
Hydrogen Sulphide Treatment Prevents Renal Ischemia-Reperfusion Injury by Inhibiting the Expression of ICAM-1 and NF-kB Concentration in Normotensive and Hypertensive Rats

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Hypertension
2.3. Exogenous Administration of NaHS
2.4. Administration of PAG
2.5. Drugs and Chemicals
2.6. Experimental Protocol
2.7. Collection of Metabolic Data
2.8. Non-Invasive Blood Pressure (NIBP) Measurment
2.9. Acute Experiment
2.10. Collection of Blood and Urine Samples
2.11. Measurement of Renal Functional Parameters
2.12. Measurement of H2S Concentration
2.13. Measurement of Oxidative Stress Markers
2.14. ICAM-1 and NF-kB Measurement in the Plasma and Kidney Tissues of WKY and L-NAME-Treated Groups
2.15. Histopathology of Rat Kidney Tissues of WKY and L-NAME Groups
2.16. Statistical Analysis
3. Results
3.1. Systemic Hemodynamic Parameters
3.2. Renal Functional Parameters
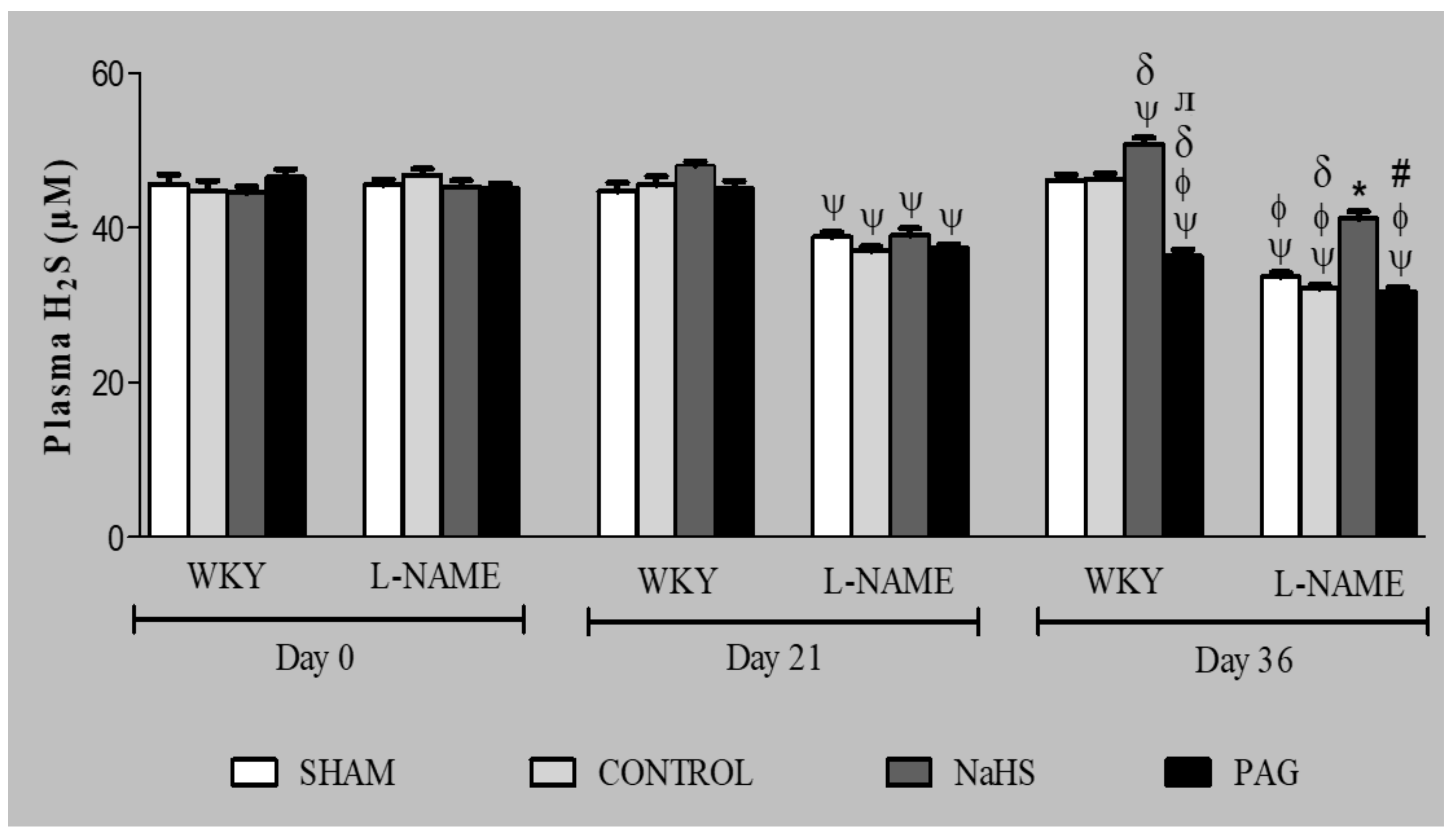
3.3. H2S Concentration in Plasma
3.4. Systemic Hemodynamics on Acute Experiment Day
3.5. Body Weight, Kidney Weight and Kidney Index
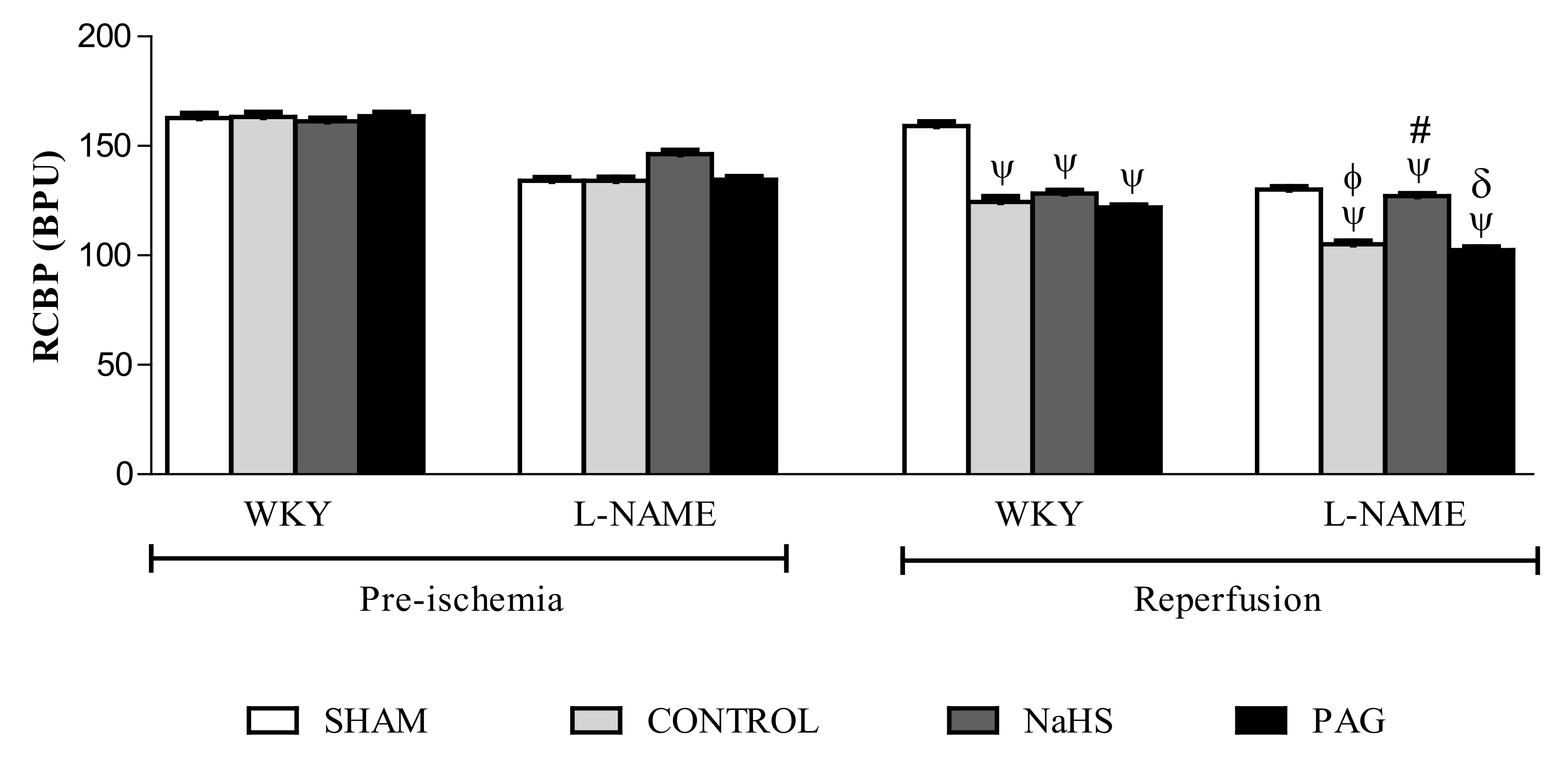
3.6. RCBP in the Pre-Ischemia and Reperfusion Phases
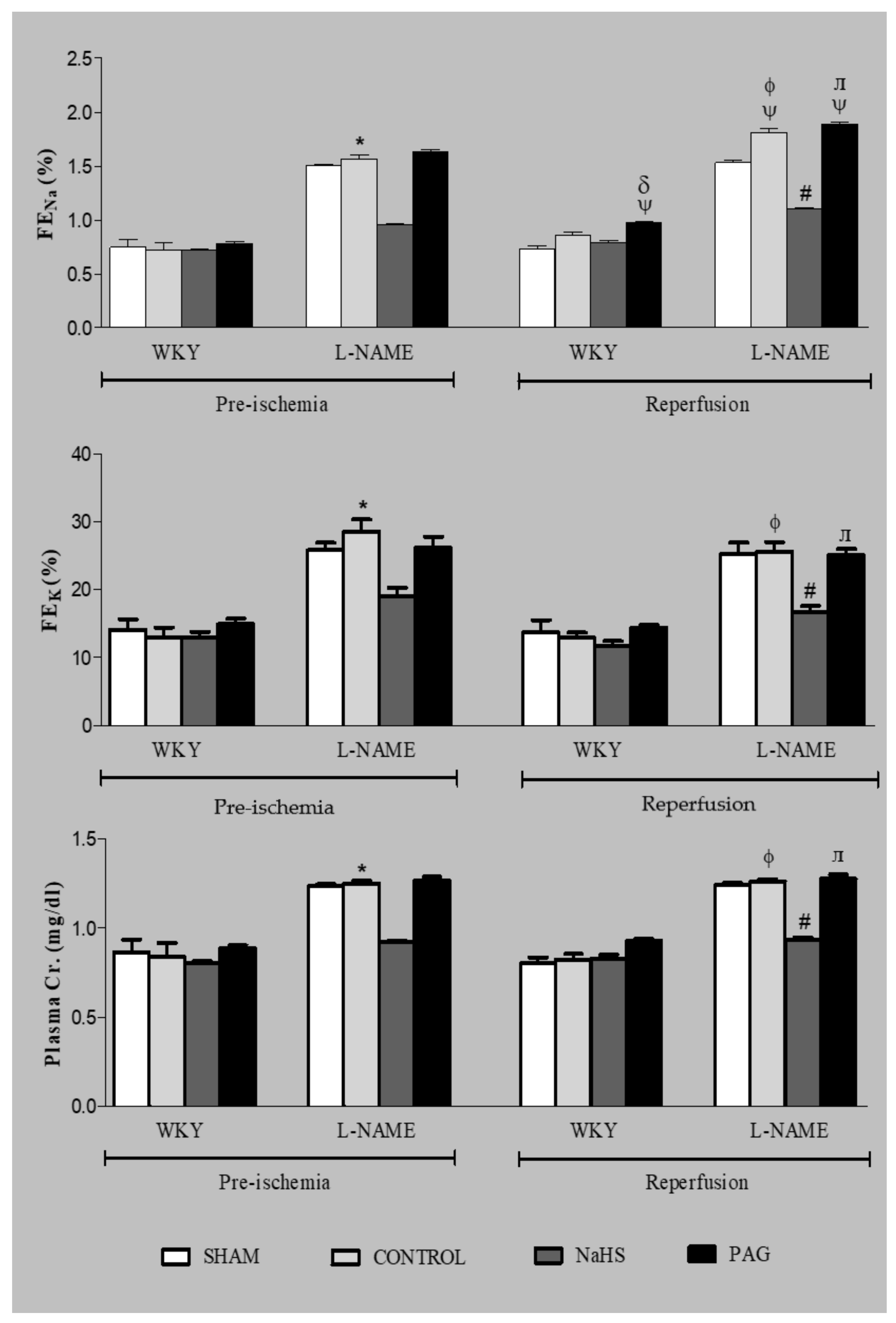
3.7. Renal Functional Parameters at the Pre-Ischemia and Reperfusion Phases
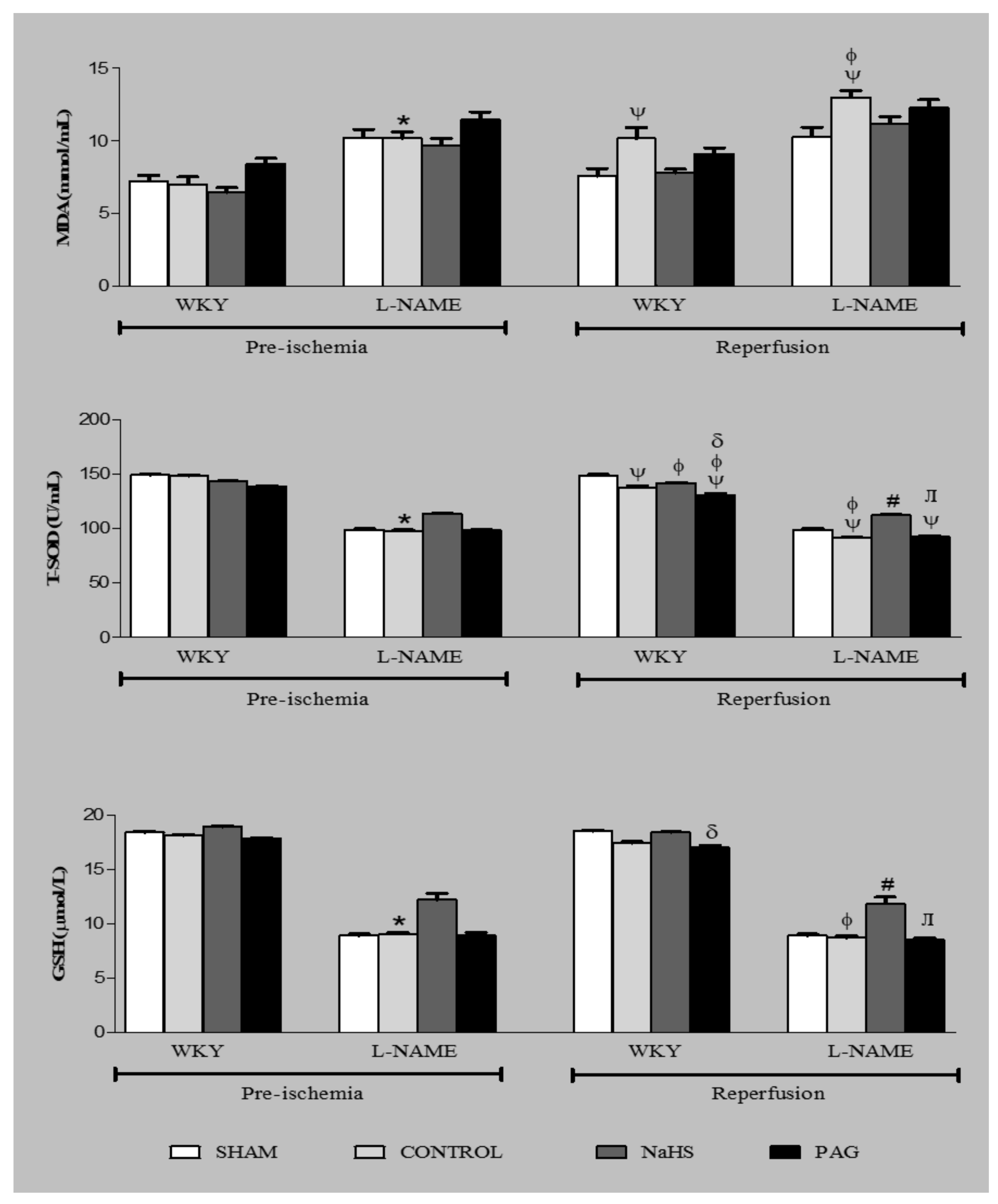
3.8. Oxidative Stress Markers at the Pre-Ischemia and Reperfusion Phases
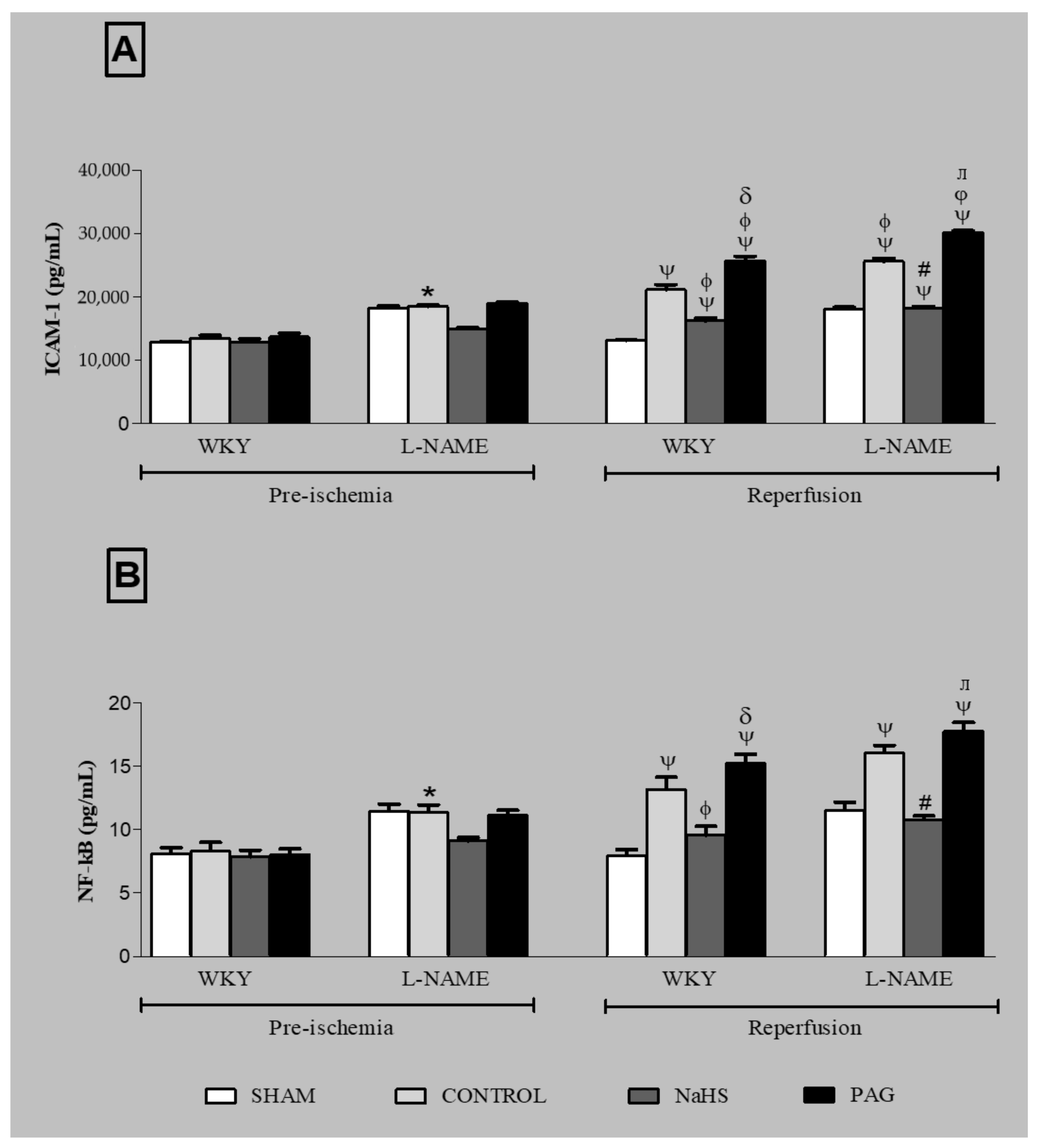
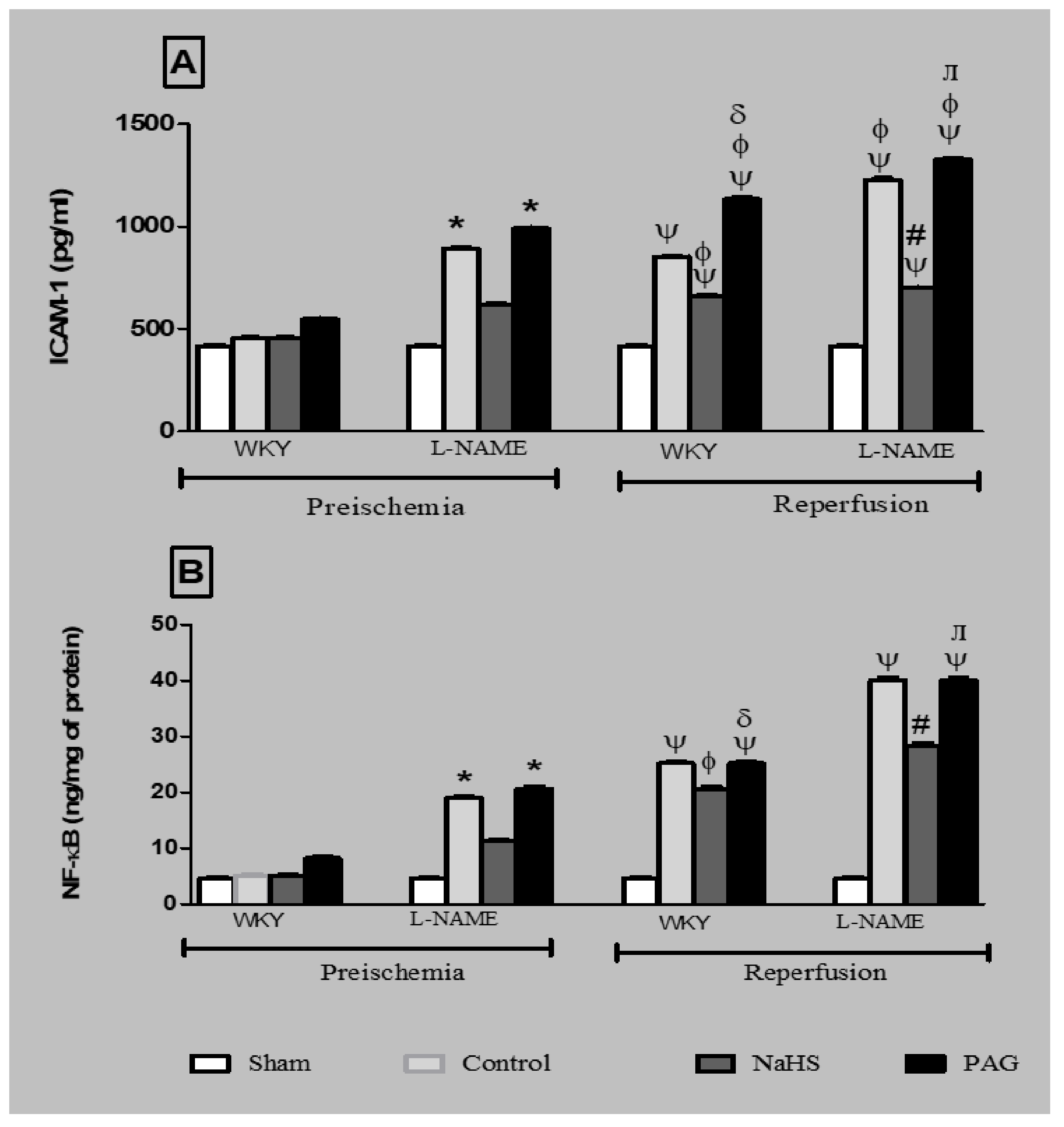
3.9. ICAM-1 Expression and NF-kB Concentration at the Pre-Ischemia and Reperfusion Phases in Plasma and Kidney Tissues of Different Groups
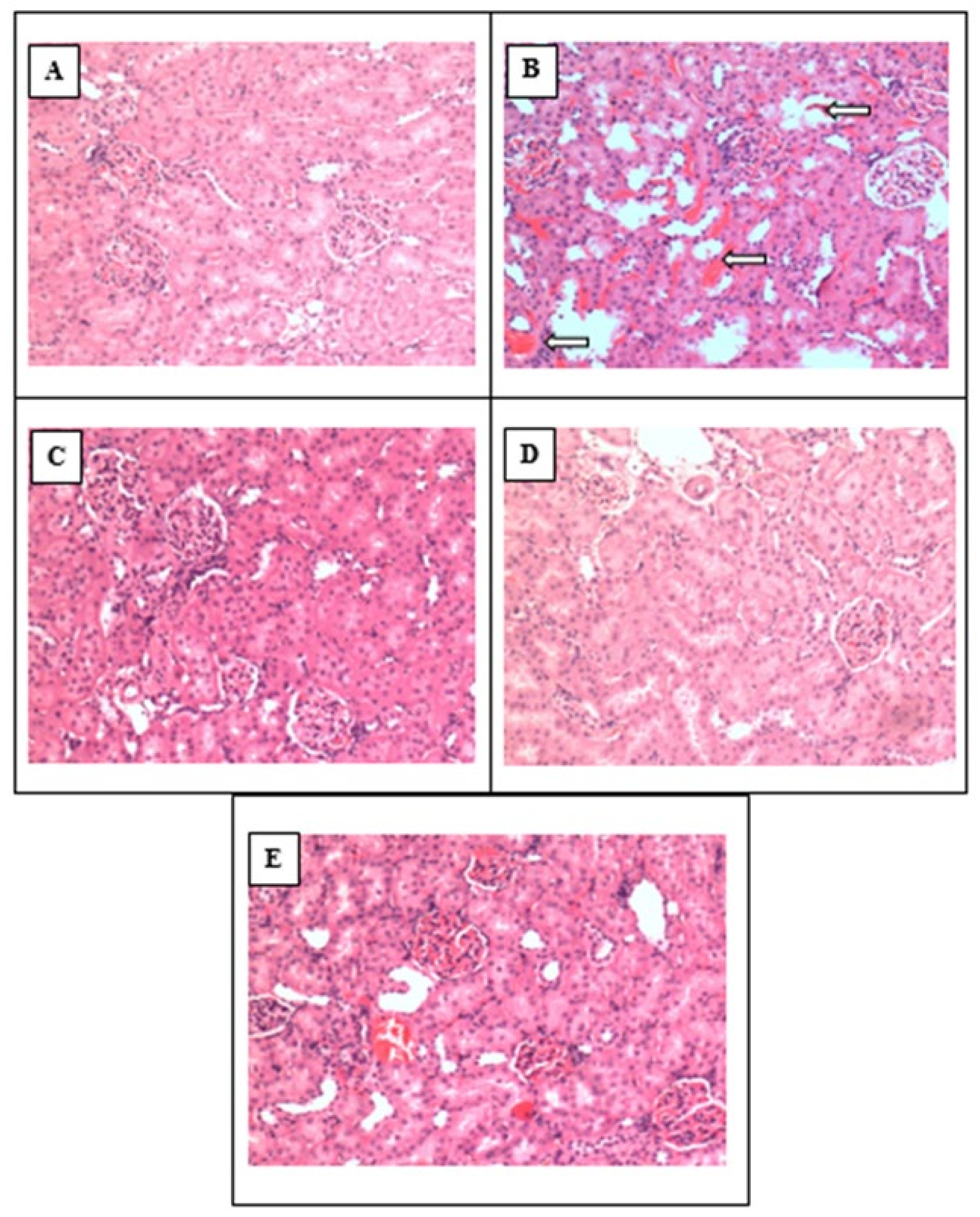
3.10. Histopathology of Kidney Tissue for All Animals in the WKY and L-NAME Groups (Sham, Control, NAHS and PAG)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Dedication
Abbreviations
| H2S | Hydrogen sulphide |
| IRI | Ischemia-reperfusion injury |
| ICAM | Intracellular adhesion molecule |
| PAG | dL-propargylglycine |
| NRF | Non-renal failure |
| g | grams |
| L-NAME | Nω-Nitro-L-arginine methyl ester hydrochloride |
| SOD | Superoxide dismutase |
| MDA | Malondialdehyde |
| TAOC | Total antioxidant capacity |
| GSH | Glutathione |
| NF-KB | Nuclear factor-kappa B |
| KI | Kidney index |
| RCBP | Renal cortical blood perfusion |
| FeNa | Fractional excretion of sodium |
| FeK | Fractional excretion of potassium |
| SBP | Systolic blood pressure |
| DBP | Diastolic blood pressure |
| HR | Heart rate |
| PWV | Pulse-wave velocity |
| NOS | Nitric oxide synthase |
| BW | Body weight |
| KW | Kidney weight |
References
- Lindner, J.R.; Coggins, M.P.; Kaul, S.; Klibanov, A.L.; Brandenburger, G.H.; Ley, K. Microbubble persistence in the microcirculation during ischemia/reperfusion and inflammation is caused by integrin-and complement-mediated adherence to activated leukocytes. Circulation 2000, 101, 668–675. [Google Scholar] [CrossRef] [Green Version]
- Kurose, I.; Anderson, D.C.; Miyasaka, M.; Tamatani, T.; Paulson, J.C.; Todd, R.F.; Rusche, J.R.; Granger, D. Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ. Res. 1994, 74, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1083–G1088. [Google Scholar] [CrossRef] [Green Version]
- Steinhoff, G.; Behrend, M.; Schrader, B.; Duijvestijn, A.; Wonigeit, K. Expression patterns of leukocyte adhesion ligand molecules on human liver endothelia. Lack of ELAM-1 and CD62 inducibility on sinusoidal endothelia and distinct distribution of VCAM-1, ICAM-1, ICAM-2, and LFA-3. Am. J. Pathol. 1993, 142, 481. [Google Scholar]
- Vollmar, B.; Glasz, J.; Menger, M.D.; Messmer, K. Leukocytes contribute to hepatic ischemia/reperfusion injury via intercellular adhesion molecule-1-mediated venular adherence. Surgery 1995, 117, 195–200. [Google Scholar] [CrossRef]
- Collins, T.; Read, M.; Neish, A.; Whitley, M.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Nichols, T.C. NF-kB and reperfusion injury. Drug News Perspect 2004, 17, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Arany, P.R.; Huang, Y.-Y.; Tomkinson, E.M.; Sharma, S.K.; Kharkwal, G.B.; Saleem, T.; Mooney, D.; Yull, F.E.; Blackwell, T.S. Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS ONE 2011, 6, e22453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Anwar, K.N.; True, A.L.; Malik, A.B. Thrombin-induced p65 homodimer binding to downstream NF-κB site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J. Immunol. 1999, 162, 5466–5476. [Google Scholar] [PubMed]
- Singh, D.; Chopra, K. The effect of naringin, a bioflavonoid on ischemia-reperfusion induced renal injury in rats. Pharmacol. Res. 2004, 50, 187–193. [Google Scholar] [CrossRef]
- Singh, D.; Chander, V.; Chopra, K. Protective effect of catechin on ischemia-reperfusion-induced renal injury in rats. Pharm. Rep 2005, 57, 70–76. [Google Scholar]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation–like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef] [Green Version]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Bos, E.M.; Leuvenink, H.G.D.; Snijder, P.M.; Kloosterhuis, N.J.; Hillebrands, J.-L.; Leemans, J.C.; Florquin, S.; van Goor, H. Hydrogen sulfide-induced hypometabolism prevents renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2009, 20, 1901–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Khan, S.A.; Lazhari, M.; Afzal, S.; Hashmi, F.; Abdullah, N.A.; Johns, E.J. A critical review of pharmacological significance of Hydrogen Sulfide in hypertension. Indian J. Pharmacol. 2015, 47, 243. [Google Scholar]
- Moore, P.K.; Bhatia, M.; Moochhala, S. Hydrogen sulfide: From the smell of the past to the mediator of the future? Trends Pharmacol. Sci. 2003, 24, 609–611. [Google Scholar] [CrossRef]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef]
- Cheng, Y.; Ndisang, J.F.; Tang, G.; Cao, K.; Wang, R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2316–H2323. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.D.; Lim, L.H.; Oh, V.M.; Yeo, S.B.; Cheong, Y.P.; Ali, M.Y.; El Oakley, R.; Lee, C.N.; Wong, P.S.; Caleb, M.G. Contractile and vasorelaxant effects of hydrogen sulfide and its biosynthesis in the human internal mammary artery. J. Pharmacol. Exp. Ther. 2008, 324, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Al-Magableh, M.R.; Hart, J.L. Mechanism of vasorelaxation and role of endogenous hydrogen sulfide production in mouse aorta. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Ping, C.; Mok, Y.Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P. Regulation of vascular nitric oxide in vitro and in vivo; A new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Zhong, G.; Chen, F.; Cheng, Y.; Tang, C.; Du, J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J. Hypertens. 2003, 21, 1879–1885. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Bond, M.; Newby, A.C.; Angelini, G.D.; Sparatore, A.; Del Soldato, P.; Jeremy, J.Y. Exogenous hydrogen sulfide inhibits superoxide formation, NOX-1 expression and Rac1 activity in human vascular smooth muscle cells. J. Vasc. Res. 2008, 45, 521–528. [Google Scholar] [CrossRef]
- Suzuki, K.; Olah, G.; Modis, K.; Coletta, C.; Kulp, G.; Gerö, D.; Szoleczky, P.; Chang, T.; Zhou, Z.; Wu, L. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 13829–13834. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Yang, Z.; Zhang, M.; Dong, Q.; Wang, X.; Lan, A.; Zeng, F.; Chen, P.; Wang, C.; Feng, J. Hydrogen sulfide protects against chemical hypoxia-induced cytotoxicity and inflammation in HaCaT cells through inhibition of ROS/NF-κB/COX-2 pathway. PLoS ONE 2011, 6, e21971. [Google Scholar] [CrossRef] [Green Version]
- Aziz, N.M.; Elbassuoni, E.A.; Kamel, M.Y.; Ahmed, S.M. Hydrogen sulfide renal protective effects: Possible link between hydrogen sulfide and endogenous carbon monoxide in a rat model of renal injury. Cell Stress Chaperones 2020, 25, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Tripatara, P.; Patel, N.S.; Collino, M.; Gallicchio, M.; Kieswich, J.; Castiglia, S.; Benetti, E.; Stewart, K.N.; Brown, P.A.; Yaqoob, M.M. Generation of endogenous hydrogen sulfide by cystathionine γ-lyase limits renal ischemia/reperfusion injury and dysfunction. Lab. Investig. 2008, 88, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobb, I.; Zhu, J.; Liu, W.; Haig, A.; Lan, Z.; Sener, A. Hydrogen sulfide treatment ameliorates long-term renal dysfunction resulting from prolonged warm renal ischemia-reperfusion injury. Can. Urol. Assoc. J. 2014, 8, E413. [Google Scholar] [CrossRef] [Green Version]
- Hosgood, S.; Nicholson, M. Hydrogen sulphide ameliorates ischaemia-reperfusion injury in an experimental model of non-heart-beating donor kidney transplantation. Br. J. Surg. 2010, 97, 202–209. [Google Scholar] [CrossRef]
- Lobb, I.; Mok, A.; Lan, Z.; Liu, W.; Garcia, B.; Sener, A. Supplemental hydrogen sulphide protects transplant kidney function and prolongs recipient survival after prolonged cold ischaemia–reperfusion injury by mitigating renal graft apoptosis and inflammation. BJU Int. 2012, 110, E1187–E1195. [Google Scholar] [CrossRef]
- Niu, X.; Smith, C.W.; Kubes, P. Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 1994, 74, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Kurose, I.; Wolf, R.; Grisham, M.B.; Aw, T.Y.; Specian, R.D.; Granger, D.N. Microvascular responses to inhibition of nitric oxide production role of active oxidants. Circ. Res. 1995, 76, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Afkir, S.; Nguelefack, T.B.; Aziz, M.; Zoheir, J.; Cuisinaud, G.; Bnouham, M.; Mekhfi, H.; Legssyer, A.; Lahlou, S.; Ziyyat, A. Arbutus unedo prevents cardiovascular and morphological alterations in L-NAME-induced hypertensive rats: Part I: Cardiovascular and renal hemodynamic effects of Arbutus unedo in L-NAME-induced hypertensive rats. J. Ethnopharmacol. 2008, 116, 288–295. [Google Scholar] [CrossRef]
- Saravanakumar, M.; Raja, B. Veratric acid, a phenolic acid attenuates blood pressure and oxidative stress in L-NAME induced hypertensive rats. Eur. J. Pharmacol. 2011, 671, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Du, J.; Tang, C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem. Biophys. Res. Commun. 2004, 313, 22–27. [Google Scholar] [CrossRef]
- Ahmad, A.; Sattar, M.A.; Azam, M.; Abdulla, M.H.; Khan, S.A.; Hashmi, F.; Abdullah, N.A.; Johns, E.J. Cystathione gamma lyase/Hydrogen Sulphide Pathway Up Regulation Enhances the Responsiveness of α1A and α1B-Adrenoreceptors in the Kidney of Rats with Left Ventricular Hypertrophy. PLoS ONE 2016, 11, e0154995. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Sattar, M.Z.; Rathore, H.A.; Akhtar, S.; Khan, M.A.; Hashmi, F.; Abdullah, N.; Johns, E.J. Impact of isoprenaline and caffeine on development of left ventricular hypertrophy and renal hemodynamic in wistar kyoto rats. Measurements 2012, 76, 1015–1026. [Google Scholar]
- Abdulla, M.H.; Sattar, M.A.; Abdullah, N.A.; Khan, M.A.H.; Abdallah, H.; Johns, E.J. Chronic treatment with losartan and carvedilol differentially modulates renal vascular responses to sympathomimetics compared to treatment with individual agents in normal Wistar Kyoto and spontaneously hypertensive rats. Eur. J. Pharmacol. 2009, 612, 69–74. [Google Scholar] [CrossRef]
- Kiew, L.V.; Munavvar, A.S.; Law, C.H.; Azizan, A.N.; Nazarina, A.R.; Sidik, K.; Johns, E.J. Effect of antisense oligodeoxynucleotides for ICAM-1 on renal ischaemia-reperfusion injury in the anaesthetised rat. J. Physiol. 2004, 557, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeling, H.; Wust, H. Colorimetric method for determination of creatinine. Arztl. Lab 1969, 15, 34. [Google Scholar]
- Ahmad, F.u.D.; Sattar, M.A.; Rathore, H.A.; Abdullah, M.H.; Tan, S.; Abdullah, N.A.; Johns, E.J. Exogenous hydrogen sulfide (H2S) reduces blood pressure and prevents the progression of diabetic nephropathy in spontaneously hypertensive rats. Ren. Fail. 2012, 34, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, J.E.; Zhao, Z.-Q.; Vinten-Johansen, J. The role of neutrophils in myocardial ischemia–reperfusion injury. Cardiovasc. Res. 1999, 43, 860–878. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Bautista, A.; Spolarics, Z.; Spitzer, J. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J. Leukoc. Biol. 1992, 52, 377–382. [Google Scholar] [CrossRef]
- Johnson, K.J.; Weinberg, J.M. Postischemic renal injury due to oxygen radicals. Curr. Opin. Nephrol. Hypertens. 1993, 2, 625–635. [Google Scholar] [CrossRef]
- Mehrabi, A.; Mood, Z.A.; Sadeghi, M.; Schmied, B.; Müller, S.; Welsch, T.; Kuttymuratov, G.; Wente, M.; Weitz, J.; Zeier, M. Thymoglobulin and ischemia reperfusion injury in kidney and liver transplantation. Nephrol. Dial. Transplant. 2007, 22, viii54–viii60. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.; Koch, M.; Mehrabi, A.; Schemmer, P.; Zeier, M.; Beimler, J.; Büchler, M.; Schmidt, J. Living-donor kidney transplantation: Risks of the donor–benefits of the recipient. Clin. Transplant. 2006, 20, 13–16. [Google Scholar] [CrossRef]
- Cerqueira, N.F.; Hussni, C.A.; Yoshida, W.B. Pathophysiology of mesenteric ischemia/reperfusion: A review. Acta Cir. Bras. 2005, 20, 336–343. [Google Scholar] [CrossRef]
- Kim, S.-R.; Bae, Y.-H.; Bae, S.-K.; Choi, K.-S.; Yoon, K.-H.; Koo, T.H.; Jang, H.-O.; Yun, I.; Kim, K.-W.; Kwon, Y.-G. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-κB activation in endothelial cells. Biochim. Biophys. Acta (Bba)—Mol. Cell Res. 2008, 1783, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Khayyal, M.T.; El-Ghazaly, M.A.; Abdallah, D.M.; Nassar, N.N.; Okpanyi, S.N.; Kreuter, M.-H. Blood pressure lowering effect of an olive leaf extract (Olea europaea) in L-NAME induced hypertension in rats. Arzneim. Forsch. 2002, 52, 797–802. [Google Scholar]
- Chia, T.Y.; Sattar, M.A.; Abdulla, M.H.; Rathore, H.A.; Ahmad, F.u.D.; Kaur, G.; Abdullah, N.A.; Johns, E.J. The effects of tempol on renal function and hemodynamics in cyclosporine-induced renal insufficiency rats. Ren. Fail. 2013, 35, 978–988. [Google Scholar] [CrossRef]
- Zicha, J.; Dobesova, Z.; Kunes, J. Antihypertensive mechanisms of chronic captopril or N-acetylcysteine treatment in L-NAME hypertensive rats. Hypertens. Res. 2006, 29, 1021–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.; Palmer, R.; Schulz, R.; Hodson, H.; Moncada, S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990, 101, 746–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biancardi, V.C.; Bergamaschi, C.T.; Lopes, O.U.; Campos, R.R. Sympathetic activation in rats with L-NAME-induced hypertension. Braz. J. Med. Biol. Res. 2007, 40, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, F.; Magalhães, A.; Gontijo, J. Effect of inhibition of nitric oxide synthase on blood pressure and renal sodium handling in renal denervated rats. Braz. J. Med Biol. Res. 2000, 33, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Muchant, D.; Beierwaltes, W.H.; Racusen, L.; Baylis, C. Evolution of chronic nitric oxide inhibition hypertension Relationship to renal function. Hypertension 1998, 31, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Snijder, P.M.; Frenay, A.-R.S.; Koning, A.M.; Bachtler, M.; Pasch, A.; Kwakernaak, A.J.; van den Berg, E.; Bos, E.M.; Hillebrands, J.-L.; Navis, G. Sodium thiosulfate attenuates angiotensin II-induced hypertension, proteinuria and renal damage. Nitric Oxide 2014, 42, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.G.; Sohn, E.J.; Lee, Y.M.; Lee, A.S.; Han, J.H.; Kim, T.Y.; Lee, H.S. Effects of bulbus Fritillaria water extract on blood pressure and renal functions in the L-NAME-induced hypertensive rats. J. Ethnopharmacol. 2004, 91, 51–56. [Google Scholar] [CrossRef]
- Wang, R. Two’s company, three’sa crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R. The gasotransmitter role of hydrogen sulfide. Antioxid. Redox Signal. 2003, 5, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.M.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yao, X.; Zhang, Y.; Li, W.; Kang, K.; Sun, L.; Sun, X. The protective role of hydrogen sulfide in myocardial ischemia–reperfusion-induced injury in diabetic rats. Int. J. Cardiol. 2011, 152, 177–183. [Google Scholar] [CrossRef]
- Kang, K.; Zhao, M.; Jiang, H.; Tan, G.; Pan, S.; Sun, X. Role of hydrogen sulfide in hepatic ischemia-reperfusion–induced injury in rats. Liver Transplant. 2009, 15, 1306–1314. [Google Scholar] [CrossRef]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.-L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Abdulla, M.H.; Khan, S.A.; Azam, M.; Abdullah, N.A.; Johns, E.J. Up Regulation of cystathione γ lyase and Hydrogen Sulphide in the Myocardium Inhibits the Progression of Isoproterenol–Caffeine Induced Left Ventricular Hypertrophy in Wistar Kyoto Rats. PLoS ONE 2016, 11, e0150137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okusa, M.D.; Linden, J.; Macdonald, T.; Huang, L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am. J. Physiol.—Ren. Physiol. 1999, 277, F404–F412. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Ye, H.; Huang, L.; Li, L.; Foss, F.W.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am. J. Physiol.—Ren. Physiol. 2006, 290, F1516–F1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ysebaert, D.K.; De Greef, K.E.; Vercauteren, S.R.; Ghielli, M.; Verpooten, G.A.; Eyskens, E.J.; De Broe, M.E. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 2000, 15, 1562–1574. [Google Scholar] [CrossRef]
- Kim, J.; Jang, H.-S.; Park, K.M. Reactive oxygen species generated by renal ischemia and reperfusion trigger protection against subsequent renal ischemia and reperfusion injury in mice. Am. J. Physiol.—Ren. Physiol. 2010, 298, F158–F166. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.-Y.; Siow, Y.L.; Karmin, O. Ischemia-reperfusion reduces cystathionine-β-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol.—Ren. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef] [Green Version]
- Eschwege, P.; Paradis, V.; Conti, M.; Holstege, A.; Richet, F.; Deteve, J.; Menager, P.; Legrand, A.; Jardin, A.; Bedossa, P. In situ detection of lipid peroxidation by-products as markers of renal ischemia injuries in rat kidneys. J. Urol. 1999, 162, 553–557. [Google Scholar] [CrossRef]
- Patel, N.S.; Sharples, E.J.; Cuzzocrea, S.; Chatterjee, P.K.; Britti, D.; Yaqoob, M.M.; Thiemermann, C. Pretreatment with EPO reduces the injury and dysfunction caused by ischemia/reperfusion in the mouse kidney in vivo. Kidney Int. 2004, 66, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Irmak, M.K.; Koltuksuz, U.; Kutlu, N.O.; Yağmurca, M.; Özyurt, H.; Karaman, A.; Akyol, Ö. The effect of caffeic acid phenethyl ester on ischemia-reperfusion injury in comparison with α-tocopherol in rat kidneys. Urol. Res. 2001, 29, 190–193. [Google Scholar] [CrossRef]
- Sener, G.; Sehirli, A.Ö.; Keyer-Uysal, M.; Arbak, S.; Ersoy, Y.; Yeğen, B.Ç. The protective effect of melatonin on renal ischemia–reperfusion injury in the rat. J. Pineal Res. 2002, 32, 120–126. [Google Scholar] [CrossRef]
- Thiemermann, C.; Patel, N.S.; Kvale, E.O.; Cockerill, G.W.; Brown, P.A.; Stewart, K.N.; Cuzzocrea, S.; Britti, D.; Mota-Filipe, H.; Chatterjee, P.K. High density lipoprotein (HDL) reduces renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2003, 14, 1833–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Şener, G.; Şehirli, Ö.; Velioğlu-Öğünç, A.; Çetinel, Ş.; Gedik, N.; Caner, M.; Sakarcan, A.; Yeğen, B.Ç. Montelukast protects against renal ischemia/reperfusion injury in rats. Pharmacol. Res. 2006, 54, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, H.; Sadeghnia, H.R.; Ziaee, T.; Danaee, A. Protective effect of aqueous saffron extract (Crocus sativus L.) and crocin, its active constituent, on renal ischemia-reperfusion-induced oxidative damage in rats. J. Pharm. Pharm. Sci. 2005, 8, 387–393. [Google Scholar] [PubMed]
- Yan, S.-K.; Chang, T.; Wang, H.; Wu, L.; Wang, R.; Meng, Q.H. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Carden, D.L.; Smith, J.K.; Korthuis, R.J. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ. Res. 1990, 66, 1436–1444. [Google Scholar] [CrossRef] [Green Version]
- Sekido, N.; Mukaida, N.; Harada, A.; Nakanishi, I.; Watanabe, Y.; Matsushima, K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 1993, 365, 654–657. [Google Scholar] [CrossRef]
- Li, Z.; Nickkholgh, A.; Yi, X.; Bruns, H.; Gross, M.L.; Hoffmann, K.; Mohr, E.; Zorn, M.; Büchler, M.W.; Schemmer, P. Melatonin protects kidney grafts from ischemia/reperfusion injury through inhibition of NF-kB and apoptosis after experimental kidney transplantation. J. Pineal Res. 2009, 46, 365–372. [Google Scholar] [CrossRef]
- Rusai, K.; Sollinger, D.; Baumann, M.; Wagner, B.; Strobl, M.; Schmaderer, C.; Roos, M.; Kirschning, C.; Heemann, U.; Lutz, J. Toll-like receptors 2 and 4 in renal ischemia/reperfusion injury. Pediatric Nephrol. 2010, 25, 853–860. [Google Scholar] [CrossRef]
- Tuuminen, R.; Nykänen, A.; Saharinen, P.; Gautam, P.; Keränen, M.; Arnaudova, R.; Rouvinen, E.; Helin, H.; Tammi, R.; Rilla, K. Donor Simvastatin Treatment Prevents Ischemia—Reperfusion and Acute Kidney Injury by Preserving Microvascular Barrier Function. Am. J. Transplant. 2013, 13, 2019–2034. [Google Scholar] [CrossRef]
- Jung, H.S.; Joo, J.-D.; Kim, D.-W.; In, J.H.; Roh, M.; Jeong, J.-T.; Noh, S.J.; Choi, J.W. Effect of milrinone on the inflammatory response and NF-kB activation in renal ischemia-reperfusion injury in mice. Korean J. Anesthesiol. 2014, 66, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Rogers, N.; Stephenson, M.; Kitching, A.; Horowitz, J.; Coates, P. Amelioration of renal ischaemia–reperfusion injury by liposomal delivery of curcumin to renal tubular epithelial and antigen-presenting cells. Br. J. Pharmacol. 2012, 166, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Seok, Y.M.; Kim, J.; Park, M.J.; Boo, Y.C.; Park, Y.K.; Park, K.M. Wen-pi-tang-Hab-Wu-ling-san attenuates kidney fibrosis induced by ischemia/reperfusion in mice. Phytother. Res. 2008, 22, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Albermann, K.; Baeuerle, P.A. Nuclear factor kB: An oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun. 1992, 17, 221–237. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Groups | Day 0 | Day 21 | Day 35 |
|---|---|---|---|---|
| SBP | WKY-SHAM | 117 ± 3 | 118 ± 3 | 119 ± 3 |
| WKY-CONTROL | 117 ± 5 | 120 ± 3 | 119 ± 2 | |
| WKY+NaHS | 121 ± 3 | 117 ± 3 | 113 ± 2 | |
| WKY+PAG | 119 ± 4 | 116 ± 3 | 117 ± 3 | |
| L-NAME-SHAM | 118 ± 4 | 162 ± 4 Ψ | 199 ± 9 Ψ,φ | |
| L-NAME-CONTROL | 119 ± 3 | 180 ± 4 Ψ | 194 ± 9 Ψ,φ,δ | |
| L-NAME+NaHS | 118 ± 2 | 139 ± 3 Ψ | 154 ± 3 Ψ,φ,ᴫ | |
| L-NAME+PAG | 116 ± 3 | 173 ± 6 Ψ | 197 ± 10 Ψ,φ | |
| MAP | WKY-SHAM | 98 ± 2 | 99 ± 2 | 100 ± 3 |
| WKY-CONTROL | 99 ± 4 | 98 ± 1 | 99 ± 1 | |
| WKY+NaHS | 100 ± 2 | 99 ± 2 | 95 ± 1 | |
| WKY+PAG | 100 ± 2 | 98 ± 1 | 98 ± 2 | |
| L-NAME-SHAM | 99 ± 2 | 142 ± 3 Ψ | 176 ± 6 Ψ,φ | |
| L-NAME-CONTROL | 99 ± 2 | 144 ± 3 Ψ | 177 ± 10 Ψ,φ,δ | |
| L-NAME+NaHS | 99 ± 1 | 120 ± 2 Ψ | 141 ± 8 Ψ,φ,ᴫ | |
| L-NAME+PAG | 98 ± 1 | 142 ± 4 Ψ | 175 ± 6 Ψ,φ | |
| HR | WKY-SHAM | 326 ± 12 | 311 ± 9 | 319 ± 8 |
| WKY-CONTROL | 326 ± 15 | 331 ± 12 | 324 ± 11 | |
| WKY+NaHS | 313 ± 14 | 304 ± 9 | 321 ± 7 | |
| WKY+PAG | 308 ± 19 | 304 ± 20 | 318 ± 12 | |
| L-NAME-SHAM | 303 ± 21 | 338 ± 32 | 384 ± 15 Ψ,φ | |
| L-NAME-CONTROL | 308 ± 12 | 340 ± 16 | 387 ± 18 Ψ,φ,δ | |
| L-NAME+NaHS | 310 ± 7 | 338 ± 7 | 355 ± 13 Ψ | |
| L-NAME+PAG | 300 ± 12 | 327 ± 10 | 394 ± 15 Ψ,φ | |
| FENa | WKY-SHAM WKY-CONTROL | 0.72 ± 0.09 0.67 ± 0.04 | 0.69 ± 0.1 0.66 ± 0.1 | 0.74 ± 0.07 0.69 ± 0.16 |
| WKY+NaHS | 0.67 ± 0.02 | 0.68 ± 0.02 | 0.69 ± 0.01 | |
| WKY+PAG | 0.65 ± 0.02 | 0.68 ± 0.02 | 0.68 ± 0.03 | |
| L-NAME-SHAM | 0.72 ± 0.02 | 0.96 ± 0.03 Ψ | 1.51 ± 0.06 Ψ,φ | |
| L-NAME-CONTROL | 0.68 ± 0.03 | 0.98 ± 0.03 Ψ | 1.56 ± 0.05 Ψ,φ,δ | |
| L-NAME+NaHS | 0.72 ± 0.02 | 0.84 ± 0.02 | 0.94 ± 0.02 Ψ,ᴫ | |
| L-NAME+PAG | 0.69 ± 0.03 | 0.96 ± 0.03 Ψ | 1.61 ± 0.08 Ψ,φ | |
| FEK | WKY-SHAM | 14 ± 3.2 | 13 ± 2.9 | 13 ± 2.0 |
| WKY-CONTROL | 13 ± 1.1 | 13 ± 5.0 | 13 ± 3.0 | |
| WKY+NaHS | 13 ± 2.8 | 12.09 ± 1.3 | 12 ± 1.6 | |
| WKY+PAG | 12 ± 1.8 | 12 ± 1.3 | 11 ± 1.1 | |
| L-NAME-SHAM | 13 ± 1.9 | 18 ± 3.0 Ψ | 27 ± 3.9 Ψ,φ | |
| L-NAME-CONTROL | 12 ± 1.8 | 18 ± 2.9 Ψ | 27 ± 4.1 Ψ,φ,δ | |
| L-NAME+NaHS | 13 ± 1.9 | 15 ± 2.2 | 19 ± 2.3 Ψ,ᴫ | |
| L-NAME+PAG | 12 ± 1.9 | 16 ± 1.4 Ψ | 25 ± 3.6 Ψ,φ | |
| Cr.Cl | WKY-SHAM | 5.36 ± 0.62 | 5.21 ± 1.02 | 4.63 ± 0.54 |
| WKY-CONTROL | 7.82 ± 1.42 | 7.12 ± 1.59 | 5.64 ± 0.58 | |
| WKY+NaHS | 8.06 ± 0.41 | 6.51 ± 2.42 | 6.71 ± 1.52 | |
| WKY+PAG | 4.70 ± 0.68 | 4.06 ± 0.55 | 5.67 ± 0.80 | |
| L-NAME-SHAM | 5.24 ± 0.3 | 2.53 ± 0.3 Ψ | 1.61 ± 0.2 Ψ | |
| L-NAME-CONTROL | 7.87 ± 3.0 | 4.49 ± 2.1 Ψ | 3.09 ± 1.3 Ψ,δ | |
| L-NAME+NaHS | 4.83 ± 0.5 | 3.41 ± 0.2 | 2.93 ± 0.3 | |
| L-NAME+PAG | 5.63 ± 0.9 | 3.45 ± 1.0 Ψ | 2.56 ± 0.6 Ψ |
| Groups | SBP (mmHg) | MAP (mmHg) | HR (rpm) | BW (g) | KW (g) | KI (%) |
|---|---|---|---|---|---|---|
| WKY-SHAM | 113 ± 5 | 100 ± 4 | 316 ± 9 | 305 ± 10 | 0.90 ± 0.01 | 0.297 ± 0.01 |
| WKY-CONTROL | 111 ± 6 | 99 ± 2 | 311 ± 14 | 315 ± 10 | 1.06 ± 0.04 Ψ | 0.336 ± 0.02 Ψ |
| WKY+NaHS | 110 ± 4 | 98 ± 3 | 300 ± 8 | 298 ± 7 # | 0.94 ± 0.01 # | 0.317 ± 0.00 |
| WKY+PAG | 113 ± 3 | 99 ± 1 | 318 ± 20 | 302 ± 5 | 1.03 ± 0.05 Ψ,δ | 0.340 ± 0.02 Ψ |
| L-NAME-SHAM | 198 ± 9 | 175 ± 6 | 374 ± 16 | 294 ± 6 | 0.98 ± 0.02 | 0.332 ± 0.01 |
| L-NAME-CONTROL | 193± 9 # | 176 ± 10 # | 379 ± 23 # | 285 ± 7 # | 1.09 ± 0.05 φ | 0.384 ± 0.02 φ,# |
| L-NAME+NaHS | 153 ± 3 φ,¥ | 140 ± 8 φ,¥ | 352 ± 18 | 297 ± 4 | 1.04 ± 0.06 | 0.349 ± 0.01 |
| L-NAME+PAG | 196 ± 10 ᴫ | 174 ± 6 ᴫ | 389 ± 14 ᴫ | 285 ± 7 | 1.174 ± 0.02 φ,¥,ᴫ | 0.412 ± 0.01 φ,ᴫ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashmi, S.F.; Rathore, H.A.; Sattar, M.A.; Johns, E.J.; Gan, C.-Y.; Chia, T.Y.; Ahmad, A. Hydrogen Sulphide Treatment Prevents Renal Ischemia-Reperfusion Injury by Inhibiting the Expression of ICAM-1 and NF-kB Concentration in Normotensive and Hypertensive Rats. Biomolecules 2021, 11, 1549. https://doi.org/10.3390/biom11101549
Hashmi SF, Rathore HA, Sattar MA, Johns EJ, Gan C-Y, Chia TY, Ahmad A. Hydrogen Sulphide Treatment Prevents Renal Ischemia-Reperfusion Injury by Inhibiting the Expression of ICAM-1 and NF-kB Concentration in Normotensive and Hypertensive Rats. Biomolecules. 2021; 11(10):1549. https://doi.org/10.3390/biom11101549
Chicago/Turabian StyleHashmi, Syed F., Hassaan Anwer Rathore, Munavvar A. Sattar, Edward J. Johns, Chee-Yuen Gan, Tan Yong Chia, and Ashfaq Ahmad. 2021. "Hydrogen Sulphide Treatment Prevents Renal Ischemia-Reperfusion Injury by Inhibiting the Expression of ICAM-1 and NF-kB Concentration in Normotensive and Hypertensive Rats" Biomolecules 11, no. 10: 1549. https://doi.org/10.3390/biom11101549