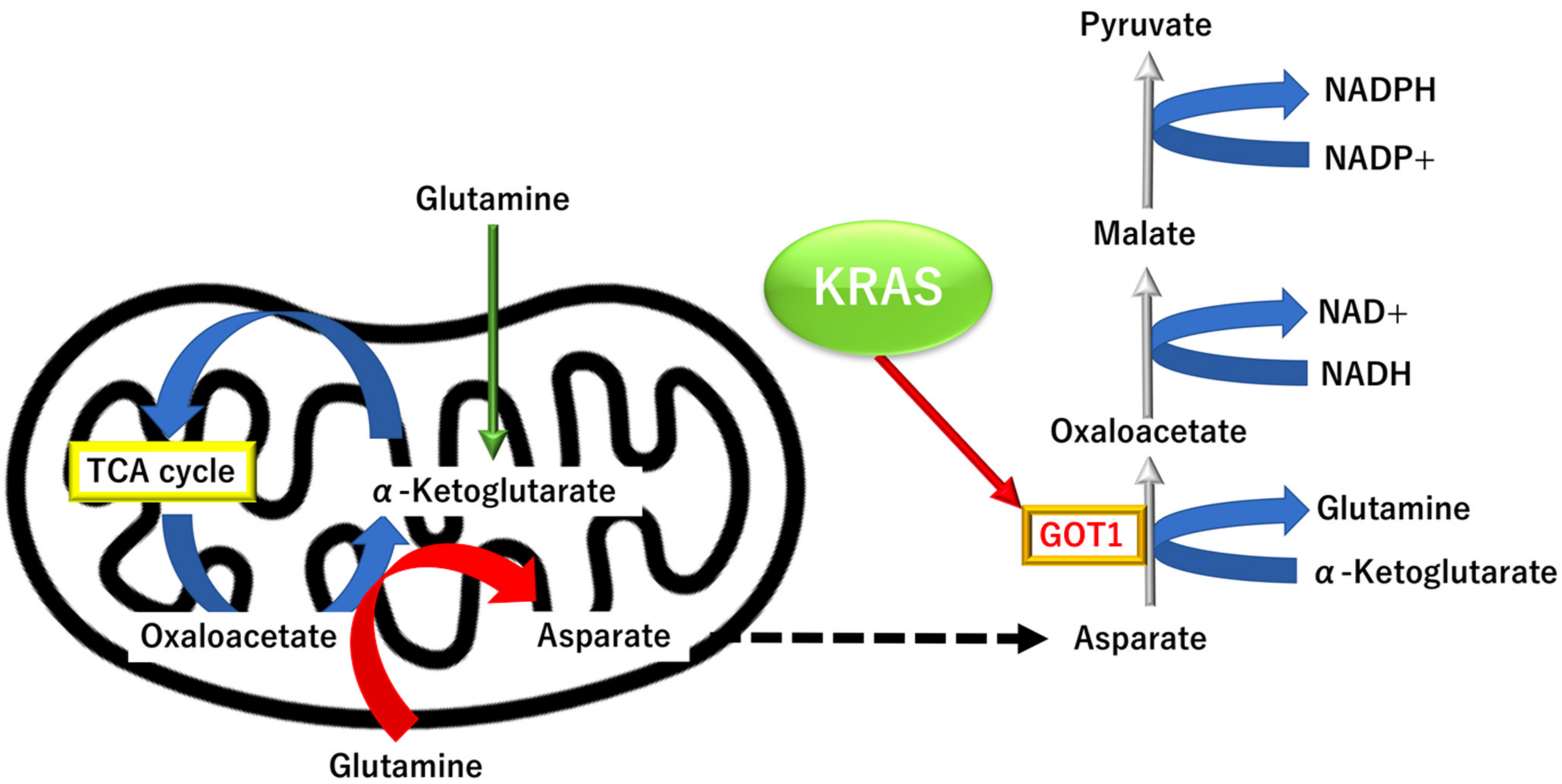
3.3.3. KRAS Proto-Oncogene and GTPase (KRAS)-Dependent Alternative Tricarboxylic Acid Pathway
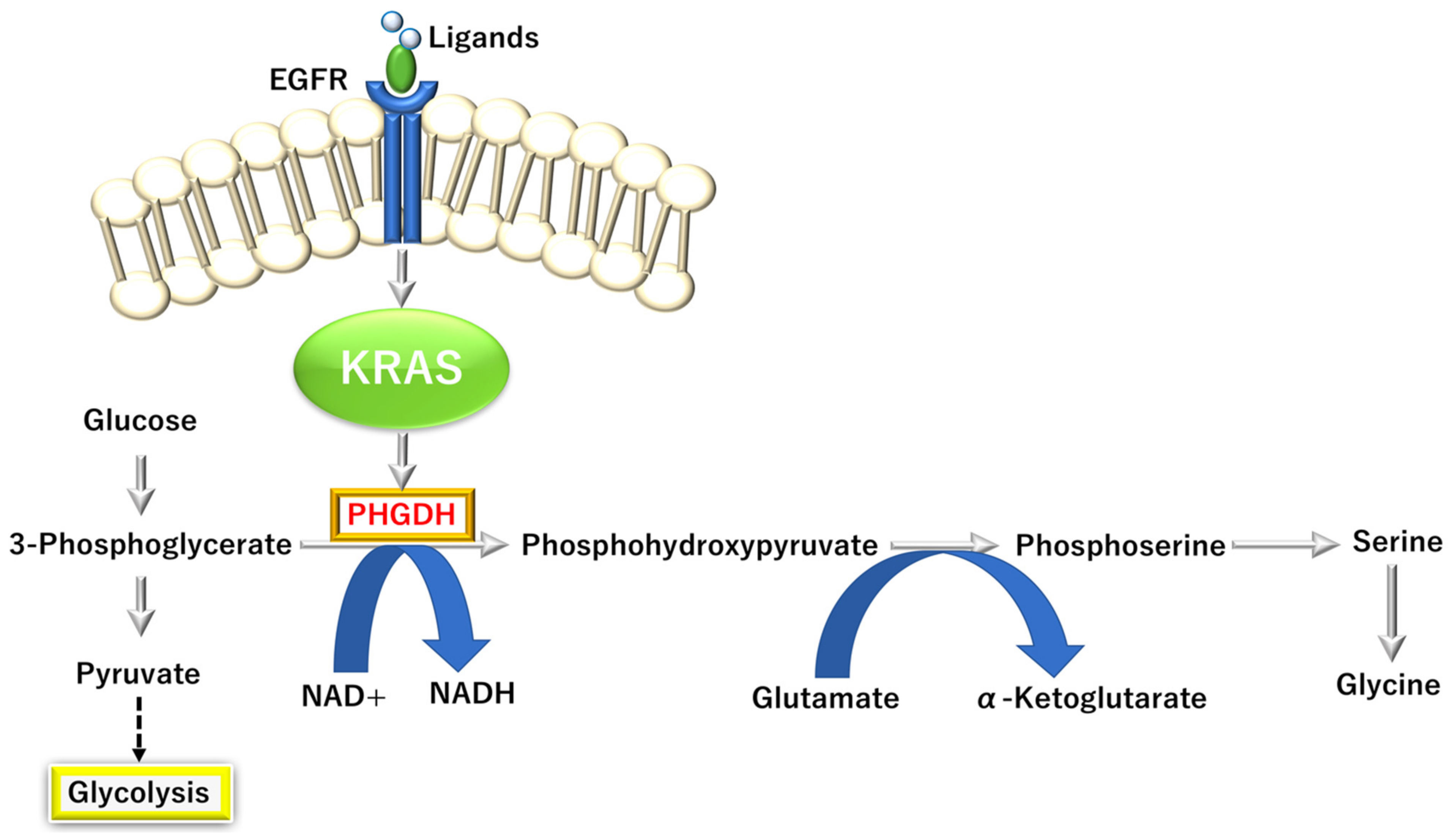
3.3.4. KRAS-Dependent One-Carbon (1C) Pathway
3.3.5. Arginase 1-Dependent Immune Suppression in PDAC
3.4. Cancer-Associated Fibroblasts in PDAC
4. PDAC Detection and Monitoring by RNA Profiling
4.1. isomiR and episomiR
4.2. circRNAs
5. PDAC Detection and Monitoring by Volatile Organic Compounds
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavenee, W.; Dryja, T.P.; Phillips, R.A.; Benedict, W.F.; Godbout, R.; Gallie, B.L.; Murphree, A.L.; Strong, L.C.; White, R.L. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983, 305, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. Tumor suppressor genes. Science 1991, 254, 1138–1146. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, C.A.; Lawrence, S.A.; Richards, A.L.; Chou, J.F.; Wong, W.; Capanu, M.; Berger, M.F.; Donoghue, M.T.A.; Yu, K.H.; Varghese, A.M.; et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer 2020, 126, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Pumarega, J.; Amaral, A.F.S.; Genkinger, J.M.; Camargo, J.; Mucci, L.; Alguacil, J.; Gasull, M.; Zhang, X.; Morales, E.; et al. PANKRAS II Study Group. Influence of KRAS mutations, persistent organic pollutants, and trace elements on survival from pancreatic ductal adenocarcinoma. Environ. Res. 2020, 190, 109781. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Muratcioglu, S.; Tsai, C.J.; Jang, H.; Gursoy, A.; Keskin, O. The Key Role of Calmodulin in KRAS-Driven Adenocarcinomas. Mol. Cancer Res. 2015, 13, 1265–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, M.; Taniguchi, M.; Ishii, H. Significant epitranscriptomes in heterogeneous cancer. Cancer Sci. 2019, 110, 2318–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Chijimatsu, R.; Ofusa, K.; Kobayashi, S.; Doki, Y.; Eguchi, H.; Ishii, H. Cancer metabolism challenges genomic instability and clonal evolution as therapeutic targets. Cancer Sci. 2022, 113, 1097–1104. [Google Scholar] [CrossRef]
- Swords, D.; Firpo, M.; Scaife, C.; Mulvihill, S.J. Biomarkers in pancreatic adenocarcinoma: Current perspectives. Onco Targets Ther. 2016, 9, 7459–7467. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.L.; Kirkwood, K.; Kim, M.P.; Maitra, A.; Koay, E.J. Intraductal papillary mucinous neoplasms of the pancreas: Current understanding and future directions for stratification of malignancy risk. Pancreas 2018, 47, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Elta, G.H.; Enestvedt, B.K.; Sauer, B.G.; Lennon, A.M. ACG clinical guideline: Diagnosis and management of pancreatic cysts. Am. J. Gastroenterol. 2018, 113, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; Vecchia, C.L.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Guler, G.D.; Ning, Y.; Ku, C.-J.; Phillips, T.; McCarthy, E.; Ellison, C.K.; Bergamaschi, A.; Collin, F.; Lloyd, P.; Scott, A.; et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat. Commun. 2020, 11, 5270. [Google Scholar] [CrossRef]
- Sammallahti, H.; Sarhadi, V.K.; Kokkola, A.; Ghanbari, R.; Rezasoltani, S.; Aghdaei, H.A.; Puolakkainen, P.; Knuutila, S. Oncogenomic Changes in Pancreatic Cancer and Their Detection in Stool. Biomolecules 2022, 12, 652. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Nishijima, S.; Kojima, Y.; Hisada, Y.; Imbe, K.; Miyoshi-Akiyama, T.; Suda, W.; Kimura, M.; Aoki, R.; Sekine, K.; et al. Metagenomic Identification of Microbial Signatures Predicting Pancreatic Cancer From a Multinational Study. Gastroenterology 2022, 163, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Mendez, R.; Kesh, K.; Arora, N.; Martino, L.D.; McAllister, F.; Merchant, N.; Banerjee, S.; Banerjee, S. Microbial Dysbiosis and Polyamine Metabolism as Predictive Markers for Early Detection of Pancreatic Cancer. Carcinogenesis 2020, 41, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.; Bieberich, C.J.; Stewart, T.M.; Casero, R.A., Jr. Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Antitumor Responses. Med. Sci. 2022, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, V.; Andl, T.; Phanstiel, O. ATP13A3 facilitates polyamine transport in human pancreatic cancer cells. Sci. Rep. 2022, 12, 4045. [Google Scholar] [CrossRef]
- Nakkina, S.P.; Gitto, S.B.; Pandey, V.; Parikh, J.G.; Geerts, D.; Maurer, H.C.; Olive, K.P.; Phanstiel, O., 4th; Altomare, D.A. Differential Expression of Polyamine Pathways in Human Pancreatic Tumor Progression and Effects of Polyamine Blockade on Tumor Microenvironment. Cancers 2021, 13, 6391. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, S.I.; Venäläinen, M.; Kumpulainen, P.; Roine, A.; Häkkinen, M.R.; Vepsäläinen, J.; Oksala, N.; Rantanen, T. Discrimination between Pancreatic Cancer, Pancreatitis and Healthy Controls Using Urinary Polyamine Panel. Cancer Control 2021, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tamari, K.; Konno, M.; Asai, A.; Koseki, J.; Hayashi, K.; Kawamoto, K.; Murai, N.; Matsufuji, S.; Isohashi, F.; Satoh, T.; et al. Polyamine flux suppresses histone lysine demethylases and enhances ID1 expression in cancer stem cells. Cell Death Discov. 2018, 4, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Nio, K.; Yamashita, T.; Okada, H.; Li, R.; Suda, T.; Li, Y.; Doan, P.T.B.; Seki, A.; Nakagawa, H.; et al. BMP9-ID1 signaling promotes EpCAM-positive cancer stem cell properties in hepatocellular carcinoma. Mol. Oncol. 2021, 15, 2203–2218. [Google Scholar] [CrossRef]
- Tamari, K.; Hayashi, K.; Ishii, H.; Kano, Y.; Konno, M.; Kawamoto, K.; Nishida, N.; Koseki, J.; Fukusumi, T.; Hasegawa, S.; et al. Identification of chemoradiation-resistant osteosarcoma stem cells using an imaging system for proteasome activity. Int. J. Oncol. 2014, 45, 2349–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Tamari, K.; Ishii, H.; Konno, M.; Nishida, N.; Kawamoto, K.; Koseki, J.; Fukusumi, T.; Kano, Y.; Nishikawa, S.; et al. Visualization and characterization of cancer stem-like cells in cervical cancer. Int. J. Oncol. 2014, 45, 2468–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijakowski, K.; Gruszczyński, D.; Kopała, D.; Surdacka, A. Salivary Metabolomics for Oral Squamous Cell Carcinoma Diagnosis: A Systematic Review. Metabolites 2022, 12, 294. [Google Scholar] [CrossRef] [PubMed]
- DeFelice, B.C.; Fiehn, O.; Belafsky, P.; Ditterich, C.; Moore, M.; Abouyared, M.; Beliveau, A.M.; Farwell, D.G.; Bewley, A.F.; Clayton, S.M.; et al. Polyamine Metabolites as Biomarkers in Head and Neck Cancer Biofluids. Diagnostics 2022, 12, 797. [Google Scholar] [CrossRef]
- Kuwabara, H.; Katsumata, K.; Iwabuchi, A.; Udo, R.; Tago, T.; Kasahara, K.; Mazaki, J.; Enomoto, M.; Ishizaki, T.; Soya, R.; et al. Salivary metabolomics with machine learning for colorectal cancer detection. Cancer Sci. 2022, 113, 3234–3243. [Google Scholar] [CrossRef]
- Cáceres, M.; Quesada, R.; Iglesias, M.; Real, F.X.; Villamonte, M.; de Villarreal, J.M.; Pérez, M.; Andaluz, A.; Moll, X.; Berjano, E.; et al. Pancreatic duct ligation reduces premalignant pancreatic lesions in a Kras model of pancreatic adenocarcinoma in mice. Sci. Rep. 2020, 10, 18344. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.N.; Jeong, M.S.; Jang, S.B. Oncogenic KRAS: Signaling and Drug Resistance. Cancers 2021, 13, 5599. [Google Scholar] [CrossRef]
- Chijimatsu, R.; Kobayashi, S.; Takeda, Y.; Kitakaze, M.; Tatekawa, S.; Arao, Y.; Nakayama, M.; Tachibana, N.; Saito, T.; Ennishi, D.; et al. Establishment of a reference single-cell RNA sequencing dataset for human pancreatic adenocarcinoma. iScience 2022, 25, 104659. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Amos, C.I.; Merriman, K.W.; Wei, Q.; Sen, S.; Killary, A.M.; Frazier, M.L. Genetic variants of p21 and p27 and pancreatic cancer risk in non-Hispanic Whites: A case-control study. Pancreas 2010, 39, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehra, S.; Singh, S.; Nagathihalli, N. Emerging Role of CREB in Epithelial to Mesenchymal Plasticity of Pancreatic Cancer. Front. Oncol. 2022, 12, 925687. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Bianchi, A.; De Castro Silva, I.; Deshpande, N.U.; Cao, L.L.; Mehra, S.; Singh, S.; Rafie, C.; Sun, X.; Chen, X.; et al. Distinct mechanisms of innate and adaptive immune regulation underlie poor oncologic outcomes associated with KRAS-TP53 co-alteration in pancreatic cancer. Oncogene 2022, 41, 3640–3654. [Google Scholar] [CrossRef] [PubMed]
- Racu, M.L.; Lebrun, L.; Schiavo, A.A.; Van Campenhout, C.; De Clercq, S.; Absil, L.; Perez, E.M.; Maris, C.; Decaestecker, C.; Salmon, I.; et al. The Role of SMAD4 Inactivation in Epithelial-Mesenchymal Plasticity of Pancreatic Ductal Adenocarcinoma: The Missing Link? Cancers 2022, 14, 973. [Google Scholar] [CrossRef]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.L.; Wang, J. KRAS gene silencing inhibits the activation of PI3K-Akt-mTOR signaling pathway to regulate breast cancer cell epithelial-mesenchymal transition, proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3085–3096. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Alzahran, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Schüle, M.; Butto, T.; Dewi, S.; Schlichtholz, L.; Strand, S.; Gerber, S.; Endres, K.; Schweiger, S.; Winter, J. mTOR Driven Gene Transcription Is Required for Cholesterol Production in Neurons of the Developing Cerebral Cortex. Int. J. Mol. Sci. 2021, 22, 6034. [Google Scholar] [CrossRef] [PubMed]
- Koh, V.; Chakrabarti, J.; Torvund, M.; Steele, N.; Hawkins, J.A.; Ito, Y.; Wang, J.; Helmrath, M.A.; Merchant, J.L.; Ahmed, S.A.; et al. Hedgehog transcriptional effector GLI mediates mTOR-Induced PD-L1 expression in gastric cancer organoids. Cancer Lett. 2021, 518, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, H.; Li, S. The role of glycometabolic plasticity in cancer. Pathol. Res. Pract. 2021, 226, 153595. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef] [Green Version]
- Koseki, J.; Colvin, H.; Fukusumi, T.; Nishida, N.; Konno, M.; Kawamoto, K.; Tsunekuni, K.; Matsui, H.; Doki, Y.; Mori, M.; et al. Mathematical analysis predicts imbalanced IDH1/2 expression associates with 2-HG-inactivating β-oxygenation pathway in colorectal cancer. Int. J. Oncol. 2015, 46, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Miyo, M.; Konno, M.; Nishida, N.; Sueda, T.; Noguchi, K.; Matsui, H.; Colvin, H.; Kawamoto, K.; Koseki, J.; Haraguchi, N.; et al. Metabolic Adaptation to Nutritional Stress in Human Colorectal Cancer. Sci. Rep. 2016, 6, 38415. [Google Scholar] [CrossRef] [Green Version]
- Sueda, T.; Sakai, D.; Kawamoto, K.; Konno, M.; Nishida, N.; Koseki, J.; Colvin, H.; Takahashi, H.; Haraguchi, N.; Nishimura, J.; et al. BRAF V600E inhibition stimulates AMP-activated protein kinase-mediated autophagy in colorectal cancer cells. Sci. Rep. 2016, 6, 18949. [Google Scholar] [CrossRef] [Green Version]
- Colyn, L.; Alvarez-Sola, G.; Latasa, M.U.; Uriarte, I.; Herranz, J.M.; Arechederra, M.; Vlachogiannis, G.; Rae, C.; Pineda-Lucena, A.; Casadei-Gardini, A.; et al. New molecular mechanisms in cholangiocarcinoma: Signals triggering interleukin-6 production in tumor cells and KRAS co-opted epigenetic mediators driving metabolic reprogramming. J. Exp. Clin. Cancer Res. 2022, 41, 183. [Google Scholar] [CrossRef] [PubMed]
- Itoyama, R.; Yasuda-Yoshihara, N.; Kitamura, F.; Yasuda, T.; Bu, L.; Yonemura, A.; Uchihara, T.; Arima, K.; Hu, X.; Jun, Z.; et al. Metabolic shift to serine biosynthesis through 3-PG accumulation and PHGDH induction promotes tumor growth in pancreatic cancer. Cancer Lett. 2021, 523, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koseki, J.; Konno, M.; Asai, A.; Colvin, H.; Kawamoto, K.; Nishida, N.; Sakai, D.; Kudo, T.; Satoh, T.; Doki, Y.; et al. Enzymes of the one-carbon folate metabolism as anticancer targets predicted by survival rate analysis. Sci. Rep. 2018, 8, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, A.; Koseki, J.; Konno, M.; Nishimura, T.; Gotoh, N.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H. Drug discovery of anticancer drugs targeting methylenetetrahydrofolate dehydrogenase 2. Heliyon 2018, 4, e01021. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Konno, M.; Koseki, J.; Taniguchi, M.; Vecchione, A.; Ishii, H. One-carbon metabolism for cancer diagnostic and therapeutic approaches. Cancer Lett. 2020, 470, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Huang, M.; Wei, Y. Diversity of the reaction mechanisms of SAM-dependent enzymes. Acta Pharm. Sin. B 2021, 11, 632–650. [Google Scholar] [CrossRef] [PubMed]
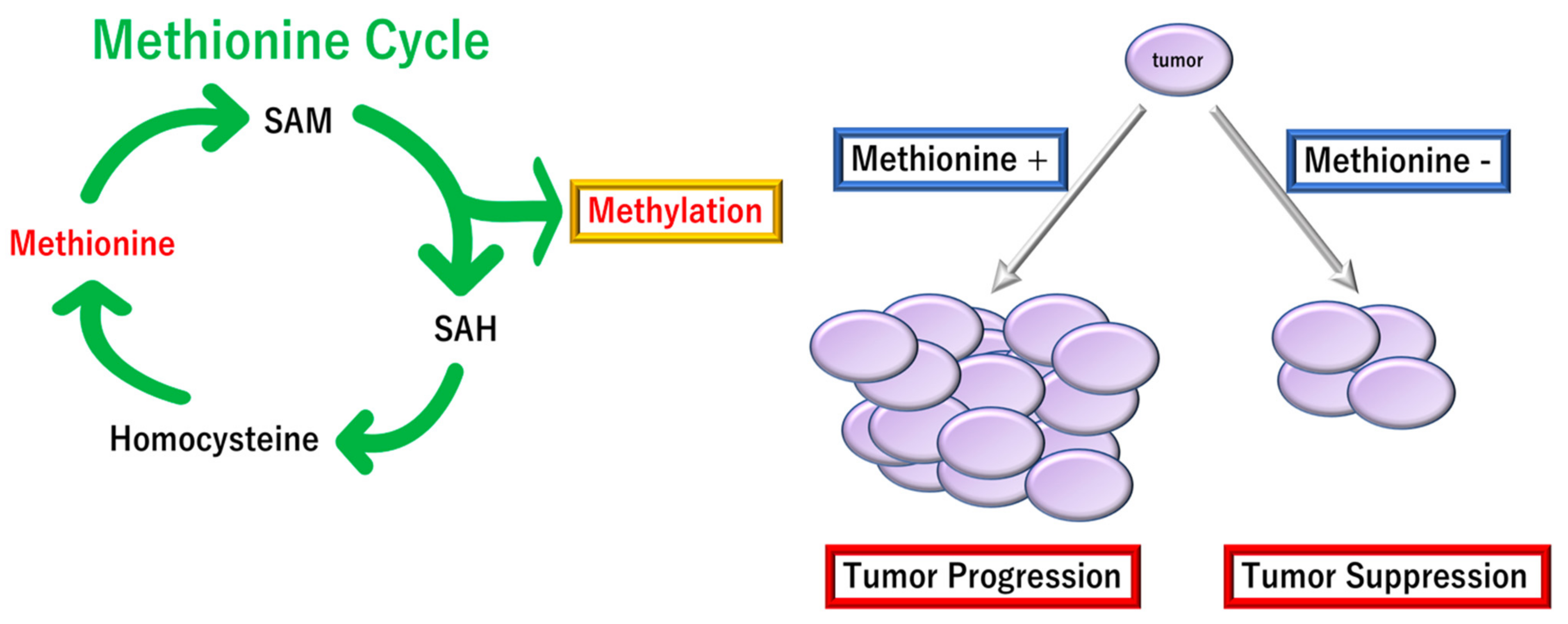
- Lauinger, L.; Kaiser, P. Sensing and Signaling of Methionine Metabolism. Metabolites 2021, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, P.H.; Wallace, C.D.; Hoffman, R.M. Altered methionine metabolism occurs in all members of a set of diverse human tumor cell lines. J. Cell. Physiol. 1984, 119, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Booher, K.; Lin, D.W.; Borrego, S.L.; Kaiser, P. Downregulation of Cdc6 and pre-replication complexes in response to methionine stress in breast cancer cells. Cell Cycle 2012, 11, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 2016, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Chung, B.P.; Kaiser, P. S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J. Cell Sci. 2014, 127, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, T.; Birnbaum, S.M.; Winitz, M.; Greenstein, J.P. Quantitative nutritional studies with water-soluble, chemically defined diets. VIII. The forced feeding of diets each lacking in one essential amino acid. Arch. Biochem. Biophys. 1959, 81, 448–455. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.; Shi, M.; Chen, Y.; Yu, D.; Zhao, C.; Gu, Y.; Yang, B.; Guo, S.; Ding, G.; et al. CD13hi Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology 2017, 6, e1258504. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Antitumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.S.; Wang, C.C.; Qiu, J.D.; Ren, B.; You, L. Arginine metabolism: A potential target in pancreatic cancer therapy. Chin. Med. J. 2020, 134, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018, 115, 6410–6415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. eBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Yosefov-Levi, O.; Kolodkin-Gal, D.; Granit, R.Z.; Peters, L.; Kalifa, R.; Xia, L.; Nasereddin, A.; Shiff, I.; Amran, O.; et al. Single-cell transcriptomes of pancreatic preinvasive lesions and cancer reveal acinar metaplastic cells’ heterogeneity. Nat. Commun. 2020, 11, 4516. [Google Scholar] [CrossRef]
- Agostini, A.; Orlacchio, A.; Carbone, C.; Guerriero, I. Understanding tricky cellular and molecular interactions in pancreatic tumor microenvironment: New food for thought. Front. Immunol. 2022, 13, 876291. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-associated fibroblast (CAF) heterogeneity and targeting therapy of CAFs in pancreatic cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Aita, A.; Millino, C.; Sperti, C.; Pacchioni, B.; Plebani, M.; De Pittà, C.; Basso, D. Serum miRNA Profiling for Early PDAC Diagnosis and Prognosis: A Retrospective Study. Biomedicines 2021, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Koh, H.; Kim, J.Y.; Lee, S.; Lee, H.; Kim, Y.; Hwang, H.K.; Kim, S.I. Tumor-Specific miRNA Signatures in Combination with CA19-9 for Liquid Biopsy-Based Detection of PDAC. Int. J. Mol. Sci. 2021, 22, 13621. [Google Scholar] [CrossRef]
- Ottaviani, S.; Stebbing, J.; Frampton, A.E.; Zagorac, S.; Krell, J.; de Giorgio, A.; Trabulo, S.M.; Nguyen, V.T.M.; Magnani, L.; Feng, H.; et al. TGF-β induces miR-100 and miR-125b but blocks let-7a through LIN28B controlling PDAC progression. Nat. Commun. 2018, 9, 1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front. Cell Dev. Biol. 2021, 9, 668648. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.; Dirksen, W.P.; Supsavhad, W.; Yilmaz, A.S.; Ozer, H.G.; Feller, J.D.; Vala, K.A.; Croce, C.M.; Rosol, T.J. Discovery and characterization of the feline miRNAome. Sci. Rep. 2017, 7, 9263. [Google Scholar] [CrossRef] [Green Version]
- Konno, M.; Koseki, J.; Asai, A.; Yamagata, A.; Shimamura, T.; Motooka, D.; Okuzaki, D.; Kawamoto, K.; Mizushima, T.; Eguchi, H.; et al. Distinct methylation levels of mature microRNAs in gastrointestinal cancers. Nat. Commun. 2019, 10, 3888. [Google Scholar] [CrossRef]
- Ohshiro, T.; Konno, M.; Asai, A.; Komoto, Y.; Yamagata, A.; Doki, Y.; Eguchi, H.; Ofusa, K.; Taniguchi, M.; Ishii, H. Single-molecule RNA sequencing for simultaneous detection of m6A and 5mC. Sci. Rep. 2021, 11, 19304. [Google Scholar] [CrossRef]
- Arao, Y.; Nakayama, M.; Tsuji, Y.; Hamano, Y.; Otsuka, C.; Vecchione, A.; Ofusa, K.; Ishii, H. EpisomiR, a New Family of miRNAs, and Its Possible Roles in Human Diseases. Biomedicines 2022, 10, 1280. [Google Scholar] [CrossRef]
- Tatekawa, S.; Tamari, K.; Chijimatsu, R.; Konno, M.; Motooka, D.; Mitsufuji, S.; Akita, H.; Kobayashi, S.; Murakumo, Y.; Doki, Y.; et al. N(6)-methyladenosine methylation-regulated polo-like kinase 1 cell cycle homeostasis as a potential target of radiotherapy in pancreatic adenocarcinoma. Sci. Rep. 2022, 12, 11074. [Google Scholar] [CrossRef]
- Kung, J.T.Y.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, S.; Yang, X.; Li, X.; Wang, J.; Gao, Y.; Shang, R.; Sun, W.; Dou, K.; Li, H. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015, 365, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Li, Y.; Luo, Y.; Zhu, J.; Zheng, H.; Gao, B.; Guo, X.; Li, Z.; Chen, R.; Chen, C. circNFIB1 inhibits lymphangiogenesis and lymphatic metastasis via the miR-486-5p/PIK3R1/VEGF-C axis in pancreatic cancer. Mol. Cancer 2020, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Shi, S.; Tan, Z.; Xu, J.; Meng, Q.; Hua, J.; Liu, J.; Zhang, B.; Wang, W.; Yu, X.; et al. Circular RNA CircEYA3 induces energy production to promote pancreatic ductal adenocarcinoma progression through the miR-1294/c-Myc axis. Mol. Cancer 2021, 20, 106. [Google Scholar] [CrossRef] [PubMed]
- Daulton, E.; Wicaksono, A.N.; Tiele, A.; Kocher, H.M.; Debernardi, S.; Crnogorac-Jurcevic, T.; Covington, J.A. Volatile organic compounds (VOCs) for the non-invasive detection of pancreatic cancer from urine. Talanta 2021, 221, 121604. [Google Scholar] [CrossRef] [PubMed]
- Bannaga, A.S.; Tyagi, H.; Daulton, E.; Covington, J.A.; Arasaradnam, R.P. Exploratory Study Using Urinary Volatile Organic Compounds for the Detection of Hepatocellular Carcinoma. Molecules 2021, 26, 2447. [Google Scholar] [CrossRef] [PubMed]
- Tyag, H.; Daulton, E.; Bannaga, A.S.; Arasaradnam, R.P.; Covington, J.A. Urinary Volatiles and Chemical Characterisation for the Non-Invasive Detection of Prostate and Bladder Cancers. Biosensors 2021, 11, 437. [Google Scholar] [CrossRef]
- Asai, A.; Konno, M.; Ozaki, M.; Kawamoto, K.; Chijimatsu, R.; Kondo, N.; Hirotsu, T.; Ishii, H. Scent test using Caenorhabditis elegans to screen for early-stage pancreatic cancer. Oncotarget 2021, 12, 1687–1696. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, H.; Sasaki, K.; Hara, T.; Tsuji, Y.; Arao, Y.; Otsuka, C.; Hamano, Y.; Ogita, M.; Kobayashi, S.; di Luccio, E.; et al. Pancreatic Cancer Research beyond DNA Mutations. Biomolecules 2022, 12, 1503. https://doi.org/10.3390/biom12101503
Sato H, Sasaki K, Hara T, Tsuji Y, Arao Y, Otsuka C, Hamano Y, Ogita M, Kobayashi S, di Luccio E, et al. Pancreatic Cancer Research beyond DNA Mutations. Biomolecules. 2022; 12(10):1503. https://doi.org/10.3390/biom12101503
Chicago/Turabian StyleSato, Hiromichi, Kazuki Sasaki, Tomoaki Hara, Yoshiko Tsuji, Yasuko Arao, Chihiro Otsuka, Yumiko Hamano, Mirei Ogita, Shogo Kobayashi, Eric di Luccio, and et al. 2022. "Pancreatic Cancer Research beyond DNA Mutations" Biomolecules 12, no. 10: 1503. https://doi.org/10.3390/biom12101503