
The Role of Epstein-Barr Virus in Modulating Key Tumor Suppressor Genes in Associated Malignancies: Epigenetics, Transcriptional, and Post-Translational Modifications

 , , and
, , and
Abstract
:1. Introduction
2. EBV Associated Malignancies
2.1. Burkitt’s Lymphoma
2.2. Nasopharyngeal Carcinoma
2.3. T-Cell Lymphoma
2.4. Gastric Cancers
2.5. Breast Cancers
3. Key Oncoprotein and Tumor Suppressors Involved in EBV-Associated Malignancies
3.1. Proto-Oncogene/Oncogene Associated with EBV-Associated Burkitt’s Lymphoma
c-Myc
3.2. Tumor Suppressor/Oncogene Associated with Nasopharyngeal Carcinoma
3.2.1. p53
3.2.2. E-Cadherin
3.3. PD-L1 Tumor Suppressor/Oncogene Associated with T-Cell Lymphoma
4. Different Ways That EBV Genes/Gene Products Modulate Tumor Suppressors
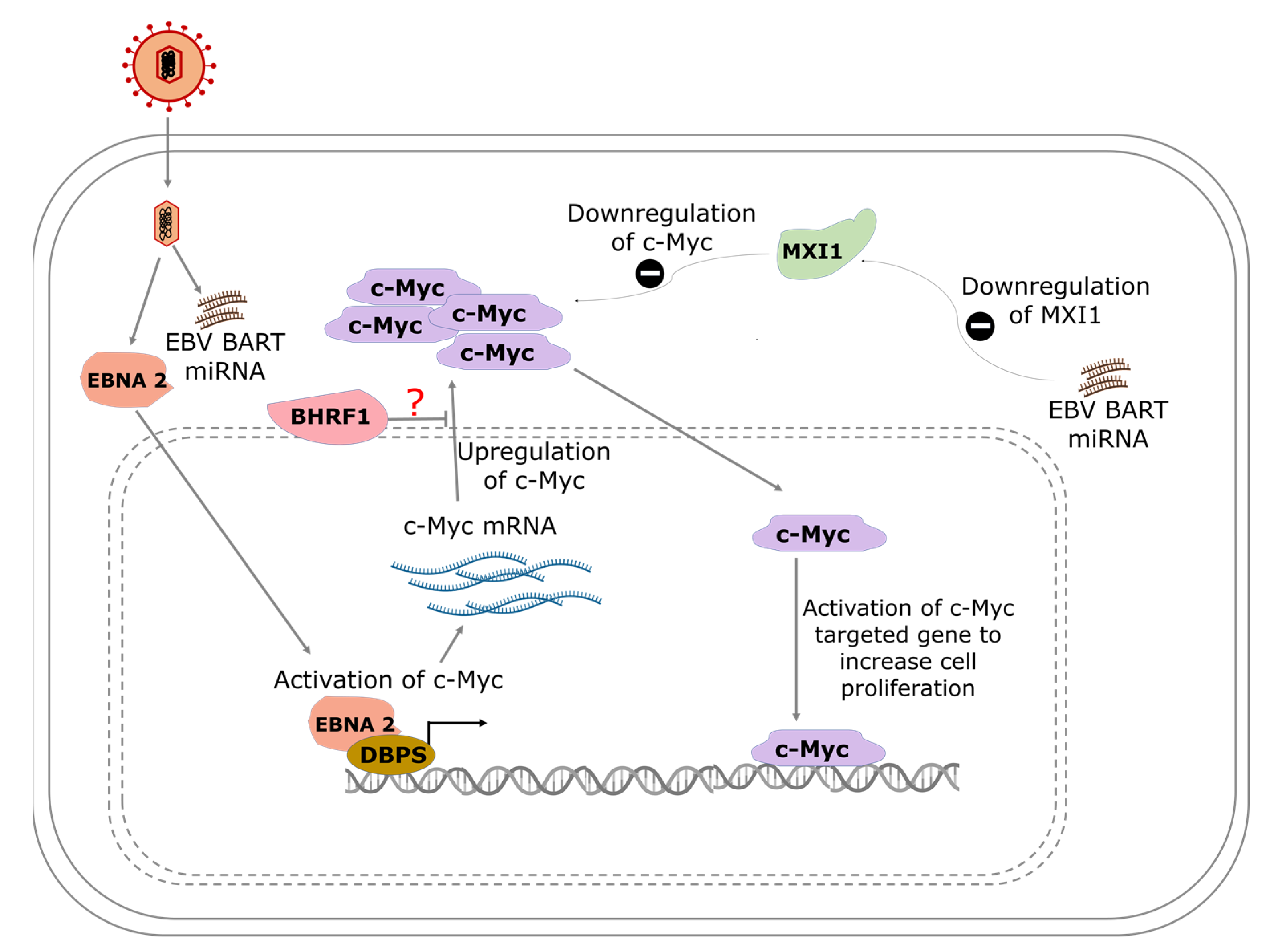
4.1. Modulation of c-Myc in BL and Its Effect on Transcription
4.1.1. The Role of EBNA-2
4.1.2. Regulation of c-Myc by EBV-Encoded MicroRNAs
4.1.3. Epigenetic Effect of EBV Modulation of c-Myc
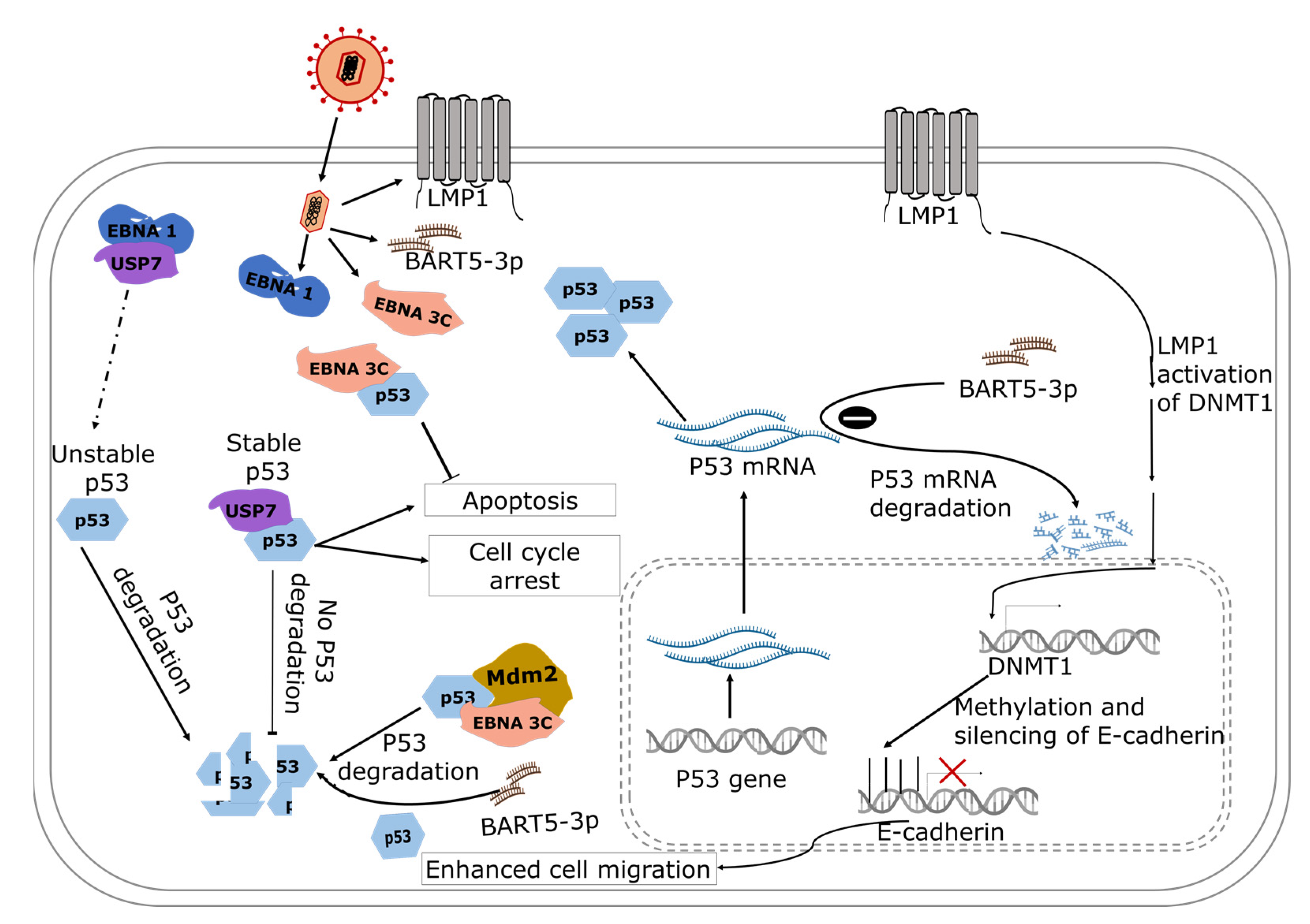
4.2. Modulation of p53 and E-Cadherin in Nasopharyngeal Carcinoma
4.2.1. The Role of miRNAs in EBV-Induced Transcriptional Regulation of p53
4.2.2. The Role of EBNA 3C in EBV-Induced Post-Translational Modification of p53
4.2.3. EBV-Induced Post-Translational Modification of p53: The Role of EBNA1
4.2.4. The Role of LMP1 in EBV-Induced Post-Translational Modification of p53
4.3. EBV-Induced Epigenetic Modulation of E-Cadherin in Nasopharyngeal Carcinoma
The Role of LMP1
4.4. Modulation of PD-L1 and c-Myc in T-Cell Lymphoma
The Role of LMP1
4.5. EBV Modulates Tumor Suppressor Genes in Gastric Cancers
4.5.1. The Role of LMP2A
4.5.2. The Role of EBV-Encoded miRNAs
4.6. EBV Modulates Tumor Suppressors in Breast Cancer
The Role of EBNA 3C
5. Expert Comments
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| EBV | Epstein-Barr virus |
| LMPs | Latent membrane proteins |
| EBNAs | Epstein-Barr virus nuclear antigens |
| NPC | Nasopharyngeal carcinoma |
| IKK | Inhibitory-κB kinase |
| NF-κB | Nuclear factor kappa B |
| CTARs | C-terminal activation regions |
| STAT | Signal transducer and activator of transcription |
| PI3K | Phosphatidylinositol 3-kinase |
| BL | Burkitt’s lymphoma |
| UICC | Union for International Cancer Control |
| AJCC | American Joint Committee on Cancer |
| TNM | Tumor, node, and metastasis |
| JAK | Janus kinase |
| mTOR | Mammalian target of rapamycin |
| EGFR | Epidermal growth factor receptor |
| MAPK | Mitogen-activated protein kinase |
| NHLs | Non-Hodgkin’s lymphomas |
| TCLs | T-cell lymphomas |
| PTCLs | Peripheral T-cell lymphomas |
| CTCLs | Cutaneous T-cell lymphomas |
| AITL | Angioimmunoblastic T-cell lymphoma |
| PD-L1 | Programmed death-ligand 1 |
| PTEN | Phosphatase and tensin homolog |
| CDKN2 | Cyclin-dependent kinase inhibitor 2 |
| Myc | Master regulator of cell cycle entry and proliferative metabolism |
| Bcl-2 | B-cell lymphoma 2 |
| Mdm2 | Mouse double minute 2 homolog |
| USP7 | Ubiquitin-specific-processing protease 7 |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| DNMT1 | DNA methyltransferase 1 |
| DKK1 | Dickkopf protein family |
References
- Smatti, M.K.; Yassine, H.M.; AbuOdeh, R.; AlMarawani, A.; Taleb, S.A.; Althani, A.A.; Nasrallah, G.K. Prevalence and molecular profiling of Epstein Barr virus (EBV) among healthy blood donors from different nationalities in Qatar. PLoS ONE 2017, 12, e0189033. [Google Scholar] [CrossRef] [Green Version]
- Santpere, G.; Darre, F.; Blanco, S.; Alcami, A.; Villoslada, P.; Albà, M.M.; Navarro, A. Genome-wide analysis of wild-type epstein-barr virus genomes derived from healthy individuals of the 1000 genomes project. Genome Biol. Evol. 2014, 6, 846–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Manners, O.; Murphy, J.C.; Coleman, A.; Hughes, D.J.; Whitehouse, A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 2018, 32, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Hutt-Fletcher, L.M. Epstein-Barr virus entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [Green Version]
- Hutt-Fletcher, L.M. The long and complicated relationship between Epstein-Barr virus and epithelial cells. J. Virol. 2017, 91, e01677-16. [Google Scholar] [CrossRef] [Green Version]
- Sathiyamoorthy, K.; Hu, Y.X.; Möhl, B.S.; Chen, J.; Longnecker, R.; Jardetzky, T.S. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 1–15. [Google Scholar] [CrossRef]
- Patrie, S.B.; Farrell, P.J. The role of Epstein-Barr virus in cancer. Expert Opin. Biol. Ther. 2006, 6, 1193–1205. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Khan, G.; Hashim, M.J. Global burden of deaths from epstein-barr virus attributable malignancies 1990–2010. Infect. Agent. Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Frappier, L. The Epstein-Barr virus EBNA1 protein. Scientifica (Cairo). 2012, 2012, 438204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentine, R.; Dawson, C.W.; Hu, C.; Shah, K.M.; Owen, T.J.; Date, K.L.; Maia, S.P.; Shao, J.; Arrand, J.R.; Young, L.S.; et al. Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-κB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol. Cancer 2010, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Pratt, Z.L.; Zhang, J.; Sugden, B. The Latent Membrane Protein 1 (LMP1) Oncogene of Epstein-Barr virus can simultaneously induce and inhibit apoptosis in B cells. J. Virol. 2012, 86, 4380–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, C.W.; Tramountanis, G.; Eliopoulos, A.G.; Young, L.S. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J. Biol. Chem. 2003, 278, 3694–3704. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, H.; Yoshizaki, T.; Miller, W.E.; Sato, H.; Furukawa, M.; Pagano, J.S.; Raab-Traub, N. Matrix metalloproteinase 9 expression Is induced by Epstein-Barr virus latent membrane protein 1 C-terminal activation regions 1 and 2. J. Virol. 1999, 73, 5548–5555. [Google Scholar] [CrossRef] [Green Version]
- Johnston, W.T.; Mutalima, N.; Sun, D.; Emmanuel, B.; Bhatia, K.; Aka, P.; Wu, X.; Borgstein, E.; Liomba, G.N.; Kamiza, S.; et al. Relationship between Plasmodium falciparum malaria prevalence, genetic diversity and endemic Burkitt lymphoma in Malawi. Sci. Rep. 2014, 4, 3741. [Google Scholar] [CrossRef] [PubMed]
- Orem, J.; Mbidde, E.K.; Lambert, B.; De Sanjose, S.; Weiderpass, E. Burkitt’s lymphoma in Africa, a review of the epidemiology and etiology. Afr. Health Sci. 2007, 7, 166–175. [Google Scholar] [CrossRef]
- Hämmerl, L.; Colombet, M.; Rochford, R.; Ogwang, D.M.; Parkin, D.M. The burden of Burkitt lymphoma in Africa. Infect. Agent. Cancer 2019, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ferry, J.A. Burkitt’s lymphoma: Clinicopathologic features and differential diagnosis. Oncologist 2006, 11, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Rochford, R.; Cannon, M.J.; Moormann, A.M. Opinion—tropical infectious diseases: Endemic Burkitt’s lymphoma: A polymicrobial disease? Nat. Rev. Microbiol. 2005, 3, 182–187. [Google Scholar] [CrossRef]
- Kalisz, K.; Alessandrino, F.; Beck, R.; Smith, D.; Kikano, E.; Ramaiya, N.H.; Tirumani, S.H. An update on Burkitt lymphoma: A review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Brady, G.; MacArthur, G.J.; Farrell, P.J. Epstein-Barr virus and Burkitt lymphoma. Postgrad. Med. J. 2008, 84, 372–377. [Google Scholar] [CrossRef]
- Ayee, R.; Ofori, M.E.O.; Wright, E.; Quaye, O. Epstein Barr virus associated lymphomas and epithelia cancers in humans. J. Cancer 2020, 11, 1737–1750. [Google Scholar] [CrossRef] [Green Version]
- Henle, W.; Henle, G.; Lennette, E.T. The Epstein-Barr virus. Sci. Am. 1979, 241, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.C.; Yuan, J.M. Epidemiology of nasopharyngeal carcinoma. Semin. Cancer Biol. 2002, 12, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Long, J.; Li, G.; Yan, H.; Feng, G.; Liu, M.; Zhu, J.; Wang, R. A new staging system for nasopharyngeal carcinoma based on intensity-modulated radiation therapy: Results of a prospective multicentric clinical study. Oncotarget 2016, 7, 15252–15261. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.L.; Wang, Y.; Liang, S.B.; He, S.S.; Chen, D.M.; Chen, H.Y.; Lu, L.X.; Chen, Y. Comparison of the seventh and eighth editions of the UICC/AJCC staging system for nasopharyngeal carcinoma: Analysis of 1317 patients treated with intensity-modulated radiotherapy at two centers. BMC Cancer 2018, 18, 1–11. [Google Scholar] [CrossRef]
- Wang, H.Y.; Chang, Y.L.; To, K.F.; Hwang, J.S.G.; Mai, H.Q.; Feng, Y.F.; Chang, E.T.; Wang, C.P.; Kam, M.K.M.; Cheah, S.L.; et al. A new prognostic histopathologic classification of nasopharyngeal carcinoma. Chin. J. Cancer 2016, 35, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.R.; Xu, Y.; Liu, J.; Zhang, W.J.; Liang, Z.H. Histopathological classification of nasopharyngeal carcinoma. Asian Pacific J. Cancer Prev. 2011, 12, 1141–1147. [Google Scholar]
- Brennan, B. Nasopharyngeal carcinoma. Orphanet J. Rare Dis. 2006, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.H.; Yu, I.T.-S.; Tse, L.A.; Au, J.S.K.; Lau, J.S.M. Occupational risk factors for nasopharyngeal carcinoma in Hong Kong Chinese: A case-referent study. Int. Arch. Occup. Environ. Health 2017, 90, 443–449. [Google Scholar] [CrossRef]
- Guo, X.; Johnson, R.C.; Deng, H.; Liao, J.; Guan, L.; Nelson, G.W.; Tang, M.; Zheng, Y.; De The, G.; O’Brien, S.J.; et al. Evaluation of nonviral risk factors for nasopharyngeal carcinoma in a high-risk population of southern China. Int. J. Cancer 2009, 124, 2942–2947. [Google Scholar] [CrossRef] [Green Version]
- Breda, E.; Catarino, R.J.F.; Azevedo, I.; Lobão, M.; Monteiro, E.; Medeiros, R. Epstein-Barr virus detection in nasopharyngeal carcinoma—Implications in a low-risk area. Braz. J. Otorhinolaryngol. 2010, 76, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Li, C.; Pan, L. Nasopharyngeal carcinoma: A review of current updates. Exp. Ther. Med. 2018, 15, 3687–3692. [Google Scholar] [CrossRef] [Green Version]
- Marquitz, A.R.; Mathur, A.; Edwards, R.H.; Raab-Traub, N. Host gene expression is regulated by two types of noncoding RNAs transcribed from the Epstein-Barr virus BamHI A rightward transcript region. J. Virol. 2015, 89, 11256–11268. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Zong, J.; Lin, W.; Wang, M.; Xu, Y.; Zhou, R.; Lin, S.; Guo, Q.; Chen, H.; Ye, Y.; et al. EBV-miR-BART8-3p induces epithelial-mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma cells through activating NF-κB and Erk1/2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef]
- Richardo, T.; Prattapong, P.; Ngernsombat, C.; Wisetyaningsih, N.; Iizasa, H.; Yoshiyama, H.; Janvilisri, T. Epstein-Barr Virus mediated signaling in nasopharyngeal carcinoma carcinogenesis. Cancers 2020, 12, 2441. [Google Scholar] [CrossRef]
- Jiang, N.; Liu, N.; Yang, F.; Zhou, Q.; Cui, R.; Jiang, W.; He, Q.; Li, W.; Guo, Y.; Zeng, J.; et al. Hotspot mutations in common oncogenes are infrequent in nasopharyngeal carcinoma. Oncol. Rep. 2014, 32, 1661–1669. [Google Scholar] [CrossRef]
- Effert, P.; McCoy, R.; Abdel-Hamid, M.; Flynn, K.; Zhang, Q.; Busson, P.; Tursz, T.; Liu, E.; Raab-Traub, N. Alterations of the p53 gene in nasopharyngeal carcinoma. J. Virol. 1992, 66, 3768–3775. [Google Scholar] [CrossRef] [Green Version]
- Armitage, J.O. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood 1997, 89, 3909–3918. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Lee Harris, N.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, M.A.; Evens, A.M.; Tallman, M.S.; Nelson, B.P.; Rosen, S.T.; Nhls, T. Review article T-cell non-Hodgkin lymphoma. Blood 2006, 107, 1255–1265. [Google Scholar] [CrossRef]
- Asano, N.; Kato, S.; Nakamura, S. Epstein-Barr virus-associated natural killer/T-cell lymphomas. Best Pract. Res. Clin. Haematol. 2013, 26, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.; Veldman, R.; Lechowicz, M.J. T-cell lymphoma epidemiology: The known and unknown. Curr. Hematol. Malig. Rep. 2016, 11, 492–503. [Google Scholar] [CrossRef]
- Bennani, N.N.; Ansell, S.M. Peripheral T-cell Lymphoma not Otherwise Specified. In The Peripheral T-Cell Lymphomas; O’Connor, O.A., Seog, K.W., Zinzani, P.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; pp. 105–114. [Google Scholar]
- Smith, A.; Crouch, S.; Lax, S.; Li, J.; Painter, D.; Howell, D.; Patmore, R.; Jack, A.; Roman, E. Lymphoma incidence, survival and prevalence 2004–2014: Sub-type analyses from the UK’s Haematological Malignancy Research Network. Br. J. Cancer 2015, 112, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Flowers, C.R.; Kadin, M.E.; Chang, E.T.; Hughes, A.M.; Ansell, S.M.; Feldman, A.L.; Lightfoot, T.; Boffetta, P.; Melbye, M.; et al. Medical history, lifestyle, family history, and occupational risk factors for peripheral T-cell lymphomas: The interlymph non-hodgkin lymphoma subtypes project. J. Natl. Cancer Inst. Monogr. 2014, 48, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Khan, G.; Norton, A.J.; Slavin, G. Epstein-Barr virus in angioimmunoblastic T-cell lymphomas. Histopathology 1993, 22, 145–150. [Google Scholar] [CrossRef]
- Delfau-Larue, M.H.; de Leval, L.; Joly, B.; Plonquet, A.; Challine, D.; Parrens, M.; Delmer, A.; Salles, G.; Morschhauser, F.; Delarue, R.; et al. Targeting intratumoral B cells with rituximab in addition to CHOP in angioimmunoblastic T-cell lymphoma. A clinicobiological study of the GELA. Haematologica 2012, 97, 1594–1602. [Google Scholar] [CrossRef]
- Ahmed, N.; Feldman, A.L. Targeting epigenetic regulators in the treatment of T-cell lymphoma. Expert Rev. Hematol. 2020, 13, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Küçük, C.; Wang, J.; Xiang, Y.; You, H. Epigenetic aberrations in natural killer/T-cell lymphoma: Diagnostic, prognostic and therapeutic implications. Ther. Adv. Med. Oncol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric cancer: Epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Nakamura, M.; Saito, M.; Sasaki, S.; Shimokuri, K.; Yanagihara, M.; Sakai, K.; Suehiro, Y.; et al. The role of epigenetic regulation in Epstein-Barr virus-associated gastric cancer. Int. J. Mol. Sci. 2017, 18, 1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Kim, S.H.; Han, S.H.; An, J.S.; Lee, E.S.; Kim, Y.S. Clinicopathological and molecular characteristics of Epstein-Barr virus-associated gastric carcinoma: A meta-analysis. J. Gastroenterol. Hepatol. 2009, 24, 354–365. [Google Scholar] [CrossRef]
- Tavakoli, A.; Monavari, S.H.; Solaymani Mohammadi, F.; Kiani, S.J.; Armat, S.; Farahmand, M. Association between Epstein-Barr virus infection and gastric cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Jácome, A.A.; de Lima, E.M.; Kazzi, A.I.; Chaves, G.F.; de Mendonça, D.C.; Maciel, M.M.; dos Santos, J.S. Epstein-Barr virus-positive gastric cancer: A distinct molecular subtype of the disease? Rev. Soc. Bras. Med. Trop. 2016, 49, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Farahmand, M.; Monavari, S.H.; Shoja, Z.; Ghaffari, H.; Tavakoli, M.; Tavakoli, A. Epstein-Barr virus and risk of breast cancer: A systematic review and meta-analysis. Futur. Oncol. 2019, 15, 2873–2885. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Torabizadeh, Z.; Nadji, A.; Naghshvar, F.; Nosrati, A.; Parsa, M. Association between Epstein-Barr virus (EBV) and breast cancer. Res. Mol. Med. 2014, 2, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Glenn, W.K.; Whitaker, N.J.; Lawson, J.S. High risk human papillomavirus and Epstein Barr virus in human breast milk. BMC Res. Notes 2012, 5, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Labrecque, L.G.; Barnes, D.M.; Fentiman, I.S.; Griffin, B.E. Epstein-Barr virus in epithelial cell tumors: A breast cancer study. Cancer Res. 1995, 55, 39–45. [Google Scholar]
- Hu, H.; Luo, M.L.; Desmedt, C.; Nabavi, S.; Yadegarynia, S.; Hong, A.; Konstantinopoulos, P.A.; Gabrielson, E.; Hines-Boykin, R.; Pihan, G.; et al. Epstein-Barr virus infection of mammary epithelial cells promotes malignant transformation. EBioMedicine 2016, 9, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadivar, M.; Monabati, A.; Joulaee, A.; Hosseini, N. Epstein-Barr virus and breast cancer: Lack of evidence for an association in Iranian women. Pathol. Oncol. Res. 2011, 17, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Morales-Sánchez, A.; Molina-Muñoz, T.; Martínez-López, J.L.E.; Hernández-Sancén, P.; Mantilla, A.; Leal, Y.A.; Torres, J.; Fuentes-Pananá, E.M. No association between Epstein-Barr virus and mouse mammary tumor virus with breast cancer in Mexican women. Sci. Rep. 2013, 3, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, A.J.; Moalwi, M.H.; Amoaten, T.; Sinclair, A.J.; Moalwi, M.H.; Accardi-Gheit, R.; Leoncini, L.; Mundo, L. Is EBV associated with breast cancer in specific geographic locations? Cancers 2021, 13, 819. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Wu, C.F.; Rajasekaran, N.; Shin, Y.K. Loss of tumor suppressor gene function in human cancer: An overview. Cell. Physiol. Biochem. 2019, 51, 2647–2693. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Inman, G.J. Developments in Burkitt’s lymphoma: Novel cooperations in oncogenic MYC signaling. Cancer Manag. Res. 2014, 6, 27–38. [Google Scholar] [PubMed] [Green Version]
- Marcu, K.B.; Patel, A.J.; Yang, Y. Differential regulation of the c-MYC P1 and P2 promoters in the absence of functional tumor suppressors: Implications for mechanisms of deregulated MYC transcription. Curr. Top. Microbiol. Immunol. 1997, 224, 47–56. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-specific DNA binding by the c-Myc protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef]
- Gerbitz, A.; Mautner, J.; Geltinger, C.; Hörtnagel, K.; Christoph, B.; Asenbauer, H.; Klobeck, G.; Polack, A.; Bornkamm, G.W. Deregulation of the proto-oncogene c-myc through t(8;22) translocation in Burkitt’s lymphoma. Oncogene 1999, 18, 1745–1753. [Google Scholar] [CrossRef] [Green Version]
- Taub, R.; Moulding, C.; Battey, J.; Murphy, W.; Vasicek, T.; Lenoir, G.M.; Leder, P. Activation and somatic mutation of the translocated c-myc gene in Burkitt lymphoma cells. Cell 1984, 36, 339–348. [Google Scholar] [CrossRef]
- Murono, S.; Yoshizaki, T.; Park, C.S.; Furukawa, M. Association of Epstein-Barr virus infection with p53 protein accumulation but not bcl-2 protein in nasopharyngeal carcinoma. Histopathology 1999, 34, 432–438. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessahaye, G.; Ibrahim, M.E. Breast Cancer as an Epstein-Barr Virus (EBV)-Associated Malignancy. In Breast Cancer-From Biology to Medicine; Van Pham, P., Ed.; IntechOpen: Lodon, UK, 2017; pp. 43–60. [Google Scholar]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riethmacher, D.; Brinkmanni, V.; Birchmeier, C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc. Natl. Acad. Sci. USA 1995, 92, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galera-Ruiz, H.; Ríos, M.J.; González-Cámpora, R.; De Miguel, M.; Carmona, M.I.; Moreno, A.M.; Galera-Davidson, H. The cadherin-catenin complex in nasopharyngeal carcinoma. Eur. Arch. Oto-Rhino-Laryngology 2011, 268, 1335–1341. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.Y.; Yi, Y.H.; Chang, K.P.; Chang, Y.S.; Chen, S.J.; Chen, H.C. The Epstein-Barr virus-encoded microRNA MiR-BART9 promotes tumor metastasis by targeting E-Cadherin in nasopharyngeal carcinoma. PLoS Pathog. 2014, 10, e1003974. [Google Scholar] [CrossRef]
- Zheng, Z.; Pan, J.; Chu, B.; Wong, Y.C.; Cheung, A.L.M.; Tsao, S.W. Downregulation and abnormal expression of E-cadherin and β-catenin in nasopharyngeal carcinoma: Close association with advanced disease stage and lymph node metastasis. Hum. Pathol. 1999, 30, 458–466. [Google Scholar] [CrossRef]
- Youn Kim, W.; Young Jung, H.; Jeong Nam, S.; Min Kim, T.; Seog Heo, D.; Kim, C.-W.; Kyung Jeon, Y. Expression of programmed cell death ligand 1 (PD-L1) in advanced stage EBV-associated extranodal NK/T cell lymphoma is associated with better prognosis. Virchows Arch. 2011, 469, 581–590. [Google Scholar] [CrossRef]
- Bi, X.; Wang, H.; Zhang, W.; Wang, J.; Liu, W.; Xia, Z.; Huang, H.; Jiang, W.; Zhang, Y.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Rossille, D.; Gressier, M.; Damotte, D.; Maucort-Boulch, D.; Pangault, C.; Semana, G.; Le Gouill, S.; Haioun, C.; Tarte, K.; Lamy, T.; et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-cell lymphoma: Results from a French multicenter clinical trial. Leukemia 2014, 28, 2367–2375. [Google Scholar] [CrossRef]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polack, A.; Hörtnagel, K.; Pajic, A.; Christoph, B.; Baier, B.; Falk, M.; Mautner, J.; Geltinger, C.; Bornkamm, G.W.; Kempkes, B. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc. Natl. Acad. Sci. USA 1996, 93, 10411–10416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, T.; Ling, P.D.; Hayward, S.D.; Peterson, M.G. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein Jκ. Science 1994, 265, 92–95. [Google Scholar] [CrossRef]
- Johannsen, E.; Koh, E.; Mosialos, G.; Tong, X.; Kieff, E.; Grossman, S.R. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J kappa and PU.1. J. Virol. 1995, 69, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.W.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [Green Version]
- Keller, A.; Rounge, T.; Backes, C.; Ludwig, N.; Gislefoss, R.; Leidinger, P.; Langseth, H.; Meese, E. Sources to variability in circulating human miRNA signatures. RNA Biol. 2017, 14, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Roser, A.E.; Gomes, L.C.; Schünemann, J.; Maass, F.; Lingor, P. Circulating miRNAs as diagnostic biomarkers for Parkinson’s disease. Front. Neurosci. 2018, 12, 1–9. [Google Scholar] [CrossRef]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating MicroRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef]
- Guay, C.; Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, Y.; Fu, M.; Zheng, B.; Qiu, Q.; Huang, Z. Implication of viral microRNAs in the genesis and diagnosis of Epstein-Barr virus-associated tumors (Review). Oncol. Lett. 2019, 18, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Ardekani, A.M.; Naeini, M.M. The role of microRNAs in human diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; van de Garde, M.D.B.; Middeldorp, J.M. Viral miRNAs exploiting the endosomal-exosomal pathway for intercellular cross-talk and immune evasion. Biochim. Biophys. Acta-Gene Regul. Mech. 2011, 1809, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Karran, L.; Gao, Y.; Smith, P.R.; Griffin, B.E. Expression of a family of complementary-strand transcripts in Epstein-Barr virus-infected cells. Proc. Natl. Acad. Sci. USA 1992, 89, 8058–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Guo, Z.; Shu, Y.; Zhou, H.; Wang, H.; Zhang, W. BART miRNAs: An unimaginable force in the development of nasopharyngeal carcinoma. Eur. J. Cancer Prev. 2017, 26, 144–150. [Google Scholar] [CrossRef]
- Yamamoto, T.; Iwatsuki, K. Diversity of Epstein-Barr virus BamHI-A rightward transcripts and their expression patterns in lytic and latent infections. J. Med. Microbiol. 2012, 61, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Giulino-Roth, L.; Wang, K.; MacDonald, T.Y.; Mathew, S.; Tam, Y.; Cronin, M.T.; Palmer, G.; Lucena-Silva, N.; Pedrosa, F.; Pedrosa, M.; et al. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood 2012, 120, 5181–5184. [Google Scholar] [CrossRef]
- Scott, R.S. Epstein-Barr virus: A master epigenetic manipulator. Curr. Opin. Virol. 2017, 26, 74–80. [Google Scholar] [CrossRef]
- Ruf, I.K.; Rhyne, P.W.; Yang, H.; Borza, C.M.; Hutt-Fletcher, L.M.; Cleveland, J.L.; Sample, J.T. Epstein-Barr virus regulates c-MYC, apoptosis, and tumorigenicity in Burkitt lymphoma. Mol. Cell. Biol. 1999, 19, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Wang, J.; Wei, L.; Peng, Q.; Gao, Y.; Fu, Y.; Lu, Y.; Qin, Z.; Zhang, X.; Lu, J.; et al. Epstein-Barr virus microRNA miR-BART5-3p inhibits p53 expression. J. Virol. 2018, 92, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.L.; Freund, S.M.V.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Regulation of DNA binding of p53 by its C-terminal domain. J. Mol. Biol. 2004, 342, 801–811. [Google Scholar] [CrossRef]
- Knight, J.S.; Sharma, N.; Robertson, E.S. SCF Skp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol. Cell. Biol. 2005, 25, 1749–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.; Murakami, M.; Kumar, P.; Bajaj, B.; Sims, K.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J. Virol. 2009, 83, 4652–4669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to epstein-barr nuclear antigen 1: Implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W.; Xiao, L.; Xu, J.; Chen, X.; Tang, M.; Dong, Z.; Tao, Q.; Cao, Y. Cell Cycle Viral oncoprotein LMP1 disrupts p53-induced cell cycle arrest and apoptosis through modulating K63-linked ubiquitination of p53 View supplementary material. Cell Cycle 2012, 11, 2327–2336. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhou, S.; Chen, X.; Guo, L.; Li, Z.; Hu, D.; Luo, X.; Ma, X.; Tang, M.; Yi, W.; et al. The activation of p53 mediated by Epstein-Barr virus latent membrane protein 1 in SV40 large T-antigen transformed cells. FEBS Lett. 2008, 582, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Kashuba, E.; Yurchenko, M.; Yenamandra, S.P.; Snopok, B.; Szekely, L.; Bercovich, B.; Ciechanover, A.; Klein, G. Epstein-Barr virus-encoded EBNA-5 forms trimolecular protein complexes with MDM2 and p53 and inhibits the transactivating function of p53. Int. J. Cancer 2011, 128, 817–825. [Google Scholar] [CrossRef]
- Tsai, C.N.; Tsai, C.L.; Tse, K.P.; Chang, H.Y.; Chang, Y.S. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the down-regulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH 2-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [Green Version]
- Bruce, J.P.; To, K.F.; Lui, V.W.Y.; Chung, G.T.Y.; Chan, Y.Y.; Tsang, C.M.; Yip, K.Y.; Ma, B.B.Y.; Woo, J.K.S.; Hui, E.P.; et al. Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-κB activation and immune escape. Nat. Commun. 2021, 12, 4193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.P.; Chang, Y.S. Epstein-Barr virus latent membrane protein 1: Structure and functions. J. Biomed. Sci. 2003, 10, 490–504. [Google Scholar] [CrossRef]
- Floettmann, J.E.; Eliopoulos, A.G.; Jones, M.; Young, L.S.; Rowe, M. Epstein-Barr virus latent membrane protein-1 (LMP1) signalling is distinct from CD40 and involves physical cooperation of its two C-terminus functional regions. Oncogene 1998, 17, 2383–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Young, L.S. LMP1 structure and signal transduction. Semin. Cancer Biol. 2001, 11, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Natkunam, Y. Extranodal NK/T-cell lymphomas: The role of natural killer cells and EBV in lymphomagenesis. Int. J. Mol. Sci. 2020, 21, 1501. [Google Scholar] [CrossRef] [Green Version]
- Fukayama, M. Epstein-Barr virus and gastric carcinoma. Pathol. Int. 2010, 60, 337–350. [Google Scholar] [CrossRef]
- Fukayama, M.; Hino, R.; Uozaki, H. Epstein-Barr virus and gastric carcinoma: Virus–host interactions leading to carcinoma. Cancer Sci. 2008, 99, 1726–1733. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Q.; Cheung, K.F.; Kang, W.; Lung, R.W.M.; Tong, J.H.M.; To, K.F.; Sung, J.J.Y.; Yu, J. Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer 2013, 119, 304–312. [Google Scholar] [CrossRef]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.W.; Choi, Y.H.; Kwon, O.K.; Lee, S.S.; Chung, H.Y.; Yu, W.; Bae, H.I.; Seo, A.N.; Kang, H.; Lee, S.K.; et al. High level of viral microRNA-BART20-5p expression is associated with worse survival of patients with Epstein-Barr virus associated gastric cancer. Oncotarget 2017, 8, 14988–14994. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Choi, H.; Lee, S.K. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015, 356, 733–742. [Google Scholar] [CrossRef]
- Min, K.; Kyeong, L.S. EBV miR-BART10-3p promotes cell proliferation and migration by targeting DKK1. Int. J. Biol. Sci. 2019, 15, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Park, M.C.; Kim, H.; Choi, H.; Chang, M.S.; Lee, S.K. Epstein-Barr virus miR-BART1-3p regulates the miR-17-92 cluster by targeting E2F3. Int. J. Mol. Sci. 2021, 22, 936. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Cotter, M.A.; Robertson, E.S. Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-H1: A molecular link to cancer metastasis. Nat. Med. 2001, 7, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Mátyási, B.; Farkas, Z.; Kopper, L.; Sebestyén, A.; Boissan, M.; Mehta, A.; Takács-Vellai, K. The function of NM23-H1/NME1 and its homologs in major processes linked to metastasis. Pathol. Oncol. Res. 2020, 26, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Kamara, S.; Cen, D.; Tang, W.; Gu, M.; Ci, X.; Chen, J.; Wang, L.; Zhu, S.; Jiang, P.; et al. Generation of novel affibody molecules targeting the EBV LMP2A N-terminal domain with inhibiting effects on the proliferation of nasopharyngeal carcinoma cells. Cell Death Dis. 2020, 11, 213, Correction Cell Death Dis. 2020, 11, 494. [Google Scholar] [CrossRef]
- Zheng, C.X.; Yan, L.; Nilsson, B.; Eklund, G.; Drettner, B. Epstein-Barr virus infection, salted fish and nasopharyngeal carcinoma: A case-control study in southern. Acta Oncol. (Madr). 1994, 33, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Bakkalci, D.; Jia, Y.; Winter, J.R.; Lewis, J.E.A.; Taylor, G.S.; Stagg, H.R. Risk factors for Epstein Barr virus-associated cancers: A systematic review, critical appraisal, and mapping of the epidemiological evidence. J. Glob. Health 2020, 10, 10405. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer | Tumor Suppressor/Oncoprotein | EBV Modulators |
|---|---|---|
| Burkitt’s lymphoma | c-Myc | EBNA-2 |
| EBV-encoded micro-RNAs | ||
| BHRF1 | ||
| Nasopharyngeal carcinoma | p53 | EBV-miR-BART5-3p |
| EBNA 3C | ||
| EBNA1 | ||
| LMP1 | ||
| EBNA2 | ||
| E-cadherin | LMP1 | |
| T-cell lymphoma | PD-L1 [74] | LMP1 |
| Gastric cancers | PTEN, CDKN2, CDH1, p15, p73, etc. | LMP2A |
| EBV-encoded microRNAs | ||
| Breast cancer | Nm23-H1 | EBNA 3C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fierti, A.O.; Yakass, M.B.; Okertchiri, E.A.; Adadey, S.M.; Quaye, O. The Role of Epstein-Barr Virus in Modulating Key Tumor Suppressor Genes in Associated Malignancies: Epigenetics, Transcriptional, and Post-Translational Modifications. Biomolecules 2022, 12, 127. https://doi.org/10.3390/biom12010127
Fierti AO, Yakass MB, Okertchiri EA, Adadey SM, Quaye O. The Role of Epstein-Barr Virus in Modulating Key Tumor Suppressor Genes in Associated Malignancies: Epigenetics, Transcriptional, and Post-Translational Modifications. Biomolecules. 2022; 12(1):127. https://doi.org/10.3390/biom12010127
Chicago/Turabian StyleFierti, Adelaide Ohui, Michael Bright Yakass, Ernest Adjei Okertchiri, Samuel Mawuli Adadey, and Osbourne Quaye. 2022. "The Role of Epstein-Barr Virus in Modulating Key Tumor Suppressor Genes in Associated Malignancies: Epigenetics, Transcriptional, and Post-Translational Modifications" Biomolecules 12, no. 1: 127. https://doi.org/10.3390/biom12010127