
Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers


1
UMR 1098 RIGHT, University Bourgogne-Franche-Comté, INSERM, EFS-BFC, F-25000 Besançon, France
2
EPIgenetics and GENe EXPression Technical Platform (EPIGENExp), University Bourgogne Franche-Comté, F-25000 Besançon, France
3
DImaCell Platform, University Bourgogne Franche-Comté, F-25000 Besançon, France
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomolecules 2021, 11(6), 782; https://doi.org/10.3390/biom11060782
Submission received: 31 March 2021
/
Revised: 18 May 2021
/
Accepted: 19 May 2021
/
Published: 22 May 2021
(This article belongs to the Special Issue EMT and Cancer)
Abstract
:Epithelial to mesenchymal transition (EMT) is a process that allows epithelial cells to progressively acquire a reversible mesenchymal phenotype. Here, we recount the main events in the history of EMT. EMT was first studied during embryonic development. Nowadays, it is an important field in cancer research, studied all around the world by more and more scientists, because it was shown that EMT is involved in cancer aggressiveness in many different ways. The main features of EMT’s involvement in embryonic development, fibrosis and cancers are briefly reviewed here.
1. Introduction
Epithelial to mesenchymal transition (EMT) is a complicated cellular phenomenon that consists in the acquisition, for a cell, of mesenchymal features in place of epithelial ones. EMT can take place in various physiological and pathological contexts. EMT can be determined by numerous molecular mechanisms. EMT can refer to different phenomena with the following common traits: the loss of epithelial features, such as cell–cell interactions and apico-basal polarity, and the gain of mesenchymal ones such as cytosolic expansions, rear-front polarity, and increased migration/invasion capacity. Due to the heterogeneity of the phenomenon called EMT, it would be more correct to use the term “an EMT” rather than “the EMT”.
EMT is involved in many physiological (embryonic development, wound-healing) and pathological (fibrosis, cancers) processes, and is generally classified according to the extracellular context rather than the molecular mechanisms. Type I EMT concerns embryologic development, type II EMT occurs during wound-healing and fibrosis, and type III EMT is found in cancers.
EMT was first described as epithelial to mesenchymal transformation [1], signifying that the switch from epithelial to mesenchymal status was definitive and that only two cellular states are possible: epithelial or mesenchymal. Nowadays, evidence shows that EMT describes a spectrum of intermediate states between an epithelial and a mesenchymal phenotype [2,3], and that cells can switch amongst these intermediate states following extracellular stimuli in a progressive and reversible manner. Here, we will discuss the history of EMT as a research field, explaining how the definitions of this term have been modified over time and highlighting the main discoveries in the domain. This review will help us to better understand actual studies on EMT by considering its history. Then, we will briefly describe the molecular aspect of EMT and the role of EMT during embryonic development and pathologies, highlighting its roles in cancers.
2. The First Steps of Research on EMT
2.1. The First Phenotypical Observations of an EMT
Elizabeth Dexter “Betty” Hay (1927–2007, Harvard Medical School) was probably the first to describe EMT and later to use this term. This American cellular and developmental biologist worked first on amphibian limb regeneration, from 1958 [4]. She especially described the dedifferentiation of cartilage cells of embryonic salamander’s limbs, which can participate in new limb formation by re-differentiating. This process resembles a kind of EMT. This work led her to study epithelial cells and embryonic development.
She then started to study the role of the extracellular matrix (ECM) in epithelial cell differentiation, showing that ECM composition (for example, collagen concentration) impacts cornea epithelial cells’ differentiation and the secretion of ECM proteins, such as collagen and glycosaminoglycans (GAG) [5].
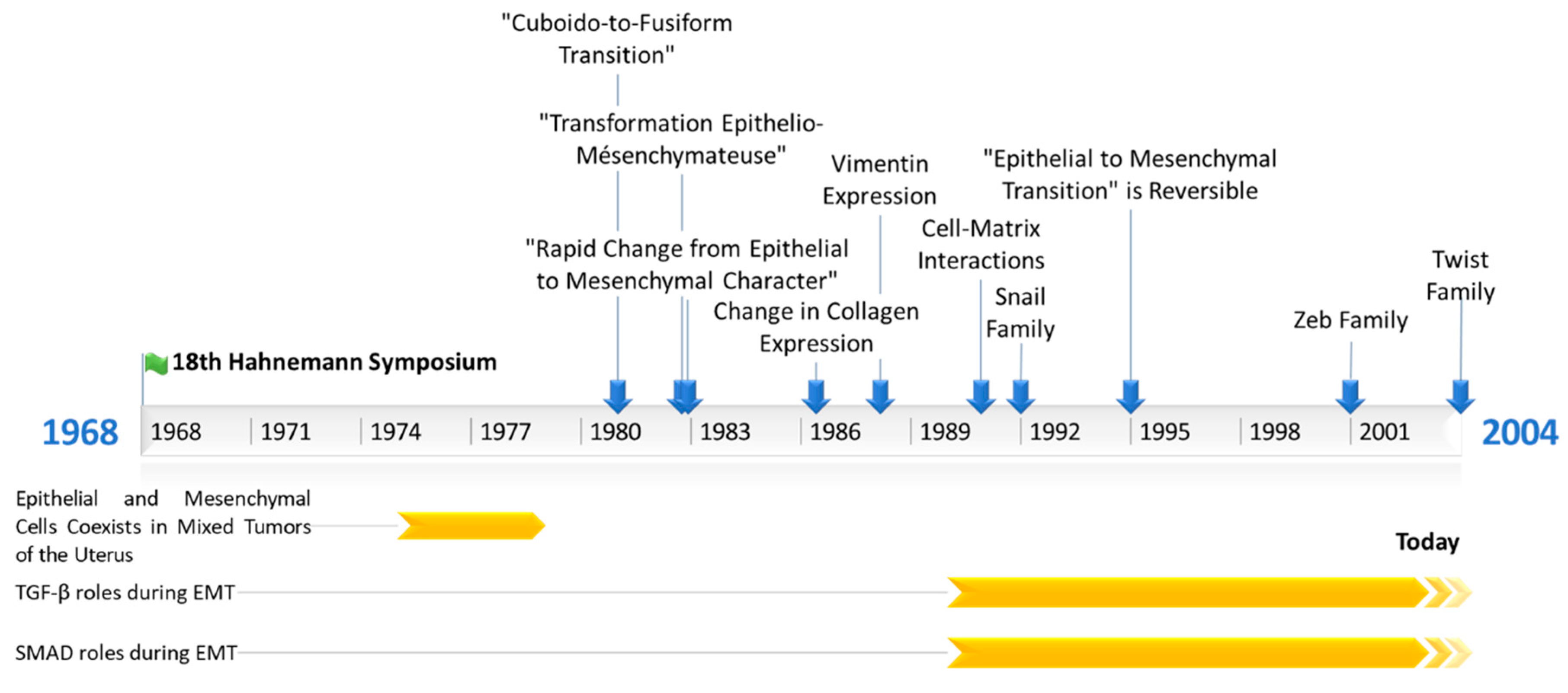
Elizabeth D. Hay then worked on embryonic development, using chick embryo models. Thanks to the very accurate descriptions of optical and electronic microscopy images obtained from embryonic tissues, she identified and listed different cellular phenotypes during their development. In 1968, she attended the 18th Hahnemann symposium in Baltimore about epithelial–mesenchymal interactions. During her speech, she described how mesenchymal tissues issued from epithelial cells during the migration of neural crest cells in neural tube formation [6]. Indeed, she was describing EMT before we had named it as such. We can probably consider the 18th Hahnemann symposium as the birthplace of EMT research.
Interestingly, in the 1970s, other teams (the team of Prof. Dr. H.-E. Stegner, University of Hamburg, Germany, and the team of Masao Sekiya, Department of Pathology, Nagaoka Red Cross Hospital, Japan) also reported that epithelial and mesenchymal cells coexist in mixed tumors of the uterus [7,8]. Then, there was a debate to determine if these cells come from the same cancer stem cell, or if mesenchymal cells can be derived directly from epithelial cells. At this time, two teams concluded on a common cancer stem cell origin, thinking it was not possible for epithelial cells to acquire a mesenchymal shape or vice versa. Moreover, they found similarities between cancer cells and stroma endometrial cells, the second of which originate the first, according to them [7,8].
Hay and her team used the term “epithelial to mesenchymal transformation” for the first time in 1982, in a publication describing for the first time adult cells undergoing an EMT. They showed that a culture of chick lens epithelial cells (adult or embryonic) suspended in collagen gels can lead to cytosolic expansion, such as in pseudopods [1]. These cells are then able to move individually in the collagen matrix, and they look like mesenchymal cells [1]. In 1981, R. Dulbecco et al. published an article in Cell Biology describing a “cuboid-to-fusiform transition” [9]. They observed that the cuboid epithelial cells of rat mammary tumors (induced by a single intravenous injection of N-nitrosomethylurea) can lead to fusiform cells looking like fibroblasts when cultured. This observation was made of many clones obtained from different rats, and also of cells similarly extracted by another group [10]. In 1982, Swiss and German researchers (team of Karl Illmensee, University of Geneva, Switzerland) showed that the first mesenchymal cells appearing during mice embryogenesis, which come from epithelial cells, lose desmosome and cytokeratin expressions and start to express vimentin [11]. They did not use the EMT term, but instead “rapid change from epithelial to mesenchymal character”.
Hay’s team continued to the work on EMT, starting with morphological studies (cytoskeleton description during EMT) using microscopy alone (optical and electronic). Since 1986, this group has been studying the molecular aspect of EMT, using the Western blotting (WB) technique. She managed to show that chicken lens epithelial cells lose type IV collagen expression (basal lamina specific) and γ-crystallin while expressing type I collagen (connective tissues) [12]. After that, this lab studied EMT in thyroid epithelial cells, which present large cytoskeleton remodeling, including a gain of vimentin expression. Moreover, Hay’s team also observed a loss of thyroglobulin expression, which is a precursor of the thyroid hormone, and highlighted cell dedifferentiation [13]. During the following years, Hay’s lab worked on embryonic development and proposed the “fixed cortex theory” to explain neural crest cells’ migration [14,15]. In a publication in 1990, Hay discussed the importance of cell–matrix interactions for mesenchymal cell migration [16].
Hay’s team next revealed that EMT is involved in another process occurring during embryo development: palatal fusion. Epithelial cells from the medial edge of the embryonic palatal epithelium migrate from each side to allow palatal fusion [17].
2.2. The First Mechanistical and Molecular Descriptions of EMT
Since 1990, a few teams have shown the great importance of TGF (transforming growth factor) family proteins (TGF-α; TGF-β1–3) during EMT. Indeed TGF-α expression was reported to lead to a mesenchymal and invasive phenotype in rat prostate cancer cells [18]. In 1991, Potts et al. showed the importance of TGF-β3 in embryonic heart endothelial cells EMT [19], and in 1994, they showed that mammary epithelial cells can undergo EMT following TGF-β treatment [20]. In the following years, thanks to the rapid evolution of the relevant techniques, studies about EMT have become more and more mechanistic. Hay’s team showed the involvement of TGF-β3 during EMT in chick embryonic palatal fusion [21]. Later, they detailed the importance of TGF-β family proteins during EMT in chicken embryonic palatal growth, especially during palatal fusion, which is mediated by EMT, itself governed by TGF-β3, and the involvement of SMAD (mothers against decapentaplegic homolog) proteins during TGF-β-induced EMT [22,23]. In 1999, the lab of Peter Ten Dijke (Ludwig Institute for Cancer Research, Uppsala, Sweden) showed for the first time the involvement of SMAD proteins in EMT induction after TGF-β receptor activation [24]. Finally, Hay’s team worked in 2008 on cancer cell lines, showing that the SNAIL family of EMT-ATFs (epithelial to mesenchymal activated transcription factors) can induce TGF-β3 expression [25]. Concurrently, other teams described EMT during the development or use of cancer cell lines, just as Hay’s team did, but using other models. Thanks to the evolution of the techniques, especially the development of immunolabeling (immunofluorescence, Western blotting), EMT studies then became focused on the biochemical mechanisms involved in this phenomenon. TGF-β was then the first molecule identified to induce EMT in the early 1990s. The main events in EMT’s history are summarized in Figure 1.
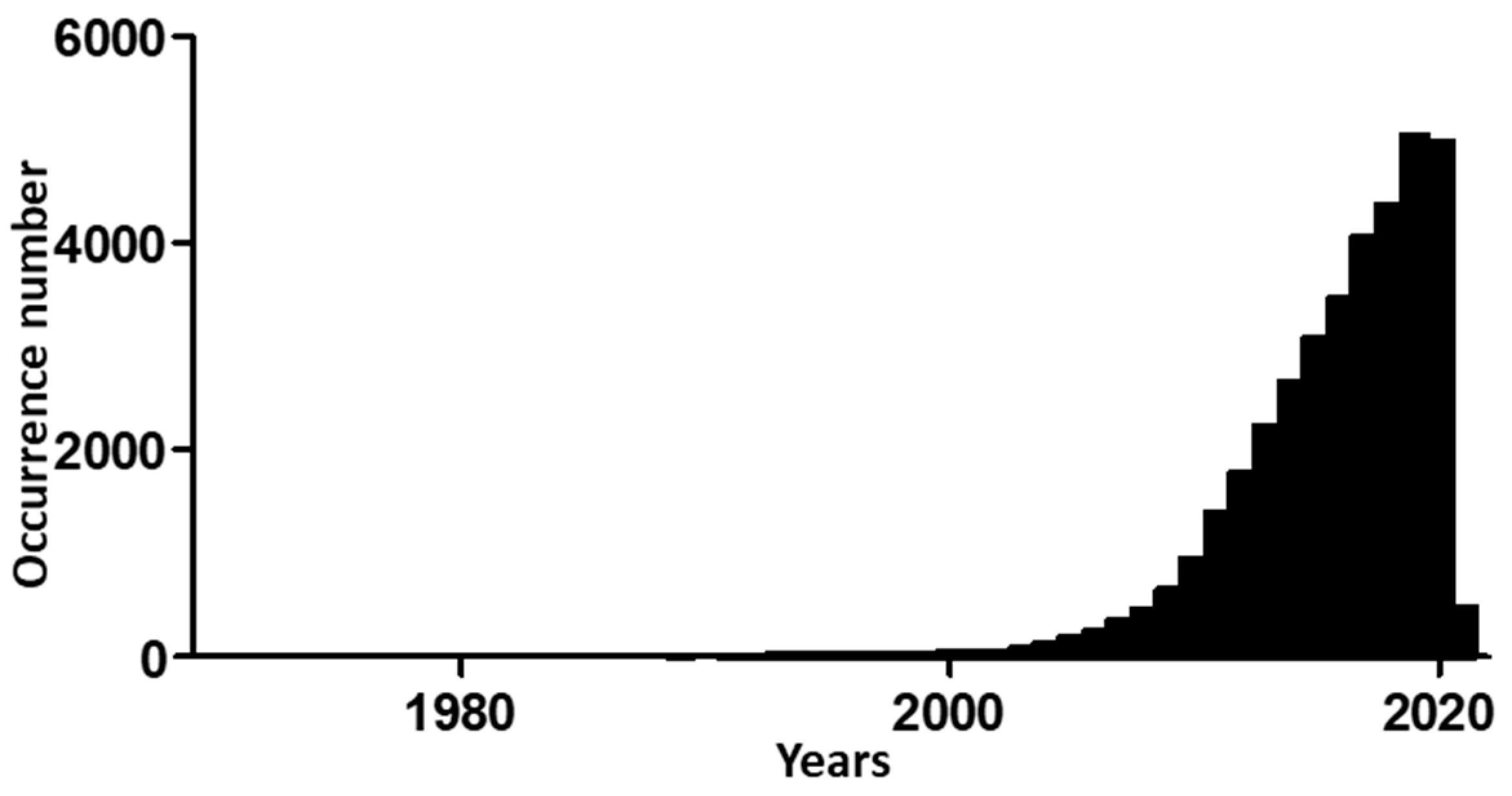
Research on EMT has become more popular since 2000. Hits when searching “epithelial to mesenchymal transition” in the PubMed database increased from 30 in 2000 to 182 in 2005, and then to 942 in 2010 and 4975 in 2020 (Figure 2).
Elizabeth Hay and her team wrote in 1995 a review named “An Overviewed of Epithelio–Mesenchymal Transformation”, in which they discussed the molecular mechanisms of EMT induction, the genes regulated by or regulating EMT, and the involvement of EMT in pathologies, especially metastases. Interestingly, they mentioned the possibility that EMT is reversible and its potential implication in therapeutics [26]. In another review article published the same year, they used for the first time the term “epithelial to mesenchymal transition”, they described some genes involved in EMT or the inverse phenomenon, mesenchymal to epithelial transition (MET) [27] (Table 1).
3. Main Molecular Aspects of EMT
The growing number of studies focused on EMT that have been described previously contributes to a view of the molecular mechanisms governing EMT. Theses mechanisms have been fully reviewed in the last decade [28,29,30,31]. Here, we will give a short summary of the state-of-the-art knowledge on molecular induction pathways, transcription factors, and RNA interference regulation in EMT.
3.1. Molecular Pathways Leading to EMT
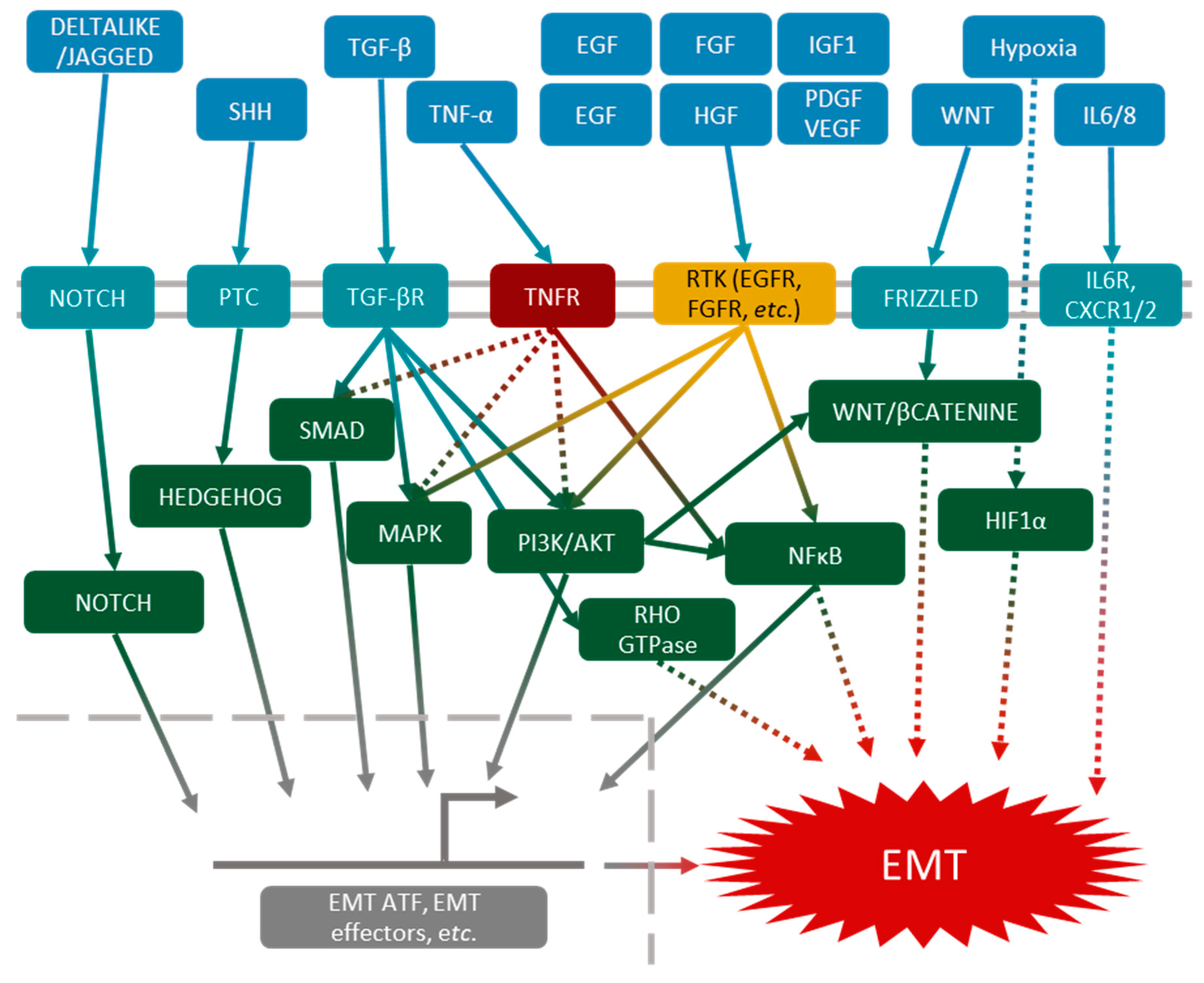
EMT can be induced by multiple molecular pathways. Here, we will summarize these pathways and briefly describe the most important, which is mediated by TGF-β.
TGF-β pathways were the first to be described in EMT induction. Nowadays, they are still the most documented [32]. TGF-β receptor can activate several intracellular pathways, such as the canonical SMAD pathway, leading to the expression of EMT-ATFs [33]. Other pathways, such as Rho-GTPase [34,35], PI3K/AKT [36] and MAPK [37,38], are activated by the TGF-β receptor, and can also induce EMT. All these pathways are redundant and can act together or separately, which explains the plurality of EMT phenotypes.
Other pathways, independently of TGF-β, can lead to EMT induction. The TNF-α-mediated NFκB pathway or EGF (epidermal growth factor) pathway may act synergistically with the TGF-β pathways [39,40], and other growth factors such as FGF (fibroblast growth factor) [41], HGF (hepatocyte growth factor) [42], IGF1 (insulin-like growth factor 1) [43], PDGF (platelet-derived growth factor) [44] and VEGF (vascular endothelial growth factor) [45] may act through the activation of PI3K/AKT and MAPK signalization. The Wnt [46], Hedgehog [47] and Notch [48,49] pathways have also been described as EMT inducers in many models.
Finally, in the cancer microenvironment, hypoxia and interleukins (mainly IL-6 and IL-8) also lead to cancer cell EMT (Figure 3).
3.2. Transcriptional Regulation of EMT
We have previously described that EMT can be induced by various molecular pathways. These pathways often lead to the activation of the expression of EMT-ATFs. The first EMT-ATFs described were Snail and Slug (later called Snail1 and Snail2) [50,51]. These transcription factors can repress or activate gene transcription in response to EMT induction. It was shown in vivo for the first time by Nieto et al. in 1994 [52] that the blockade of Snail or Slug expression can inhibit EMT. Since then, many studies have reported these observations [53,54].
There are two other main families of EMT-ATFs. The ZEB (Zinc finger E-box-binding homeobox) family was described in 2001 [55], and it includes two members, ZEB1 and ZEB2, which are both activators or repressors of transcription. The Twist family (Twist1 and Twist2) of transcription factors was described in 2004 [56].
3.3. Non-Coding RNA Regulation of EMT
EMT is largely regulated by non-coding RNA. Here, we will cite two regulation loops mediated by non-coding RNA that are essential during EMT regulation. First, miR-200 can induce ZEB1/2 and Snail1/2 mRNA degradation, and thus a decrease in their protein levels in cells. Actually, the forced expression of miR-200 is sufficient to blockade TGF-β-induced EMT [62,63,64,65]. Moreover, ZEB and Snail family members are able to repress miR-200 expression, forming a regulatory loop [66].
In the same way, miR-34 can also target Snail family mRNA and Snail1/2, directly decreasing the expression of miR-34 and thus constituting a similar regulatory loop [28].
4. EMT during Embryonic Development
EMT was first observed during embryonic development. Here, we report the main steps of development by which an EMT takes place.
4.1. Gastrulation
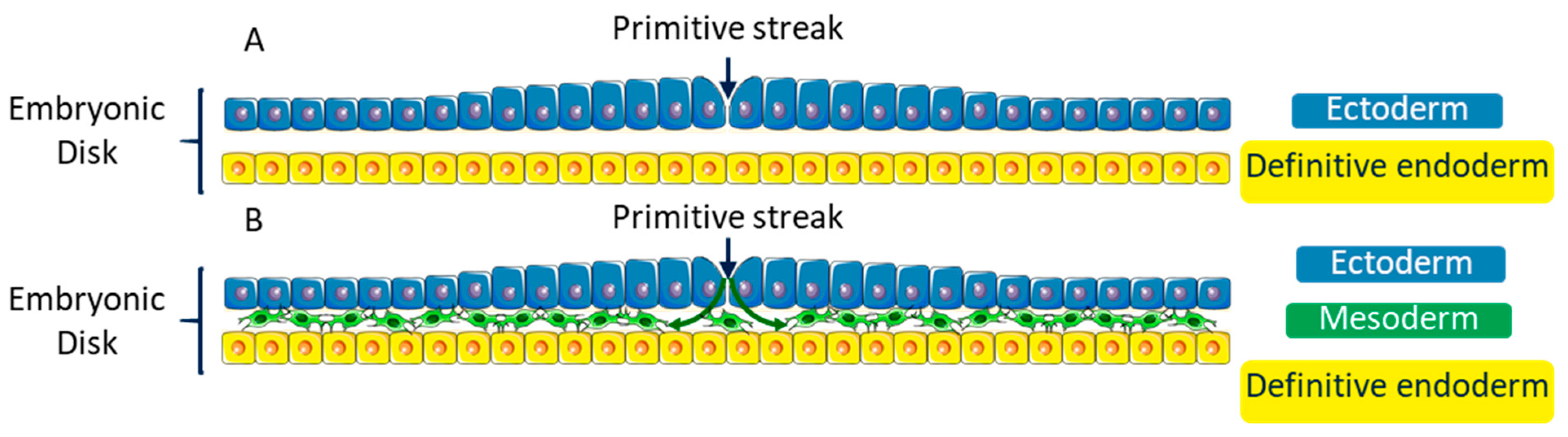
At the beginning of the third week of the development, the embryo consists of an embryonic disk including two layers: the hypoblast (primitive endoderm) and the epiblast (primitive ectoderm) (Figure 4A). Some epiblast cells undergo an EMT and move between the two layers to form a third one: the mesoderm [67,68,69]. This step of the development is called gastrulation (Figure 4B). This third layer extends entirely between the two other layers of the embryonic disk, except in two points: the cloacal membrane and the pharyngeal membrane, which are the first indication of future caudal extremity, and the first indication of the cranial extremity of the alimentary tract, respectively.
4.2. Neural Tube Formation
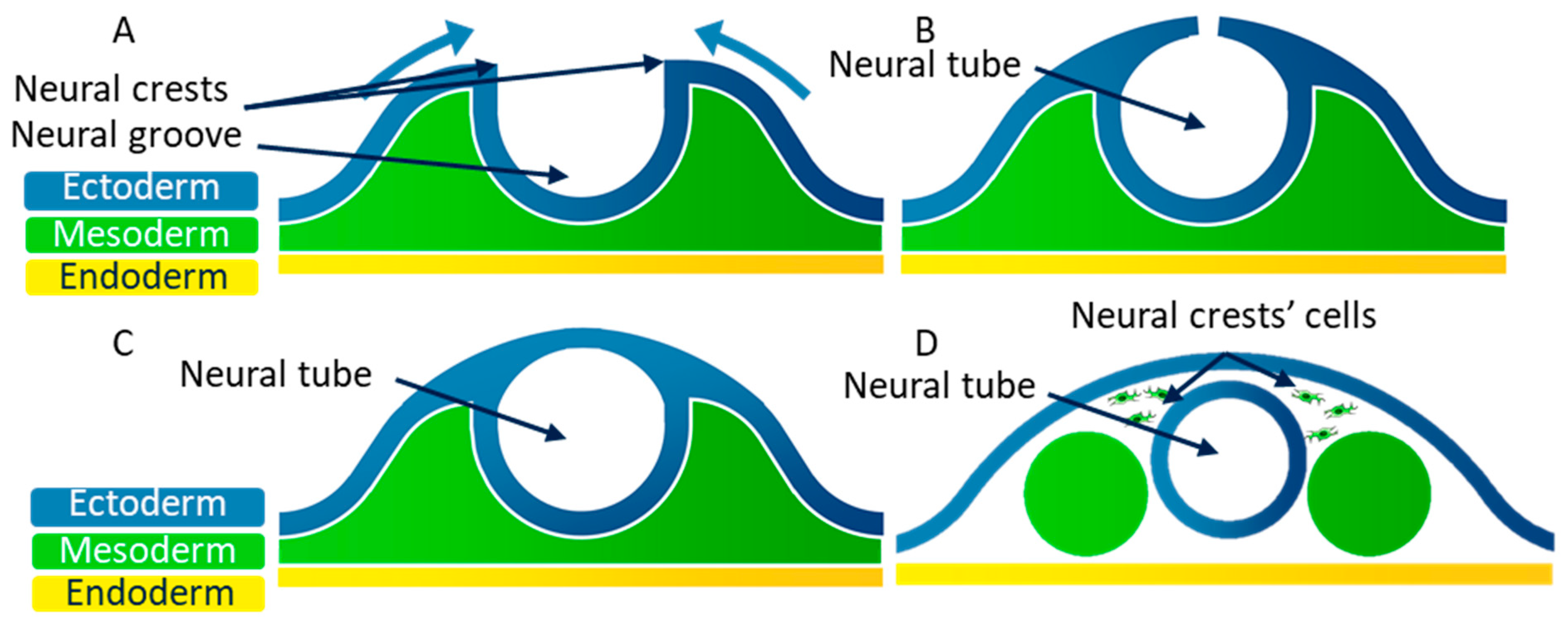
At the end of the third week of embryonic development, the embryo consists of three embryonic layers, as was previously described: the ectoderm, the mesoderm, and the endoderm. The neural plate corresponds to a thick area of the endoderm where cells divide rapidly. The edges of this plate grow to form the neural groove (Figure 5A). The neural crests are located on both sides of the neural groove. The neural crest cells then move to the medial edge, which allows the fusion of the neural crests (Figure 5B). During this fusion, the neural crest cells undergo an EMT, and they scatter in the mesoderm [70,71] (Figure 5C). The neural tube then splits from the ectoderm thanks to the scattering of the neural crest cells (Figure 5D).
4.3. Embryonic Palatal Fusion
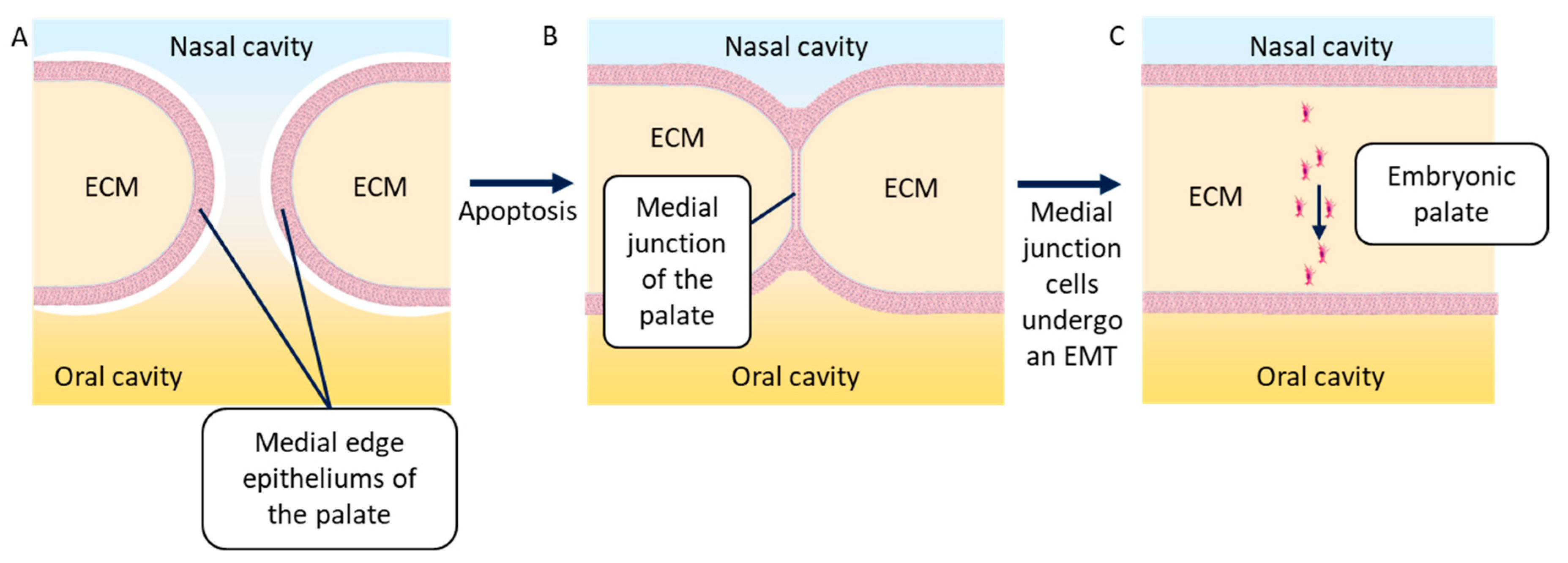
During the ninth week of embryonic development, the embryonic palatal shelves (one each side) move to meet on the medial edge and fuse. First, the epithelia of both shelves stick to each other to form the medial edge epithelium of the embryonic palate. This epithelium then disappears when its epithelial cells undergo an EMT and scatter in the neighboring mesenchyme [72]. Several researchers have described the apoptosis that may be responsible for the disappearance of the medial edge epithelium of the palate [73,74]; however, others showed that the cells undergoing apoptosis were part of the superficial layer of this epithelium. This superficial layer, by undergoing apoptosis, allows cells from the basal layers to fuse and undergo an EMT [17,22]. Recently, it has been shown that these cells do not scatter, but migrate together in the direction of the oral cavity. These cells may undergo a partial EMT, leading to the maintenance of some cell–cell junctions. The most mesenchymal among these cells can lead the migration of the group of cells [75] (Figure 6). This phenomenon is also observed during the formation of metastasis by cancer cells.
Gastrulation, neural tube formation and embryonic palatal fusion are the main steps of embryonic development in which an EMT is described, but EMT is also involved in additional processes. Indeed, EMT and MET control somite formation [76,77,78] and heart valve formation from the embryonic endocardium [19,79,80].
5. EMT in Fibrosis
One of the first description of EMT in fibrosis was made by the team of Eric Neilson in 2002 (Nashville, TN, USA) [81]. Using a kidney fibrosis model, they showed that fibroblast-like cells can appear locally as a result of EMT during fibrosis. Later, the team of Raghu Kalluri (Harvard Medical School, Boston, MA, USA) provided evidence that EMT (or endothelial to mesenchymal transition) also contributes to fibroblast-like cells’ appearance in liver or cardiac fibrosis, using lineage-tracing experiments [82,83]. More recently, Kalluri’s lab suggested that myofibroblasts do not derive from epithelial renal cells during renal fibrosis [84]. However, other works from Kalluri’s team, and Angela Nieto’s lab especially (Instituto de Neurociencias, Sant Joan d’Alacant, Spain), provided new data explaining that renal epithelial cells undergo a partial EMT, which is necessary for the recruitment of bone marrow-derived mesenchymal cells and myofibroblasts involved in fibrosis [85,86,87]. The main fibrosis treatment strategies thus target EMT [28].
6. EMT in Cancers
EMT’s involvement in cancers has been controversial, but there is growing evidence that its role is central in cancer aggressiveness, more so than in tumor development. Aggressiveness is exhibited by many cancer cell properties, such as treatment resistance, immune escape, cancer stem cell formation and metastasis. Indeed, tumor aggressiveness is an important factor in mortality for cancer patients, and that is why EMT is currently an important topic in anticancer treatment research. The involvement of EMT in cancers has been well reviewed [90,91].
For example, the analysis of circulating tumor cells (CTCs) is a good way to assess the importance of EMT in cancers—breast cancer CTCs express both epithelial and mesenchymal genes, showing they undergo EMT at multiple levels [92].
6.1. EMT Induction Factors of Cancer Cells
Many extra-cellular factors that can induce EMT are present in the tumor microenvironment. They have two main sources of origin: hypoxia [93] and inflammation [94].
Inflammation at the tumor site is characterized by the presence of immune cells, such as MDSCs (myeloid-derived suppressor cells) and macrophages, which secrete growth factors (TGF-β, HGF [94]) and cytokines (TNF-α (tumor necrosis factor α), IL (Interleukin)-1, IL-6, and IL-8) able to induce an EMT in cancer cells [95,96,97]. Moreover, since the SNAIL1 EMT-ATF can also induce pro-inflammatory cytokine expressions (IL-1, IL-6, and IL-8), this promotes a positive regulation loop [98]. Interestingly, it has been shown in spontaneous skin tumor mouse models that immune infiltration is associated with advanced EMT areas in the tumor [3].
When a tumor is growing, some cancer cells exist transiently in a hypoxia state. It has been shown that the transcription factor linked to hypoxia HIF1-α (hypoxia-inducible factor 1-α) can directly induce the expression of SNAIL1 EMT-ATF [99]. EMT induction due to hypoxia in tumors is also linked to TGF-β pathways [100].
6.2. EMT and Anticancer Therapy Resistance
Various teams have shown that EMT can induce resistance to classical anticancer treatments. For example, breast cancer patients resistant to chemotherapies have more CTCs and more mesenchymal cells than women with a good response to treatment [101]. A similar resistance to treatment has been associated with EMT in breast and pancreas cancers [102,103].
At a molecular level, EMT-ATFs can induce the expression of genes involved in DDR (DNA damage response) systems, which could counterbalance the effects of chemo- and radiotherapies [104,105]. SNAIL1 and TWIST EMT-ATFs also lead to an overexpression of ABC (adenosine triphosphate-binding cassette) transporters, which are responsible for multiple chemotherapy resistances. In the breast cancer cell line MCF7, TWIST directly binds to ABCC4 and ABCC5 promoters [106]. Finally, in HER2 (human EGF receptor 2)-positive breast cancers, the overexpression of miR-21 leads to resistance to chemotherapy and immunotherapy targeting HER2 in an EMT-inducing manner [107].
6.3. Immune Escape
Immune escape linked to EMT has recently been reviewed by Terry et al. [108]. SNAIL1-expressing mouse melanoma cells, when co-cultured with spleen cells, can induce the proliferation of immunosuppressive Treg (regulatory T cells) cells via TGF-β secretion. The silencing of SNAIL1 in these cells reduces Treg proliferation and invasion of melanoma cells [109].
EMT can also lead to a reduced expression of MHC class 1 (major histocompatibility complex) in cancer cells, and so to a diminution of the formation of immune synapses with cytotoxic T cells or NK (natural killer) cells decreasing cancer cell destruction by the immune system [110,111].
Moreover, EMT can induce an overexpression of PD-L1 (programmed death ligand 1), allowing cancer cells to inactivate immune cells harboring the associated receptor PD1 (programmed death 1). EMT induction in lung cancer cells leads to PD-L1 overexpression, and allows the inactivation of cytotoxic T cells and immune escape [112]. The EMT-ATF ZEB1 is involved in PD-L1 expression in MCF7 cells and a mouse lung adenocarcinoma model [113,114].
6.4. Metastasis Formation
Various studies showed the involvement of EMT during metastasis, based on tumor cell xenografts in immunodeficient mice and/or the overexpression of EMT-ATFs [115]. SNAIL1/2 is overexpressed in mouse spontaneous mammary tumors, but not in bone marrow metastasis. These proteins are important for local tissue invasion, but not in metastatic colonization. Inhibiting SNAIL1/2 expression in this model prevents metastasis formation [54,116].
Some pioneers’ studies have shown the importance of cell plasticity during the metastatic cascade, describing that a fine regulation of EMT is required to allow cancer cells to metastasize. In other words, cells need “a good” epithelial or mesenchymal shape at each step of this process to metastasize efficiently [117,118,119]. In a model of spontaneous mammary mouse tumors, a few cells undergo EMT, invade neighboring tissues then blood vessels, and then establish in other organs (lung, liver, and lymph nodes). In the secondary organ, cells can undergo an MET to develop a metastasis [120]. However, the involvement of EMT during metastasis formation is still a controversial field. Two main studies suggest that EMT is not essential for this phenomenon, showing that metastatic cells from mouse breast cancer never express vimentin, and that blocking EMT by the overexpression of miR-200 does not reduce metastatic formation in this model [102]. However, this work showed that there is an enrichment of mesenchymal cells in CTCs, suggesting that these cells could be essential for intravasation and could facilitate the intravasation of epithelial tumor cells by collective migration [3,28]. This could explain why metastasis are only formed by epithelial cells in this model. Then, it was demonstrated that the loss of expression of SNAIL1 or TWIST1 does not prevent metastasis formation in mice pancreatic ductal adenocarcinoma [103]. However, other EMT-ATFs could induce EMT instead of SNAIL1 and TWIST1 in these cancers [28]. Moreover, these two papers highlighted that the authors did not achieve the complete blockade of EMT or track all forms of EMT programs. This confirms that these works are not sufficient to prove that EMT is not required in the metastasis process in these models [121,122].
In a model of spontaneous mammary mice tumors, a partial EMT gives cells with both epithelial and mesenchymal features. This seems to facilitate metastasis development, and promotes the collective migration of cells, while full mesenchymal cells migrate alone [2,3]. In this way, the co-expression of epithelial and mesenchymal markers could indicate a poor prognosis in various cancers (breast, liver, and brain) [123,124,125].
6.5. Cancer Stem Cells
CSCs (cancer stem cells) are cancer cells with stem characteristics and high clonogenic potential [2]. CSCs are defined as the source of tumor development and/or being capable of being so. They have been described to express a high level of CD44 and a low level of CD24, or to highly express ALDH1 (aldehyde dehydrogenase 1). These features are generated during EMT in breast cancer cells [115,116,126]. The specific stem cell transcription factors OCT4 and NANOG are also frequently expressed during EMT. Both phenotypes are mechanistically linked [127]. In pancreas cancers, EMT is linked to stem cell properties because ZEB1 represses the expression of miRNAs that normally inhibit the stem cell phenotype [128]. SNAIL1 is also responsible for the symmetric division of colorectal CSCs, increasing their pool [129].
However, in oral squamous cell carcinomas, three categories of CSC have been identified according to the expression of CD44, ESA (epidermal surface antigen) and ALDH1 (aldehyde dehydrogenase 1). The CD44high/ESAlow cells are identified as mesenchymal CSCs while the CD44high/ESAhigh are epithelial. Moreover, the same team have shown that all epithelial CSCs can reconstitute tumor heterogeneity (epithelial and mesenchymal cells), while only a few mesenchymal CSCs can do the same. This heterogeneity in mesenchymal CSC population is characterized by ALDH1 expression. Indeed, CD44high/ESAlow/ALDH1high cells are mesenchymal CSCs able to restore tumor heterogeneity, while CD44high/ESAlow/ALDH1low cells cannot [130]. This could explain why very mesenchymal cells could not systematically metastasize more than strongly epithelial cells. Finally, in a model of mice spontaneous skin tumors, tumor cells with a mixed epithelial/mesenchymal phenotype can give rise to all the different tumor cell phenotypes when they are injected subcutaneously into other animals, while the more epithelial or mesenchymal cells failed [3].
6.6. Anticancer Treatments Targeting EMT
EMT can be promoted by multiple molecular pathways at the same time, and these pathways can be made redundant by finally converging in the same pathway. The therapeutic targeting of this phenomenon should affect different levels. The targeting of extracellular stimuli, EMT-ATFs and mesenchymal effectors seems to be a good strategy [131].
Concerning extracellular stimuli, some clinical trials are testing TGF-βRI (TGF-βReceptor I) (LY2157299) inhibitor molecule efficacy in pancreas cancers, hepatocarcinoma, glioma and glioblastoma [131,132,133,134], and anti-TGF-β antibody (Fresolimumab) efficacy in combination with radiotherapy in breast metastatic cancers [135]. Moreover, an inhibitor of the EGF receptor (erlotinib) has been approved for advanced lung cancer treatment [136]. EMT induction by hypoxia has also been targeted, at the preclinical level for now. An indirect inhibitor of HIF1α activity was shown to decrease metastasis formation in a mouse model of tumor dissemination [137].
The second class of EMT targets are the EMT-ATFs. The direct inhibition of these transcription factors is not well documented. However, several molecules can inhibit EMT-ATFs indirectly: CDK4/6 inhibitors or STAT3 inhibitors [131,138,139,140]. Immunotherapies targeting EMT-ATFs are under development. An anti-TWIST1 vaccine is effective in mice, helping to prevent tumor growth and metastasis formation in a 4T1 breast cancer model [141]. Another vaccine, targeting the EMT-ATF Brachyury, is currently under phase I and II clinical trials in different advanced cancers (NCT02383498) [142].
The third targets that could help blockade EMT are mesenchymal or epithelial effectors. Various pre-clinical studies tried to re-express the epithelial protein E-Cadherin in tumor cells. This is a difficult strategy because E-Cadherin expression has also been linked to tumor growth [131]. Re-expressing E-Cadherin can be achieved by targeting DNA methylation and histone deacetylation at its promoter during EMT [131,143]. Epigenetic mechanisms are a promising approach for cancer therapies targeting EMT. Indeed, not only E-Cadherin but many EMT-related genes are regulated epigenetically. For example, in breast, kidney or lung cancer cells, MMP9 (matrix metalloproteinase 9) and ADAM19 (Adam metallopeptidase 19) metalloproteinase expression during EMT are modulated by changes in histone methylation marks [144]. Finally, the silencing of vimentin or N-Cadherin expression could decrease the invasion and migration of tumor cell lines or mice models. The activity of these mesenchymal effectors has been modulated by specific antibodies or by chemical compounds. EMT targeting has been exhaustively reviewed by Malek et al. [131].
An EMT blockade in cancer cells can sensitize them to immunotherapies. This is why the association of these two strategies is promising and is being tested in various clinical trials. TGF-βRI inhibitors and anti-TGF-β antibodies (including anti-PD-L1 and anti-TGF-βRII fusion antibody) are currently being tested [145].
7. Conclusions
The increasing number of EMT studies is linked to the involvement of this phenomenon in cancers. However, EMT was discovered through Elizabeth Hay’s team’s work on chicken embryonic development. Nowadays, EMT is thought to be involved in development, wound-healing, fibrosis, and cancers. It was first considered as a binary phenomenon (called epithelial to mesenchymal transformation) transforming a full epithelial cell into a full mesenchymal one. Then, the binary nature of EMT was challenged with the identification of the inverse phenomenon: MET (mesenchymal to epithelial transition). Now, it is considered that EMT can lead to a spectrum of intermediate states between a full epithelial phenotype and a full mesenchymal one, mainly due to the work of Pastushenko et al. [2,3]. The better comprehension of this phenomenon allows us to imagine more effective cancer therapies targeting multiple features of EMT.
Author Contributions
Conceptualization, C.L.; writing—original draft preparation, C.L.; writing—review and editing, E.H. and P.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from institutional grants from INSERM (Institut National de la Santé et de la Recherche Médicale), EFS (Etablissement Français du Sang), Univ. Bourgogne Franche-Comté and Région Bourgogne Franche-Comté.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABC | Adenosine triphosphate-binding cassette |
| ADAM19 | Adam metallopeptidase 19 |
| ALDH1 | Aldehyde dehydrogenase 1 |
| CTC | Circulating tumor cells |
| DDR | DNA damage response |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EMT | Epithelial–mesenchymal transition |
| EMT-ATF | Epithelial to mesenchymal transition-activated transcription factor |
| ESA | Epidermal surface antigen |
| FGF | Fibroblast growth factor |
| GAG | Glycosaminoglycans |
| HER2 | Human EGF receptor 2 |
| HGF | Hepatocyte growth factor |
| HIF1-α | Hypoxia inducible factor 1-α |
| IGF1 | Insluin-like growth factor 1 |
| IL | Interleukin |
| MDSC | Myeloid-derived suppressor cells |
| MET | Mesenchymal to epithelial transition |
| MHC | Major histocompatibility complex |
| MMP9 | Matrix metalloproteinase 9 |
| NK | Natural killer |
| PD1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| PDGF | Platelet-derived growth factor |
| SMAD | Mothers against decapentaplegic homolog |
| TGF | Tranforming growth gactor |
| TGF-βR | TGF-β receptor |
| TNF | Tumor necrosis factor |
| Treg | Regulatory T cell |
| VEGF | Vascular endothelial growth factor |
| WB | Western blotting |
| ZA | Zonula adherens |
References
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Hay, E.D. The Fine Structure of Blastema Cells and Differentiating Cartilage Cells in Regenerating Limbs of Amblystoma Larvae. J. Cell Biol. 1958, 4, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Meier, S.; Hay, E.D. Control of corneal differentiation by extracellular materials. Collagen as a promoter and stabilizer of epithelial stroma production. Dev. Biol. 1974, 38, 249–270. [Google Scholar] [CrossRef]
- Hay, E.D. Organization and Fine Structure of Epithelium and Mesenchyme in the Developing Chick Embryo. In Epithelial-Mesenchymal Intercations. 18th Hahnemann Symposium; Fleischmajer, R., Billingham, R., Eds.; The Williams and Wilkins Company: Baltimore, MD, USA, 1968; pp. 31–55. [Google Scholar]
- Böcker, W.; Stegner, H.-E. A light and electron microscopic study of endometrial sarcomas of the uterus. Virchows Arch. A Path. Anat. Histol. 1975, 368, 141–156. [Google Scholar] [CrossRef]
- Ishikawa, S.; Kaneko, H.; Sumida, T.; Sekiya, M. Ultrastructure of Mesodermal Mixed Tumor of the Uterus. Pathol. Int. 1979, 29, 801–809. [Google Scholar] [CrossRef]
- Dulbecco, R.; Henahan, M.; Bowman, M.; Okada, S.; Battifora, H.; Unger, M. Generation of fibroblast-like cells from cloned epithelial mammary cells in vitro: A possible new cell type. Proc. Natl. Acad. Sci. USA 1981, 78, 2345–2349. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.C.; Peachey, L.A.; Durbin, H.; Rudland, P.S. A possible mammary stem cell line. Cell 1978, 15, 283–298. [Google Scholar] [CrossRef]
- Franke, W.W.; Grund, C.; Kuhn, C.; Jackson, B.W.; Illmensee, K. Formation of cytoskeletal elements during mouse embryogenesis. III. Primary mesenchymal cells and the first appearance of vimentin filaments. Differentiation 1982, 23, 43–59. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E. Cytodifferentiation and tissue phenotype change during transformation of embryonic lens epithelium to mesenchyme-like cells in vitro. Dev. Biol. 1986, 115, 363–379. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E. Cytoskeleton and thyroglobulin expression change during transformation of thyroid epithelium to mesenchyme-like cells. Development 1988, 102, 605–622. [Google Scholar] [CrossRef]
- Bilozur, M.E.; Hay, E.D. Cell migration into neural tube lumen provides evidence for the “fixed cortex” theory of cell motility. Cell Motil. 1989, 14, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Bilozur, M.E.; Hay, E.D. Neural crest migration in 3D extracellular matrix utilizes laminin, fibronectin, or collagen. Dev. Biol. 1988, 125, 19–33. [Google Scholar] [CrossRef]
- Hay, E.D. Role of cell-matrix contacts in cell migration and epithelial-mesenchymal transformation. Cell Differ. Dev. 1990, 32, 367–375. [Google Scholar] [CrossRef]
- Fitchett, J.E.; Hay, E.D. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 1989, 131, 455–474. [Google Scholar] [CrossRef]
- Gavrilovic, J.; Moens, G.; Thiery, J.P.; Jouanneau, J. Expression of transfected transforming growth factor alpha induces a motile fibroblast-like phenotype with extracellular matrix-degrading potential in a rat bladder carcinoma cell line. Cell Regul. 1990, 1, 1003–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potts, J.D.; Dagle, J.; Walder, J.A.; Weeks, D.L.; Runyan, R.B. Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta 3. Proc. Natl. Acad. Sci. USA 1991, 88, 1516–1520. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Vanderburg, C.; Odierna, G.; Hay, E. TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development 1998, 125, 95–105. [Google Scholar] [CrossRef]
- Nawshad, A.; LaGamba, D.; Hay, E. Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch. Oral Biol. 2004, 49, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Nawshad, A.; Medici, D.; Liu, C.-C.; Hay, E.D. TGFβ3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J. Cell Sci. 2007, 120, 1646–1653. [Google Scholar] [CrossRef] [Green Version]
- Piek, E.; Moustakas, A.; Kurisaki, A.; Heldin, C.; Dijke, P.T. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 1999, 112, 4557–4568. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug Promote Epithelial-Mesenchymal Transition through β-Catenin–T-Cell Factor-4-dependent Expression of Transforming Growth Factor-β3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Dijke, P.T. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Tavares, A.L.P.; Mercado-Pimentel, M.E.; Runyan, R.B.; Kitten, G.T. TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev. Dyn. 2006, 235, 1589–1598. [Google Scholar] [CrossRef]
- Cho, H.J.; Yoo, J. Rho activation is required for transforming growth factor-beta-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol. Int. 2007, 31, 1225–1230. [Google Scholar] [CrossRef]
- Kattla, J.J.; Carew, R.M.; Heljić, M.; Godson, C.; Brazil, D.P. Protein kinase B/Akt activity is involved in renal TGF-β1-driven epithelial-mesenchymal transition in vitro and in vivo. Am. J. Physiol. Ren. Physiol. 2008, 295, F215–F225. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-β1-Induced EMT In Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002, 115, 3193–3206. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-J.; Luo, J.; Li, D.; Zhou, Y.-H.; Yan, B.; Wei, J.-J.; Tu, J.-C.; Li, Y.-R.; Zhang, G.-M.; Feng, Z.-H. TGF-β1 and TNF-α synergistically induce epithelial to mesenchymal transition of breast cancer cells by enhancing TAK1 activation. J. Cell Commun. Signal. 2019, 13, 369–380. [Google Scholar] [CrossRef]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar]
- Sun, X.; Meyers, E.N.; Lewandoski, M.; Martin, G.R. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999, 13, 1834–1846. [Google Scholar] [CrossRef] [Green Version]
- Grotegut, S.; Von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like Growth Factor-I–Dependent Up-regulation of ZEB1 Drives Epithelial-to-Mesenchymal Transition in Human Prostate Cancer Cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Liu, Z.-R. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.J.; et al. Vascular Endothelial Growth Factor Receptor-1 Activation Mediates Epithelial to Mesenchymal Transition in Human Pancreatic Carcinoma Cells. Cancer Res. 2006, 66, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.; del Amo, F.F.; Gridley, T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: Its expression pattern suggests multiple roles during postimplantation development. Development 1992, 116, 1033–1039. [Google Scholar] [CrossRef]
- Nieto, M.; Bennett, M.; Sargent, M.; Wilkinson, D. Cloning and developmental expression of Sna, a murine homologue of the Drosophila snail gene. Development 1992, 116, 227–237. [Google Scholar] [CrossRef]
- Nieto, M.Á.; Sargent, M.G.; Wilkinson, D.; Cooke, J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 1994, 264, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The Mouse Snail Gene Encodes a Key Regulator of the Epithelial-Mesenchymal Transition. Mol. Cell. Biol. 2001, 21, 8184–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 Expression Is Necessary for Metastatic Competence in Breast Cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The Two-Handed E Box Binding Zinc Finger Protein SIP1 Downregulates E-Cadherin and Induces Invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Girelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; de Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Birchmeier, C.; Brand-Saheri, B.; Birchmeier, W.; Brand-Saberi, B. Epithelial-Mesenchymal Transitions in Cancer Progression. Cells Tissues Organs 1996, 156, 217–226. [Google Scholar] [CrossRef]
- Perl, A.T.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nat. Cell Biol. 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Xu, L.-Y.; Li, E.-M. A Family of Pleiotropically Acting MicroRNAs in Cancer Progression, miR-200: Potential Cancer Therapeutic Targets. Curr. Pharm. Des. 2014, 20, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, P.P.L.; Williams, E.A.; Chan, W.Y. Gastrulation in the mouse embryo: Ultrastructural and molecular aspects of germ layer morphogenesis. Microsc. Res. Tech. 1993, 26, 301–328. [Google Scholar] [CrossRef]
- Nakaya, Y.; Sheng, G. Epithelial to mesenchymal transition during gastrulation: An embryological view. Dev. Growth Differ. 2008, 50, 755–766. [Google Scholar] [CrossRef]
- Tam, P.; Beddington, R. The formation of mesodermal tissues in the mouse embryo during gastrulation and early organogenesis. Development 1987, 99, 109–126. [Google Scholar] [CrossRef]
- Rogers, C.D.; Saxena, A.; Bronner, M.E. Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J. Cell Biol. 2013, 203, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Bronner, M.E. Formation and migration of neural crest cells in the vertebrate embryo. Histochem. Cell Biol. 2012, 138, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Gibbins, J.R.; Manthey, A.; Tazawa, Y.M.; Scott, B.; Bloch-Zupan, A.; Hunter, N. Midline fusion in the formation of the secondary palate anticipated by upregulation of keratin K5/6 and localized expression of vimentin mRNA in medial edge epithelium. Int. J. Dev. Biol. 1999, 43, 237–244. [Google Scholar] [PubMed]
- Cuervo, R.; Covarrubias, L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 2004, 131, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, C.; Nakamura, N.; Okamoto, Y.; Osawa, M.; Shiota, K. Cytochemical identification of programmed cell death in the fusing fetal mouse palate by specific labelling of DNA fragmentation. Anat. Embryol. 1994, 190, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.M.; Benson, M.D. Medial epithelial seam cell migration during palatal fusion. J. Cell. Physiol. 2020, 235, 1417–1424. [Google Scholar] [CrossRef]
- Duband, J.L.; Dufour, S.; Hatta, K.; Takeichi, M.; Edelman, G.M.; Thiery, J.P. Adhesion molecules during somitogenesis in the avian embryo. J. Cell Biol. 1987, 104, 1361–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, Y.; Kuroda, S.; Katagiri, Y.T.; Kaibuchi, K.; Takahashi, Y. Mesenchymal-Epithelial Transition during Somitic Segmentation Is Regulated by Differential Roles of Cdc42 and Rac1. Dev. Cell 2004, 7, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Sato, Y.; Suetsugu, R.; Nakaya, Y. Mesenchymal-to-Epithelial Transition during Somitic Segmentation: A Novel Approach to Studying the Roles of Rho Family GTPases in Morphogenesis. Cells Tissues Organs 2005, 179, 36–42. [Google Scholar] [CrossRef]
- Person, A.D.; Klewer, S.E.; Runyan, R.B. Cell Biology of Cardiac Cushion Development. Int. Rev. Cytol. 2005, 243, 287–335. [Google Scholar]
- Runyan, R.B.; Heimark, R.L.; Camenisch, T.D.; Klewer, S.E. Epithelial-Mesenchymal Transformation in the Embryonic Heart. In Rise and Fall of Epithelial Phenotype: Concepts of Epithelial-Mesenchymal Transition; Savagner, P., Ed.; Springer: Boston, MA, USA, 2005; pp. 40–55. [Google Scholar]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts Derive from Hepatocytes in Liver Fibrosis via Epithelial to Mesenchymal Transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; de Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M. Ángela Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Borges, F.T.; Melo, S.A.; Özdemir, B.C.; Kato, N.; Revuelta, I.; Miller, C.A.; Ii, V.H.G.; LeBleu, V.S.; Kalluri, R. TGF-β1–Containing Exosomes from Injured Epithelial Cells Activate Fibroblasts to Initiate Tissue Regenerative Responses and Fibrosis. J. Am. Soc. Nephrol. 2012, 24, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Strippoli, R.; Moreno-Vicente, R.; Battistelli, C.; Cicchini, C.; Noce, V.; Amicone, L.; Marchetti, A.; del Pozo, M.A.; Tripodi, M. Molecular Mechanisms Underlying Peritoneal EMT and Fibrosis. Stem Cells Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Khoo, B.L.; Lee, S.C.; Kumar, P.; Tan, T.Z.; Warkiani, M.E.; Ow, S.G.; Nandi, S.; Lim, C.T.; Thiery, J.P. Short-term expansion of breast circulating cancer cells predicts response to anti-cancer therapy. Oncotarget 2015, 6, 15578–15593. [Google Scholar] [CrossRef] [Green Version]
- Eskiizmir, G.; Özgür, E. Epithelial-Mesenchymal Transition in Tumor Microenvironment Induced by Hypoxia. Cancer Metastasis 2018. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Mittal, V. Tumor Microenvironment Regulates Epithelial—Mesenchymal Transitions in Mestastasis. Expert. Rev. Anticancer Ther. 2012, 12, 857–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.-C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.-P.; Abastado, J.-P. Mesenchymal Transition and Dissemination of Cancer Cells Is Driven by Myeloid-Derived Suppressor Cells Infiltrating the Primary Tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ieda, T.; Tazawa, H.; Okabayashi, H.; Yano, S.; Shigeyasu, K.; Kuroda, S.; Ohara, T.; Noma, K.; Kishimoto, H.; Nishizaki, M.; et al. Visualization of epithelial-mesenchymal transition in an inflammatory microenvironment–colorectal cancer network. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Lyons, J.G.; Patel, V.; Roue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail Up-regulates Proinflammatory Mediators and Inhibits Differentiation in Oral Keratinocytes. Cancer Res. 2008, 68, 4525–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Wang, J.; Li, J.; Post, M. Mouse Snail Is a Target Gene for HIF. Mol. Cancer Res. 2011, 9, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.; Choi, H.; Rayes, T.E.; Ryu, S.; Troeger, J.; et al. EMT is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. EMT Program is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.S.-S.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Chang, S.-Y.; Tsai, T.-L.; Chang, C.-C.; Tzeng, C.-H.; Wu, K.-J.; Kao, J.-Y.; et al. Regulation of Excision Repair Cross-Complementation Group 1 by Snail Contributes to Cisplatin Resistance in Head and Neck Cancer. Clin. Cancer Res. 2010, 16, 4561–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; A Stephens, M.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [Green Version]
- De Mattos-Arruda, L.; Bottai, G.; Nuciforo, P.G.; di Tommaso, L.; Giovannetti, E.; Peg, V.; Losurdo, A.; Pérez-Garcia, J.; Masci, G.; Corsi, F.; et al. MicroRNA-21 links epithelial-to-mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2-positive breast cancer patients. Oncotarget 2015, 6, 37269–37280. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; de Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell–Mediated Lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. OncoImmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nat. Cell Biol. 2015, 525, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celià-Terrassa, T.; Meca-Cortés, Ó.; Mateo, F.; de Paz, A.M.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [Green Version]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudernaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nat. Cell Biol. 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nat. Cell Biol. 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat. Commun. 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, H.; Huang, Y.; Xiang, L.; Chen, Z.; Fang, Y.; Lin, Y.; Cui, Z.; Yu, S.; Li, X.; Yang, D. Circulating Tumor Cell Phenotype Indicates Poor Survival and Recurrence After Surgery for Hepatocellular Carcinoma. Dig. Dis. Sci. 2018, 63, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Polioudaki, H.; Agelaki, S.; Chiotaki, R.; Politaki, E.; Mavroudis, D.; Matikas, A.; Georgoulias, V.; Theodoropoulos, P.A. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer 2015, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to Mesenchymal Transition Is Mechanistically Linked with Stem Cell Signatures in Prostate Cancer Cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef]
- Wellner, U.F.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Hwang, W.-L.; Jiang, J.-K.; Yang, S.-H.; Huang, T.-S.; Lan, H.-Y.; Teng, H.-W.; Yang, C.-Y.; Tsai, Y.-P.; Lin, C.-H.; Yang, M.-H. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef]
- Biddle, A.; Liang, X.; Gammon, L.; Fazil, B.; Harper, L.J.; Emich, H.; Costea, D.E.; MacKenzie, I.C. Cancer Stem Cells in Squamous Cell Carcinoma Switch between Two Distinct Phenotypes That Are Preferentially Migratory or Proliferative. Cancer Res. 2011, 71, 5317–5326. [Google Scholar] [CrossRef] [Green Version]
- Malek, R.; Wang, H.; Taparra, K.; Tran, P.T. Therapeutic Targeting of Epithelial Plasticity Programs: Focus on the Epithelial-Mesenchymal Transition. Cells Tissues Organs 2017, 203, 114–127. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFβ receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S.; et al. Biomarkers and overall survival in patients with advanced hepatocellular carcinoma treated with TGF-βRI inhibitor galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formenti, S.C.; Hawtin, R.E.; Dixit, N.; Evensen, E.; Lee, P.; Goldberg, J.D.; Li, X.; Vanpouille-Box, C.; Schaue, D.; McBride, W.H.; et al. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J. Immunother. Cancer 2019, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Szczésna, A.; Juhász, E.; Esteban, E.; Molinier, O.; Brugger, W.; Melezínek, I.; et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: A multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010, 11, 521–529. [Google Scholar] [CrossRef]
- Goto, Y.; Zeng, L.; Yeom, C.J.; Zhu, Y.; Morinibu, A.; Shinomiya, K.; Kobayashi, M.; Hirota, K.; Itasaka, S.; Yoshimura, M.; et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1α. Nat. Commun. 2015, 6, 6153. [Google Scholar] [CrossRef] [Green Version]
- Arima, Y.; Hayashi, H.; Sasaki, M.; Hosonaga, M.; Goto, T.M.; Chiyoda, T.; Kuninaka, S.; Shibata, T.; Ohata, H.; Nakagama, H.; et al. Induction of ZEB Proteins by Inactivation of RB Protein Is Key Determinant of Mesenchymal Phenotype of Breast Cancer. J. Biol. Chem. 2012, 287, 7896–7906. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep. 2016, 5, 17663. [Google Scholar] [CrossRef] [Green Version]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W.; et al. Phase I study of OPB -51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef]
- Ardiani, A.; Gameiro, S.R.; Palena, C.; Hamilton, D.H.; Kwilas, A.; King, T.H.; Schlom, J.; Hodge, J.W. Vaccine-mediated immunotherapy directed against a transcription factor driving the metastatic process. Cancer Res. 2014, 74, 1945–1957. [Google Scholar] [CrossRef] [Green Version]
- Heery, C.R.; Singh, B.H.; Rauckhorst, M.; Marté, J.L.; Donahue, R.N.; Grenga, I.; Rodell, T.C.; Dahut, W.L.; Arlen, P.M.; Madan, R.A.; et al. Phase I Trial of a Yeast-Based Therapeutic Cancer Vaccine (GI-6301) Targeting the Transcription Factor Brachyury. Cancer Immunol. Res. 2015, 3, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone deacetylase inhibitors: Current status and overview of recent clinical trials. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Timeline representing the main events in Epithelial to Mesenchymal Transition’s (EMT) research history. TGF-β = Transforming growth factor β; SMAD = Mothers against decapentaplegic homolog.
Figure 1.
Timeline representing the main events in Epithelial to Mesenchymal Transition’s (EMT) research history. TGF-β = Transforming growth factor β; SMAD = Mothers against decapentaplegic homolog.

Figure 2.
Time evolution of occurrences for “Epithelial to Mesenchymal Transition” search in PubMed database (National Center for Biotechnology Information, Bethesda, MD, USA), stopped on 27 January 2021.
Figure 2.
Time evolution of occurrences for “Epithelial to Mesenchymal Transition” search in PubMed database (National Center for Biotechnology Information, Bethesda, MD, USA), stopped on 27 January 2021.

Figure 3.
Molecular aspects of EMT induction. EMT induction can result from the activation, simultaneously or not, of one or several pathways: Notch, Hedgehog, SMAD, MAPK, PI3K/AKT, Rho GTPase, NFκB, Wnt and Hypoxia inducible factor 1α (HIF1α).
Figure 3.
Molecular aspects of EMT induction. EMT induction can result from the activation, simultaneously or not, of one or several pathways: Notch, Hedgehog, SMAD, MAPK, PI3K/AKT, Rho GTPase, NFκB, Wnt and Hypoxia inducible factor 1α (HIF1α).

Figure 4.
Simplified diagram of a transversal cross-section of the embryonic disk at the third week of development showing the gastrulation step. (A) At the beginning of the third week of development, the embryonic disk consists of two back-to-back layers: the primitive ectoderm (or epiblast) and the definitive endoderm (or hypoblast). (B) During the third week of embryonic development, some cells from the primitive streak of the primitive ectoderm undergo an EMT and move between the ectoderm and the endoderm to create a third mesenchymal embryonic layer: the mesoderm.
Figure 4.
Simplified diagram of a transversal cross-section of the embryonic disk at the third week of development showing the gastrulation step. (A) At the beginning of the third week of development, the embryonic disk consists of two back-to-back layers: the primitive ectoderm (or epiblast) and the definitive endoderm (or hypoblast). (B) During the third week of embryonic development, some cells from the primitive streak of the primitive ectoderm undergo an EMT and move between the ectoderm and the endoderm to create a third mesenchymal embryonic layer: the mesoderm.

Figure 5.
Simplified diagram of a transversal cross-section of the embryonic disk at the end of the third week of development showing the closing of the neural tube. (A) At the end of the third week of embryonic development, the ectoderm forms the neural groove, which creates neural crests. (B) Neural crest cells move to the medial edge to close the neural tube. (C) During the neural crests’ fusion, epithelial cells from these crests undergo an EMT to scatter in the mesoderm. (D) The neural tube splits from the ectoderm thanks to the scattering of the neural crest cells.
Figure 5.
Simplified diagram of a transversal cross-section of the embryonic disk at the end of the third week of development showing the closing of the neural tube. (A) At the end of the third week of embryonic development, the ectoderm forms the neural groove, which creates neural crests. (B) Neural crest cells move to the medial edge to close the neural tube. (C) During the neural crests’ fusion, epithelial cells from these crests undergo an EMT to scatter in the mesoderm. (D) The neural tube splits from the ectoderm thanks to the scattering of the neural crest cells.

Figure 6.
Simplified diagram of a frontal cross-section of the embryonic palatal fusion. (A) Medial edge epithelia of the right and left palatal shelves migrate in the direction of the medial axis of the embryo, marking out the nasal and the oral cavities. (B) The medial edge epithelia of the palatal shelves stick with each other to create the medial junction of the palatal shelves, thanks to the apoptosis of the cells composing the superficial layer of these epithelia. (C) The cells of the medial junction of the palatal shelves undergo an EMT and migrate together in the direction of the oral cavity, allowing for palatal ECM continuity. ECM = extracellular matrix.
Figure 6.
Simplified diagram of a frontal cross-section of the embryonic palatal fusion. (A) Medial edge epithelia of the right and left palatal shelves migrate in the direction of the medial axis of the embryo, marking out the nasal and the oral cavities. (B) The medial edge epithelia of the palatal shelves stick with each other to create the medial junction of the palatal shelves, thanks to the apoptosis of the cells composing the superficial layer of these epithelia. (C) The cells of the medial junction of the palatal shelves undergo an EMT and migrate together in the direction of the oral cavity, allowing for palatal ECM continuity. ECM = extracellular matrix.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Genes that promote EMT or Mesenchymal to epithelial transition (MET), adapted from [27].
Table 1.
Genes that promote EMT or Mesenchymal to epithelial transition (MET), adapted from [27].
| Genes That Promote | ||
|---|---|---|
| EMT | MET | |
| Cell surface programs | α5β1 integrin | L-CAM; E-cadherin; α6 integrin; Syndecan 1; Laminin/nidogen |
| Oncogenes | v-src; c-fos; v-ras; v-mos | c-met |
| Growth factors | TGF-β1-3; MIF; TGF-α; aFGF | HGF/SF |
| Other genes | wnt-1; wnt-4; pax 2; E1a | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lachat, C.; Peixoto, P.; Hervouet, E. Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules 2021, 11, 782. https://doi.org/10.3390/biom11060782
AMA Style
Lachat C, Peixoto P, Hervouet E. Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules. 2021; 11(6):782. https://doi.org/10.3390/biom11060782
Chicago/Turabian StyleLachat, Camille, Paul Peixoto, and Eric Hervouet. 2021. "Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers" Biomolecules 11, no. 6: 782. https://doi.org/10.3390/biom11060782
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.