Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease


Abstract
:1. Basic Information on Tissue-nonspecific Alkaline Phosphatase (TNAP/Tnap)
1.1. Genetic Information on ALPL
1.2. Biochemical Information on TNAP
2. Hypophosphatasia—Diagnosis and Treatment (an Update)
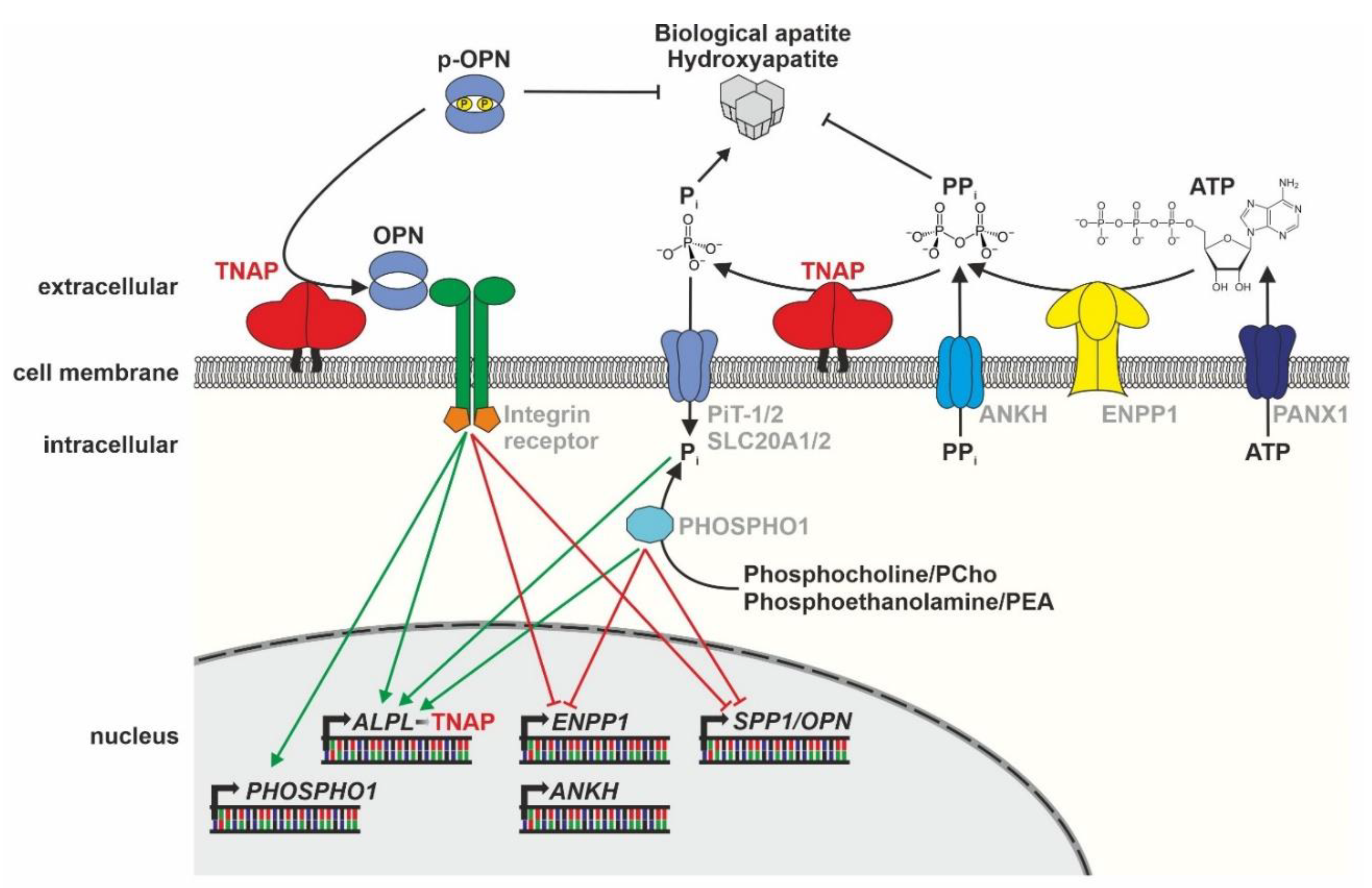
3. The Molecular Role of TNAP in Bone Mineralization
3.1. Cellular Function and Regulation
3.2. Animal Models of HPP
3.3. Combinatory Effects of TNAP in Murine Animal Models
4. The Role of TNAP in Teeth
4.1. TNAP Function in Human Dentition
4.2. Vertebrate TNAP Dentition Models
5. TNAP and Its Role in Pathologies Like Craniosynostosis and Atherosclerosis
5.1. Craniosynostosis
5.2. Vascular Calcification
6. TNAP beyond Mineralization—TNAP and Its Molecular Role in General Brain Function, Pathology, Behavior, and Pain
6.1. The Role of TNAP in the Development of the Nervous System
6.2. Neurological Symptoms of HPP
6.3. Localization of TNAP/Tnap in the Nervous System
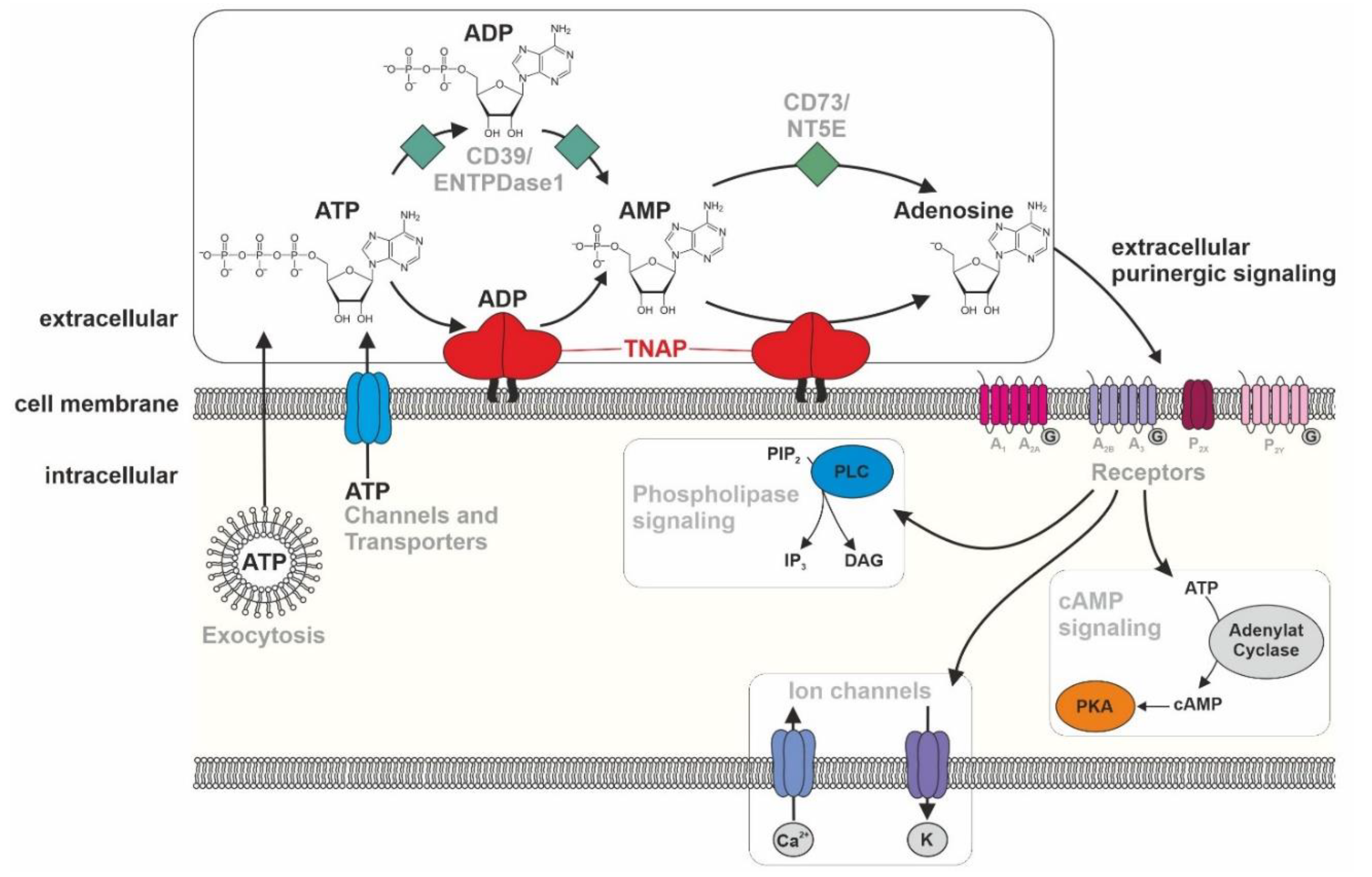
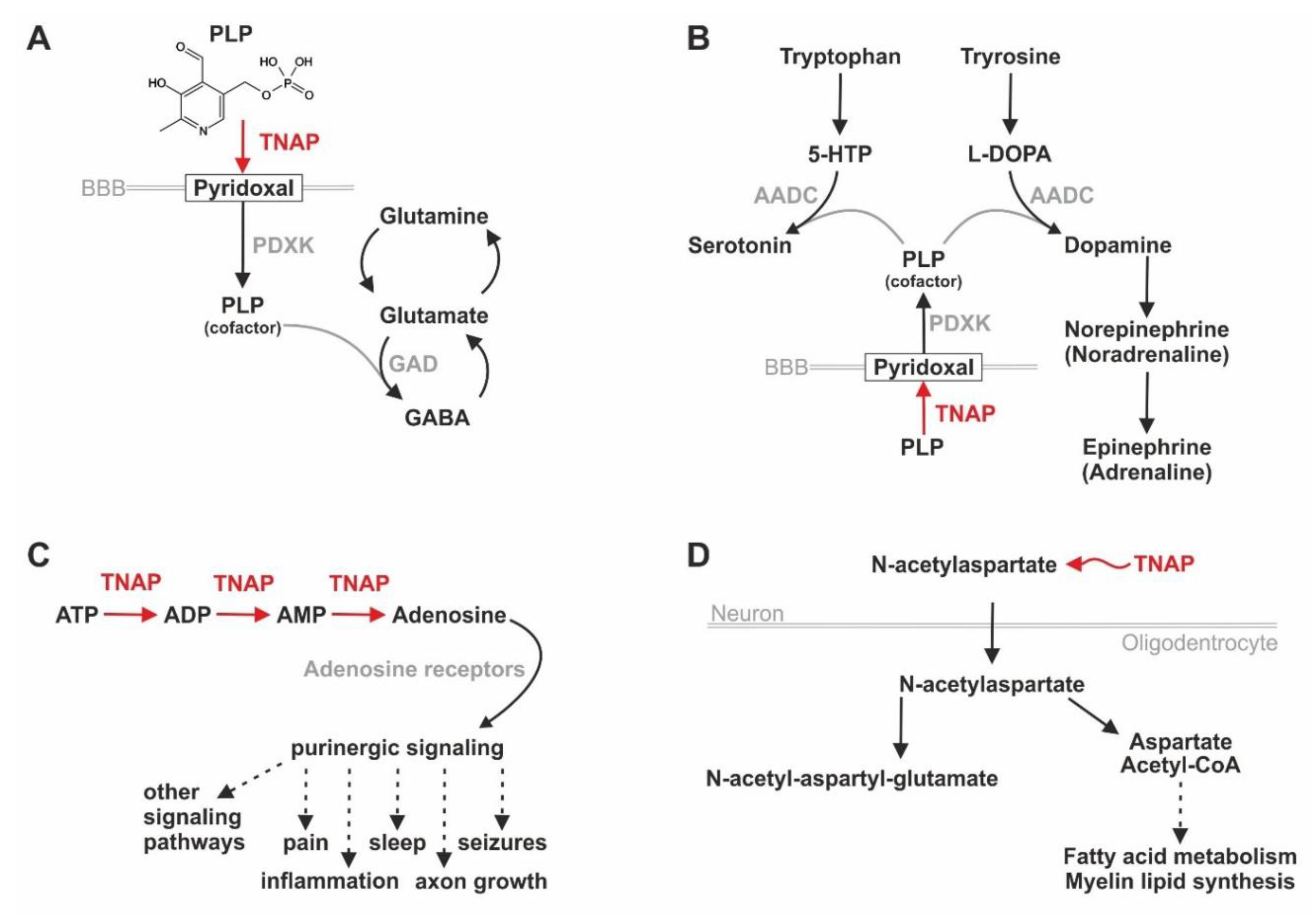
6.4. TNAP/Tnap’s Influence on Pain, Inflammation, Purinergic Signaling, and Neurotransmitter Synthesis
7. TNAP beyond Mineralization—The Role of TNAP for Sensory Perception
7.1. Visual Perception
7.2. Sound Perception
7.3. Olfactory Perception
8. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Millán, J.L.; Whyte, M.P. Alkaline Phosphatase and Hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef] [Green Version]
- Simão, A.M.S.; Bolean, M.; Hoylaerts, M.F.; Millán, J.L.; Ciancaglini, P. Effects of pH on the Production of Phosphate and Pyrophosphate by Matrix Vesicles’ Biomimetics. Calcif. Tissue Int. 2013, 93, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Narisawa, S.; Yadav, M.C.; Millán, J.L. In Vivo Overexpression of Tissue-Nonspecific Alkaline Phosphatase Increases Skeletal Mineralization and Affects the Phosphorylation Status of Osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef] [Green Version]
- Harris, H. The human alkaline phosphatases: What we know and what we don’t know. Clin. Chim. Acta 1990, 186, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Kishi, F.; Kajii, T. Characterization of a 5′-flanking region of the human liver/bone/kidney alkaline phosphatase gene: Two kinds of mRNA from a single gene. Biochem. Biophys. Res. Commun. 1990, 168, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [PubMed]
- Ohlebusch, B.; Borst, A.; Frankenbach, T.; Klopocki, E.; Jakob, F.; Liedtke, D.; Graser, S. Investigation of alpl expression and Tnap-activity in zebrafish implies conserved functions during skeletal and neuronal development. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Wandler, A.M.; Postlethwait, J.H.; Guillemin, K. Dynamic Evolution of the LPS-Detoxifying Enzyme Intestinal Alkaline Phosphatase in Zebrafish and Other Vertebrates. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Choida, V.; Bubbear, J. Update on the management of hypophosphatasia. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720. [Google Scholar] [CrossRef]
- Weiss, M.J.; Henthorn, P.S.; Lafferty, M.A.; Slaughter, C.; Raducha, M.; Harris, H. Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc. Natl. Acad. Sci. USA 1986, 83, 7182–7186. [Google Scholar] [CrossRef] [Green Version]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millán, J.L. Concerted Regulation of Inorganic Pyrophosphate and Osteopontin by Akp2, Enpp1, and Ank. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Favarin, B.; Bolean, M.; Ramos, A.; Magrini, A.; Rosato, N.; Millán, J.; Bottini, M.; Costa-Filho, A.; Ciancaglini, P. Lipid composition modulates ATP hydrolysis and calcium phosphate mineral propagation by TNAP-harboring proteoliposomes. Arch. Biochem. Biophys. 2020, 691. [Google Scholar] [CrossRef]
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.F.; Simão, A.M.S.; Bolean, M.; Hoylaerts, M.F.; Millán, J.L.; Ciancaglini, P.; Costa-Filho, A.J. Effects of GPI-anchored TNAP on the dynamic structure of model membranes. Phys. Chem. Chem. Phys. 2015, 17, 26295–26301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mauro, S.; Manes, T.; Hessle, L.; Kozlenkov, A.; Pizauro, J.M.; Hoylaerts, M.F.; Millán, J.L. Kinetic Characterization of Hypophosphatasia Mutations With Physiological Substrates. J. Bone Miner. Res. 2002, 17, 1383–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaru, K.; Ishida, Y.; Amaya, Y.; Goseki-Sone, M.; Orimo, H.; Oda, K. Novel aggregate formation of a frame-shift mutant protein of tissue-nonspecific alkaline phosphatase is ascribed to three cysteine residues in the C-terminal extension. FEBS J. 2005, 272, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Ciancaglini, P.; Simão, A.; Camolezi, F.; Millán, J.; Pizauro, J.M. Contribution of matrix vesicles and alkaline phosphatase to ectopic bone formation. Braz. J. Med. Biol. Res. 2006, 39, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Lehto, M.T.; Sharom, F.J. PI-Specific Phospholipase C Cleavage of a Reconstituted GPI-Anchored Protein: Modulation by the Lipid Bilayer†. Biochemistry 2002, 41, 1398–1408. [Google Scholar] [CrossRef]
- Fujihara, Y.; Ikawa, M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2015, 57, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Low, M.G.; Huang, K.S. Factors affecting the ability of glycosylphosphatidylinositol-specific phospholipase D to degrade the membrane anchors of cell surface proteins. Biochem. J. 1991, 279, 483–493. [Google Scholar] [CrossRef]
- Sykes, E.; Ghag, S.; Epstein, E.; Kiechle, F.L. Effect of insulin, S-adenosylhomocysteine, phospholipase C, n-butanol and Triton X-114 on alkaline phosphatase from isolated rat adipocyte plasma membranes. Clin. Chim. Acta 1987, 169, 133–139. [Google Scholar] [CrossRef]
- Hofmann, C.; Seefried, L.; Jakob, F. Asfotase alfa: Enzyme replacement for the treatment of bone disease in hypophosphatasia. Drugs Today 2016, 52, 217. [Google Scholar] [CrossRef] [PubMed]
- Millán, J.L. Alkaline Phosphatases. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Du, M.-H.; Stigbrand, T.; Taussig, M.J.; Menez, A.; Stura, E.A. Crystal Structure of Alkaline Phosphatase from Human Placenta at 1.8 A Resolution: IMPLICATION FOR A SUBSTRATE SPECIFICITY. J. Biol. Chem. 2000, 276, 9158–9165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoylaerts, M.F.; Van Kerckhoven, S.; Kiffer-Moreira, T.; Sheen, C.; Narisawa, S.; Millán, J.L. Functional Significance of Calcium Binding to Tissue-Nonspecific Alkaline Phosphatase. PLoS ONE 2015, 10, e0119874. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Rathbun, J.C. "Hypophosphatasia". Am. J. Dis. Child. 1948, 75, 822–831. [Google Scholar] [CrossRef]
- Mornet, E.; Yvard, A.; Taillandier, A.; Fauvert, D.; Simon-Bouy, B. A Molecular-Based Estimation of the Prevalence of Hypophosphatasia in the European Population. Ann. Hum. Genet. 2011, 75, 439–445. [Google Scholar] [CrossRef]
- Mornet, E.; Hofmann, C.; Bloch-Zupan, A.; Girschick, H.; Le Merrer, M. Clinical utility gene card for: Hypophosphatasia—Update 2013. Eur. J. Hum. Genet. 2013, 22, 572. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.L.; Guañabens, N.; Hofmann, C.; Jakob, F.; Roux, C.; Zillikens, M.C. Hypophosphatasia in adolescents and adults: Overview of diagnosis and treatment. Osteoporos. Int. 2020, 31, 1445–1460. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Girschick, H.J.; Mentrup, B.; Graser, S.; Seefried, L.; Liese, J.; Jakob, F. Clinical Aspects of Hypophosphatasia: An Update. Clin. Rev. Bone Miner. Metab. 2013, 11, 60–70. [Google Scholar] [CrossRef]
- Vogt, M.; Girschick, H.; Schweitzer, T.; Benoit, C.; Holl-Wieden, A.; Seefried, L.; Jakob, F.; Hofmann, C. Pediatric hypophosphatasia: Lessons learned from a retrospective single-center chart review of 50 children. Orphanet J. Rare Dis. 2020, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Leung, E.; Wilcox, W.R.; Liese, J.; Argente, J.; Martos-Moreno, G.Á.; Reeves, A.; Fujita, K.P.; Moseley, S.; Hofmann, C.; et al. Natural History of Perinatal and Infantile Hypophosphatasia: A Retrospective Study. J. Pediatr. 2019, 209, 116–124.e4. [Google Scholar] [CrossRef] [Green Version]
- Christine, H.; Girschick, H.; Mornet, E.; Schneider, D.; Jakob, F.; Mentrup, B. Unexpected high intrafamilial phenotypic variability observed in hypophosphatasia. Eur. J. Hum. Genet. 2014, 22, 1160–1164. [Google Scholar] [CrossRef]
- Whyte, M.P.; Wenkert, D.; Zhang, F. Hypophosphatasia: Natural history study of 101 affected children investigated at one research center. Bone 2016, 93, 125–138. [Google Scholar] [CrossRef]
- Genest, F.; Seefried, L. Subtrochanteric and diaphyseal femoral fractures in hypophosphatasia—Not atypical at all. Osteoporos. Int. 2018, 29, 1815–1825. [Google Scholar] [CrossRef]
- Collmann, H.; Mornet, E.; Gattenlöhner, S.; Beck, C.; Girschick, H. Neurosurgical aspects of childhood hypophosphatasia. Child’s Nerv. Syst. 2008, 25, 217–223. [Google Scholar] [CrossRef]
- Rockman-Greenberg, C. Hypophosphatasia. Pediatr. Endocrinol. Rev. 2013, 10, 380–388. [Google Scholar] [PubMed]
- Whyte, M.P.; Mahuren, J.D.; Vrabel, L.A.; Coburn, S.P. Markedly increased circulating pyridoxal-5’-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J. Clin. Investig. 1985, 76, 752–756. [Google Scholar] [CrossRef] [Green Version]
- Beck, C.; Morbach, H.; Wirth, C.; Beer, M.; Girschick, H.J. Whole-body MRI in the childhood form of hypophosphatasia. Rheumatol. Int. 2010, 31, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Girschick, H.J.; Schneider, P.; Haubitz, I.; Hiort, O.; Collmann, H.; Beer, M.; Shin, Y.S.; Seyberth, H.W. Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia. Orphanet J. Rare Dis. 2006, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Kishnani, P.S.; Rush, E.T.; Arundel, P.; Bishop, N.; Dahir, K.; Fraser, W.; Harmatz, P.; Linglart, A.; Munns, C.F.; Nunes, M.E.; et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol. Genet. Metab. 2017, 122, 4–17. [Google Scholar] [CrossRef] [Green Version]
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Tsuruoka, S. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model. J. Pathol. 2020, 250, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone 2020, 137. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.H.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-Replacement Therapy in Life-Threatening Hypophosphatasia. New Engl. J. Med. 2012, 366, 904–913. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, C.; Harmatz, P.; Vockley, J.; Högler, W.; Nakayama, H.; Bishop, N.; Martos-Moreno, G.Á.; Moseley, S.; Fujita, K.P.; Liese, J.; et al. Efficacy and Safety of Asfotase Alfa in Infants and Young Children With Hypophosphatasia: A Phase 2 Open-Label Study. J. Clin. Endocrinol. Metab. 2019, 104, 2735–2747. [Google Scholar] [CrossRef] [Green Version]
- Whyte, M.P.; Simmons, J.H.; Moseley, S. Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single-arm, open-label, phase 2 extension trial. Lancet Diabetes Endo. 2019, 7, E2. [Google Scholar] [CrossRef]
- Whyte, M.P.; Madson, K.L.; Phillips, D.; Reeves, A.L.; McAlister, W.H.; Yakimoski, A.; Mack, K.E.; Hamilton, K.; Kagan, K.; Fujita, K.P.; et al. Asfotase alfa therapy for children with hypophosphatasia. JCI Insight 2016, 1, e85971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; Rockman-Greenberg, C.; Ozono, K.; Riese, R.; Moseley, S.; Melian, A.; Thompson, D.D.; Bishop, N.; Hofmann, C. Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J. Clin. Endocrinol. Metab. 2016, 101, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishnani, P.S.; Rockman-Greenberg, C.; Rauch, F.; Bhatti, M.T.; Moseley, S.; Denker, A.E.; Watsky, E.; Whyte, M.P. Five-year efficacy and safety of asfotase alfa therapy for adults and adolescents with hypophosphatasia. Bone 2019, 121, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Millán, J.L. The Role of Phosphatases in the Initiation of Skeletal Mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, P.; Toroian, D.; Chan, W.S. Tissue-nonspecific Alkaline Phosphatase Is Required for the Calcification of Collagen in Serum. J. Biol. Chem. 2008, 284, 4594–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastián-Serrano, Á.; De Diego-García, L.; Henshall, D.C.; Engel, T.; Díaz-Hernández, M. Haploinsufficient TNAP Mice Display Decreased Extracellular ATP Levels and Expression of Pannexin-1 Channels. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Abitbol, J.M.; O’Donnell, B.L.; Wakefield, C.B.; Jewlal, E.; Kelly, J.J.; Barr, K.; Willmore, K.E.; Allman, B.L.; Penuela, S. Double deletion of Panx1 and Panx3 affects skin and bone but not hearing. J. Mol. Med. 2019, 97, 723–736. [Google Scholar] [CrossRef]
- Yadav, M.C.; Simão, A.M.S.; Narisawa, S.; Huesa, C.; McKee, M.D.; Farquharson, C.; Millán, J.L. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: A unified model of the mechanisms of initiation of skeletal calcification. J. Bone Miner. Res. 2011, 26, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Yoshiko, Y.; Candeliere, G.A.; Maeda, N.; Aubin, J.E. Osteoblast Autonomous Pi Regulation via Pit1 Plays a Role in Bone Mineralization. Mol. Cell. Biol. 2007, 27, 4465–4474. [Google Scholar] [CrossRef] [Green Version]
- Couasnay, G.; Bon, N.; Devignes, C.-S.; Sourice, S.; Bianchi, A.; Véziers, J.; Weiss, P.; Elefteriou, F.; Provot, S.; Guicheux, J.; et al. PiT1/Slc20a1 Is Required for Endoplasmic Reticulum Homeostasis, Chondrocyte Survival, and Skeletal Development. J. Bone Miner. Res. 2019, 34, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Shou, P.; Zhang, L.; Xu, C.; Zheng, C.; Han, Y.; Li, W.; Huang, Y.; Zhang, X.; Shao, C.; et al. An Osteopontin-Integrin Interaction Plays a Critical Role in Directing Adipogenesis and Osteogenesis by Mesenchymal Stem Cells. STEM CELLS 2014, 32, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kläning, E.; Christensen, B.; Bajic, G.; Hoffmann, S.V.; Jones, N.C.; Callesen, M.M.; Andersen, G.; Sørensen, E.S.; Vorup-Jensen, T. Multiple low-affinity interactions support binding of human osteopontin to integrin α X β 2. Biochim. et Biophys. Acta Proteins Proteom. 2015, 1854, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Chellaiah, M.A.; Hruska, K. The Integrin {alpha} v {beta} 3 and CD44 Regulate the Actions of Osteopontin on Osteoclast Motility. Calcif. Tissue Int. 2003, 72, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nakamura-Takahashi, A.; Kasahara, M.; Yamaguchi, A.; Azuma, T. Tissue-nonspecific alkaline phosphatase promotes the osteogenic differentiation of osteoprogenitor cells. Biochem. Biophys. Res. Commun. 2020, 524, 702–709. [Google Scholar] [CrossRef]
- Gao, M.; Su, Q.; Liang, T.; Ma, J.; Stoddart, M.; Richards, R.; Zhou, Z.; Zou, X. Transcriptional activation of ENPP1 by osterix in osteoblasts and osteocytes. eCM 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Komori, T. Signaling networks in RUNX2-dependent bone development. J. Cell. Biochem. 2011, 112, 750–755. [Google Scholar] [CrossRef]
- Yadav, M.C.; Huesa, C.; Narisawa, S.; Hoylaerts, M.F.; Moreau, A.; Farquharson, C.; Millán, J.L. Ablation of Osteopontin Improves the Skeletal Phenotype ofPhospho1−/−Mice. J. Bone Miner. Res. 2014, 29, 2369–2381. [Google Scholar] [CrossRef] [Green Version]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non–specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B–6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef]
- Narisawa, S.; Fröhlander, N.; Millán, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446. [Google Scholar] [CrossRef]
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millan, J.L.; MacGregor, G.R.; et al. Alkaline Phosphatase Knock-Out Mice Recapitulate the Metabolic and Skeletal Defects of Infantile Hypophosphatasia. J. Bone Miner. Res. 1999, 14, 2015–2026. [Google Scholar] [CrossRef] [Green Version]
- Bessueille, L.; Briolay, A.; Como, J.; Mebarek, S.; Mansouri, C.; Gleizes, M.; El Jamal, A.; Buchet, R.; Dumontet, C.; Matera, E.; et al. Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Durussel, J.; Liu, J.; Campbell, C.; Nam, H.K.; Hatch, N.E. Bone mineralization-dependent craniosynostosis and craniofacial shape abnormalities in the mouse model of infantile hypophosphatasia. Dev. Dyn. 2016, 245, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, B.; Sheen, C.; Hatch, N.; Liu, J.; Cory, E.; Narisawa, S.; Kiffer-Moreira, T.; Sah, R.; Whyte, M.; Somerman, M.; et al. Periodontal Defects in the A116T Knock-in Murine Model of Odontohypophosphatasia. J. Dent. Res. 2015, 94, 706–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabrautzki, S.; Rubio-Aliaga, I.; Hans, W.; Fuchs, H.; Rathkolb, B.; Calzada-Wack, J.; Cohrs, C.M.; Klaften, M.; Seedorf, H.; Eck, S.; et al. New mouse models for metabolic bone diseases generated by genome-wide ENU mutagenesis. Mamm. Genome 2012, 23, 416–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aigner, B.; Rathkolb, B.; Klaften, M.; Sedlmeier, R.; Klempt, M.; Wagner, S.; Michel, D.; Mayer, U.; Klopstock, T.; De Angelis, M.H.; et al. Generation ofN-ethyl-N-nitrosourea-induced mouse mutants with deviations in plasma enzyme activities as novel organ-specific disease models. Exp. Physiol. 2009, 94, 412–421. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.H.; Flaswinkel, H.; Fuchs, H.; Rathkolb, B.; Soewarto, D.; Marschall, S.; Heffner, S.; Pargent, W.; Wuensch, K.; Jung, M.; et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 2000, 25, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Forero, L.L.; Winkler, C.; Schulte-Merker, S. Zebrafish and medaka as models for biomedical research of bone diseases. Dev. Biol. 2020, 457, 191–205. [Google Scholar] [CrossRef]
- Kwon, R.Y.; Watson, C.J.; Karasik, D. Using zebrafish to study skeletal genomics. Bone 2019, 126, 37–50. [Google Scholar] [CrossRef]
- Tonelli, F.; Cotti, S.; Leoni, L.; Besio, R.; Gioia, R.; Marchese, L.; Giorgetti, S.; Villani, S.; Gistelinck, C.; Wagener, R.; et al. Crtap and p3h1 knock out zebrafish support defective collagen chaperoning as the cause of their osteogenesis imperfecta phenotype. Matrix Biol. 2020, 90, 40–60. [Google Scholar] [CrossRef]
- Tonelli, F.; Bek, J.W.; Besio, R.; De Clercq, A.; Leoni, L.; Salmon, P.; Coucke, P.J.; Willaert, A.; Forlino, A. Zebrafish: A Resourceful Vertebrate Model to Investigate Skeletal Disorders. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef]
- Kague, E.; Roy, P.; Asselin, G.; Hu, G.; Simonet, J.; Stanley, A.; Albertson, C.; Fisher, S. Osterix/Sp7 limits cranial bone initiation sites and is required for formation of sutures. Dev. Biol. 2016, 413, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Yan, Y.-L.; Liu, C.; Bo, L.; Li, G.-F.; Wang, H.; Xu, Y. Therapeutic Effect of Deferoxamine on Iron Overload-Induced Inhibition of Osteogenesis in a Zebrafish Model. Calcif. Tissue Int. 2014, 94, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cui, Y.; Zhou, X.; Han, J. Phosphate/Pyrophosphate and MV-related Proteins in Mineralisation: Discoveries from Mouse Models. Int. J. Biol. Sci. 2012, 8, 778–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.L. Can biological calcification occur in the presence of pyrophosphate? Arch. Biochem. Biophys. 1984, 231, 1–8. [Google Scholar] [CrossRef]
- Harmey, D.; Johnson, K.A.; Zelken, J.; Camacho, N.P.; Hoylaerts, M.F.; Noda, M.; Terkeltaub, R.; Millán, J.L. Elevated Skeletal Osteopontin Levels Contribute to the Hypophosphatasia Phenotype in Akp2−/− Mice. J. Bone Miner. Res. 2006, 21, 1377–1386. [Google Scholar] [CrossRef]
- Roberts, S.; Narisawa, S.; Harmey, D.; Millán, J.L.; Farquharson, C. Functional Involvement of PHOSPHO1 in Matrix Vesicle-Mediated Skeletal Mineralization. J. Bone Miner. Res. 2007, 22, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Murshed, M.; Harmey, D.; Millán, J.L.; McKee, M.D.; Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005, 19, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Addison, W.N.; Azari, F.; Sørensen, E.S.; Kaartinen, M.T.; McKee, M.D. Pyrophosphate Inhibits Mineralization of Osteoblast Cultures by Binding to Mineral, Up-regulating Osteopontin, and Inhibiting Alkaline Phosphatase Activity. J. Biol. Chem. 2007, 282, 15872–15883. [Google Scholar] [CrossRef] [Green Version]
- Narisawa, S.; Wennberg, C.; Millán, J.L. Abnormal vitamin B6 metabolism in alkaline phosphatase knock-out mice causes multiple abnormalities, but not the impaired bone mineralization. J. Pathol. 2001, 193, 125–133. [Google Scholar] [CrossRef]
- De Oliveira, F.A.; Narisawa, S.; Bottini, M.; Millán, J.L. Visualization of Mineral-Targeted Alkaline Phosphatase Binding to Sites of Calcification In Vivo. J. Bone Miner. Res. 2020, 35, 1765–1771. [Google Scholar] [CrossRef]
- Boskey, A.; Maresca, M.; Ullrich, W.; Doty, S.; Butler, W.; Prince, C. Osteopontin-hydroxyapatite interactions in vitro: Inhibition of hydroxyapatite formation and growth in a gelatin-gel. Bone Miner. 1993, 22, 147–159. [Google Scholar] [CrossRef]
- Hunter, G.K.; Kyle, C.L.; Goldberg, H.A. Modulation of crystal formation by bone phosphoproteins: Structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem. J. 1994, 300, 723–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huesa, C.; Houston, D.A.; Kiffer-Moreira, T.; Yadav, M.C.; Millán, J.L.; Farquharson, C. The functional co-operativity of tissue-nonspecific alkaline phosphatase (TNAP) and PHOSPHO1 during initiation of skeletal mineralization. Biochem. Biophys. Rep. 2015, 4, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Huang, J.; Pinkerton, A.B.; Millan, J.L.; Van Zelst, B.D.; Levine, M.A.; Sundberg, J.P.; Uitto, J. Inhibition of Tissue-Nonspecific Alkaline Phosphatase Attenuates Ectopic Mineralization in the Abcc6 Mouse Model of PXE but Not in the Enpp1 Mutant Mouse Models of GACI. J. Investig. Dermatol. 2019, 139, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gámez-Belmonte, R.; Hernández-Chirlaque, C.; De Medina, F.S.; Martínez-Augustin, O. Experimental acute pancreatitis is enhanced in mice with tissue nonspecific alkaline phoshatase haplodeficiency due to modulation of neutrophils and acinar cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, M.J.; Dennis, C.; Yang, X.B.; Kirkham, J. Tissue non-specific alkaline phosphatase production by human dental pulp stromal cells is enhanced by high density cell culture. Cell Tissue Res. 2015, 361, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Kramer, K.; Chavez, M.; Tran, A.; Farah, F.; Tan, M.; Kolli, T.; Dos Santos, E.L.; Wimer, H.; Millán, J.; Suva, L.; et al. Dental defects in the primary dentition associated with hypophosphatasia from biallelic ALPL mutations. Bone 2020, 10. [Google Scholar] [CrossRef]
- Foster, B.; Nagatomo, K.; Tso, H.; Tran, A.; Nociti, F.H.; Narisawa, S.; Yadav, M.; McKee, M.; Millán, J.; Somerman, M. Tooth root dentin mineralization defects in a mouse model of hypophosphatasia. J. Bone Miner. Res. 2012, 28, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Reibel, A.; Maniere, M.C.; Clauss, F.; Droz, D.; Alembik, Y.; Mornet, E.; Bloch-Zupan, A. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet. J. Rare Dis. 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Olsson, A.; Matsson, L.; Blomquist, H.K.; Larsson, A.; Sjodin, B. Hypophosphatasia affecting the permanent dentition. J. Oral Pathol. Med. 1996, 25, 343–347. [Google Scholar] [CrossRef]
- Goseki-Sone, M.; Iimura, T.; Takeda, K.; Nifuji, A.; Ogata, Y.; Yanagishita, M.; Oida, S. Expression of mRNA encoding tissue-nonspecific alkaline phosphatase in human dental tissues. Calcif. Tissue Int. 1999, 64, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.L.; Foster, B.L.; Silvério, K.G.; Martins, L.; Casati, M.Z.; Sallum, E.A.; Somerman, M.J.; Nociti, F.H. Hypophosphatasia-associated Deficiencies in Mineralization and Gene Expression in Cultured Dental Pulp Cells Obtained from Human Teeth. J. Endod. 2012, 38, 907–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melms, H.; Herrmann, M.; Förstner, K.; Bharti, R.; Schneider, D.; Mentrup, B.; Rudert, M.; Schlagenhauf, U.; Jakob, F.; Graser, S. Novel molecular cues for dental defects in hypophosphatasia. Exp. Cell Res. 2020, 392. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, J.; Lei, H.; Zhu, T.; Gan, Y.; Ge, L. Genetic Etiology and Dental Pulp Cell Deficiency of Hypophosphatasia. J. Dent. Res. 2010, 89, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Umeda, M.; Seki, T.; Ishikawa, I. Clinical and Laboratory Studies of Severe Periodontal Disease in an Adolescent Associated With Hypophosphatasia. A Case Report. J. Periodontol. 1993, 64, 174–180. [Google Scholar] [CrossRef]
- Chu, E.; Vo, T.; Chavez, M.; Nagasaki, A.; Mertz, E.; Nociti, F.; Aitken, S.; Kavanagh, D.; Zimmerman, K.; Li, X.; et al. Genetic and pharmacologic modulation of cementogenesis via pyrophosphate regulators. Bone 2020, 136. [Google Scholar] [CrossRef]
- Thumbigere-Math, V.; Alqadi, A.; Chalmers, N.; Chavez, M.; Chu, E.; Collins, M.; Ferreira, C.; Fitzgerald, K.; Gafni, R.; Gahl, W.; et al. Hypercementosis Associated with ENPP1 Mutations and GACI. J. Dent. Res. 2017, 97, 432–441. [Google Scholar] [CrossRef]
- Zweifler, L.E.; Patel, M.K., Jr.; Wimer, H.F.; Millan, J.L.; Somerman, M.J.; Foster, B.L. Counter-regulatory phosphatases TNAP and NPP1 temporally regulate tooth root cementogenesis. Int. J. Oral Sci. 2015, 7, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Anderson, H.C.; Hsu, H.H.; Morris, D.C.; Fedde, K.N.; Whyte, M.P. Matrix vesicles in osteomalacic hypophosphatasia bone contain apatite-like mineral crystals. Am. J. Pathol. 1997, 151, 1555–1561. [Google Scholar]
- Anderson, H.C.; Sipe, J.B.; Hessle, L.; Dhamyamraju, R.; Atti, E.; Camacho, N.P.; Millán, J.L. Impaired Calcification Around Matrix Vesicles of Growth Plate and Bone in Alkaline Phosphatase-Deficient Mice. Am. J. Pathol. 2004, 164, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.C.; De Oliveira, R.C.; Foster, B.L.; Fong, H.; Cory, E.; Narisawa, S.; Sah, R.L.; Somerman, M.J.; Whyte, M.P.; Millán, J.L. Enzyme replacement prevents enamel defects in hypophosphatasia mice. J. Bone Miner. Res. 2012, 27, 1722–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotton, D.; Mauro, N.; Lézot, F.; Forest, N.; Berdal, A. Differential expression and activity of tissue-nonspecific alkaline phosphatase (TNAP) in rat odontogenic cells in vivo. J. Histochem. Cytochem. 1999, 47, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, M.D.; Yadav, M.; Foster, B.; Somerman, M.; Farquharson, C.; Millán, J. Compounded PHOSPHO1/ALPL deficiencies reduce dentin mineralization. J. Dent. Res. 2013, 92, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huysseune, A.; Van Der Heyden, C.; Sire, J.-Y. Early development of the zebrafish (Danio rerio) pharyngeal dentition (Teleostei, Cyprinidae). Brain Struct. Funct. 1998, 198, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Bruneel, B.; Mathä, M.; Paesen, R.; Ameloot, M.; Weninger, W.J.; Huysseune, A. Imaging the Zebrafish Dentition: From Traditional Approaches to Emerging Technologies. Zebrafish 2015, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sadier, A.; Jackman, W.R.; Laudet, V.; Gibert, Y. The Vertebrate Tooth Row: Is It Initiated by a Single Organizing Tooth? BioEssays 2020, 42, e1900229. [Google Scholar] [CrossRef]
- Van der Heyden, C.; Huysseune, A. Dynamics of tooth formation and replacement in the zebrafish (Danio rerio) (Teleostei, Cyprinidae). Dev. Dyn. 2000, 219, 486–496. [Google Scholar] [CrossRef]
- Neel, E.A.A.; Aljabo, A.; Strange, A.; Ibrahim, S.; Coathup, M.; Young, A.M.; Bozec, L.; Mudera, V. Demineralization–remineralization dynamics in teeth and bone. Int. J. Nanomed. 2016, 11, 4743–4763. [Google Scholar] [CrossRef]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhao, X.; Wu, H. Transcriptional Programming in Arteriosclerotic Disease: A Multifaceted Function of the Runx2 (Runt-Related Transcription Factor 2). Arterioscler. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Orriss, I.R.; Neven, E.; D’Haese, P.C.; Verhulst, A. Extracellular Nucleotides Regulate Arterial Calcification by Activating Both Independent and Dependent Purinergic Receptor Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 7636. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.; Singh, N.; Richtsmeier, J.T. Understanding craniosynostosis as a growth disorder. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 429–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Xu, H.; Yao, Y.; Xu, T.; Yuan, M.; Zhang, X.; Lv, Z.; Wu, M. BMP Signaling in the Development and Regeneration of Cranium Bones and Maintenance of Calvarial Stem Cells. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, R.; Kaucka, M.; Kastriti, M.E.; Petersen, J.; Chontorotzea, T.; Englmaier, L.; Akkuratova, N.; Yang, Y.; Häring, M.; Dyachuk, V.; et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364, eaas9536. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Li, Q.; Yuan, Q.; Zhang, S. Spatial Distributions, Characteristics, and Applications of Craniofacial Stem Cells. Stem Cells Int. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Siismets, E.M.; Hatch, N.E. Cranial Neural Crest Cells and Their Role in the Pathogenesis of Craniofacial Anomalies and Coronal Craniosynostosis. J. Dev. Biol. 2020, 8, 18. [Google Scholar] [CrossRef]
- Doro, D.; Liu, A.; Grigoriadis, A.E.; Liu, K.J. The Osteogenic Potential of the Neural Crest Lineage May Contribute to Craniosynostosis. Mol. Syndr. 2018, 10, 48–57. [Google Scholar] [CrossRef]
- Goos, J.A.; Mathijssen, I.M. Genetic Causes of Craniosynostosis: An Update. Mol. Syndr. 2018, 10, 6–23. [Google Scholar] [CrossRef]
- Liu, J.; Nam, H.K.; Campbell, C.; Gasque, K.C.D.S.; Millán, J.L.; Hatch, N.E. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl−/− mouse model of infantile hypophosphatasia. Bone 2014, 67, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Campbell, C.; Nam, H.K.; Caron, A.; Yadav, M.C.; Millán, J.L.; Hatch, N.E. Enzyme replacement for craniofacial skeletal defects and craniosynostosis in murine hypophosphatasia. Bone 2015, 78, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Nam, H.K.; Veselá, I.; Schutte, S.D.; Hatch, N.E. Viral delivery of tissue nonspecific alkaline phosphatase diminishes craniosynostosis in one of two FGFR2C342Y/+ mouse models of Crouzon syndrome. PLoS ONE 2020, 15, e0234073. [Google Scholar] [CrossRef] [PubMed]
- Topczewska, J.M.; Shoela, R.A.; Tomaszewski, J.P.; Mirmira, R.B.; Gosain, A.K. The Morphogenesis of Cranial Sutures in Zebrafish. PLoS ONE 2016, 11, e0165775. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.P.; Fenwick, A.L.; Brockop, M.S.; McGowan, S.J.; Goos, J.A.; Hoogeboom, A.J.; Brady, A.F.; Jeelani, N.O.; Lynch, S.A.; Mulliken, J.B.; et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet. 2013, 45, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Bialek, P.; Kern, B.; Yang, X.; Schrock, M.; Sosic, D.; Hong, N.; Wu, H.; Yu, K.; Ornitz, D.M.; Olson, E.N.; et al. A Twist Code Determines the Onset of Osteoblast Differentiation. Dev. Cell 2004, 6, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Teng, C.S.; Ting, M.-C.; Farmer, D.T.; Brockop, M.; Maxson, R.E.; Crump, J.G. Altered bone growth dynamics prefigure craniosynostosis in a zebrafish model of Saethre-Chotzen syndrome. eLife 2018, 7. [Google Scholar] [CrossRef]
- Blümel, R.; Zink, M.; Klopocki, E.; Liedtke, D. On the traces of tcf12: Investigation of the gene expression pattern during development and cranial suture patterning in zebrafish (Danio rerio). PLoS ONE 2019, 14, e0218286. [Google Scholar] [CrossRef]
- Laue, K.; Pogoda, H.-M.; Daniel, P.B.; Van Haeringen, A.; Alanay, Y.; Von Ameln, S.; Rachwalski, M.; Morgan, T.; Gray, M.J.; Breuning, M.H.; et al. Craniosynostosis and Multiple Skeletal Anomalies in Humans and Zebrafish Result from a Defect in the Localized Degradation of Retinoic Acid. Am. J. Hum. Genet. 2011, 89, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Isoherranen, N.; Zhong, G. Biochemical and physiological importance of the CYP26 retinoic acid hydroxylases. Pharmacol. Ther. 2019, 204. [Google Scholar] [CrossRef]
- Yashiro, K.; Zhao, X.; Uehara, M.; Yamashita, K.; Nishijima, M.; Nishino, J.; Saijoh, Y.; Sakai, Y.; Hamada, H. Regulation of Retinoic Acid Distribution Is Required for Proximodistal Patterning and Outgrowth of the Developing Mouse Limb. Dev. Cell 2004, 6, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R. New insights into endogenous mechanisms of protection against arterial calcification. Atherosclerosis 2020, 306, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Van Rosendael, A.R.; Shaw, L.J.; Xie, J.X.; Dimitriu-Leen, A.C.; Smit, J.M.; Scholte, A.J.; Van Werkhoven, J.M.; Callister, T.Q.; Delago, A.; Berman, D.S.; et al. Superior Risk Stratification With Coronary Computed Tomography Angiography Using a Comprehensive Atherosclerotic Risk Score. JACC Cardiovasc. Imaging 2019, 12, 1987–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, B.Y.C.; Wilson, J.M.; Spann, S.J.; Pettigrew, R.I.; Preventza, O.A.; Muthupillai, R. Coronary artery calcium scoring: An evidence-based guide for primary care physicians. J. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Orekhov, A.N.; Sobenin, I.; Chistiakov, D.A. Role of Bone-Type Tissue-Nonspecific Alkaline Phosphatase and PHOSPO1 in Vascular Calcification. Curr. Pharm. Des. 2014, 20, 5821–5828. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Hilaire, C.S.; Huang, Y.; Yang, D.; Dmitrieva, N.I.; Negro, A.; Schwartzbeck, R.; Liu, Y.; Yu, Z.; Walts, A.; et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal. 2016, 9, ra121. [Google Scholar] [CrossRef] [Green Version]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.A.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Hear. Assoc. 2015, 4, e002499. [Google Scholar] [CrossRef] [Green Version]
- Hanics, J.; Barna, J.; Xiao, J.; Millán, J.L.; Fonta, C.; Négyessy, L. Ablation of TNAP function compromises myelination and synaptogenesis in the mouse brain. Cell Tissue Res. 2012, 349, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, C.; Liese, J.G.; Schwarz, T.; Kunzmann, S.; Wirbelauer, J.; Nowak, J.; Hamann, J.; Girschick, H.J.; Graser, S.; Dietz, K.; et al. Compound heterozygosity of two functional null mutations in the ALPL gene associated with deleterious neurological outcome in an infant with hypophosphatasia. Bone 2013, 55, 150–157. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef]
- Street, S.E.; Kramer, N.J.; Walsh, P.L.; Taylor-Blake, B.; Yadav, M.C.; King, I.F.; Vihko, P.; Wightman, R.M.; Millán, J.L.; Zylka, M.J. Tissue-nonspecific alkaline phosphatase acts redundantly with PAP and NT5E to generate adenosine in the dorsal spinal cord. J. Neurosci. 2013, 33, 11314–11322. [Google Scholar] [CrossRef] [Green Version]
- Morello, S.; Turiello, R.; Madonna, G.; Pinto, A.; Ascierto, P.A.; Capone, M. Enzyme activity of circulating CD73 in human serum. Method. Enzymolog. 2019, 629, 257–267. [Google Scholar] [CrossRef]
- Ricatti, M.J.; Alfie, L.D.; Lavoie, É.G.; Sévigny, J.; Schwarzbaum, P.J.; Faillace, M.P. Immunocytochemical localization of NTPDases1 and 2 in the neural retina of mouse and zebrafish. Synapse 2009, 63, 291–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, U.; Machida, T.; Broekman, M.J.; Marcus, A.J.; Levi, R. Targeted Deletion of Ectonucleoside Triphosphate Diphosphohydrolase 1/CD39 Leads to Desensitization of Pre- and Postsynaptic Purinergic P2 Receptors. J. Pharmacol. Exp. Ther. 2007, 322, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Kukulski, F.; Lévesque, S.A.; Sévigny, J. Impact of Ectoenzymes on P2 and P1 Receptor Signaling. Stud. Surface Sci. Catalys. 2011, 61, 263–299. [Google Scholar] [CrossRef]
- Zimmermann, H.; Langer, D. Tissue-Nonspecific Alkaline Phosphatase in the Developing Brain and in Adult Neurogenesis. Subcell. Biochem. 2015, 76, 61–84. [Google Scholar] [CrossRef]
- Langer, D.; Ikehara, Y.; Takebayashi, H.; Hawkes, R.; Zimmermann, H. The ectonucleotidases alkaline phosphatase and nucleoside triphosphate diphosphohydrolase 2 are associated with subsets of progenitor cell populations in the mouse embryonic, postnatal and adult neurogenic zones. Neuroscience 2007, 150, 863–879. [Google Scholar] [CrossRef]
- Yang, H.K.; Sundholm-Peters, N.L.; Goings, G.E.; Walker, A.S.; Hyland, K.; Szele, F.G. Distribution of doublecortin expressing cells near the lateral ventricles in the adult mouse brain. J. Neurosci. Res. 2004, 76, 282–295. [Google Scholar] [CrossRef]
- Kermer, V.; Ritter, M.; Albuquerque, B.; Leib, C.; Stanke, M.; Zimmermann, H. Knockdown of tissue nonspecific alkaline phosphatase impairs neural stem cell proliferation and differentiation. Neurosci. Lett. 2010, 485, 208–211. [Google Scholar] [CrossRef]
- Narisawa, S.; Hasegawa, H.; Watanabe, K.; Millán, J.L. Stage-specific expression of alkaline phosphatase during neural development in the mouse. Dev. Dyn. 1994, 201, 227–235. [Google Scholar] [CrossRef]
- Mitrovic, N.; Zarić, M.; Drakulić, D.; Martinovic, J.; Sévigny, J.; Stanojlović, M.; Nedeljković, N.; Grkovic, I. 17β-Estradiol-Induced Synaptic Rearrangements Are Accompanied by Altered Ectonucleotidase Activities in Male Rat Hippocampal Synaptosomes. J. Mol. Neurosci. 2016, 61, 412–422. [Google Scholar] [CrossRef]
- Mitrovic, N.; Zarić, M.; Drakulić, D.; Martinovic, J.; Sévigny, J.; Stanojlović, M.; Nedeljković, N.; Grkovic, I. Erratum to: 17β-Estradiol-Induced Synaptic Rearrangements Are Accompanied by Altered Ectonucleotidase Activities in Male Rat Hippocampal Synaptosomes. J. Mol. Neurosci. 2017, 61, 423–424. [Google Scholar] [CrossRef] [Green Version]
- Thisse, B.; Thisse, C. Fast Release Clones: A High Throughput Expression Analysis. ZFIN Direct Data Submission. 2004. Available online: http://zfin.org (accessed on 26 October 2020).
- Steiner, A.B.; Kim, T.; Cabot, V.; Hudspeth, A.J. Dynamic gene expression by putative hair-cell progenitors during regeneration in the zebrafish lateral line. Proc. Natl. Acad. Sci. USA 2014, 111, E1393–E1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Zhao, X.-F.; Prat, C.R.; Narawane, S.; Suh, C.S.; Gharbi, N.; Ellingsen, S.; Fjose, A. Zebrafish transgenic lines co-expressing a hybrid Gal4 activator and eGFP in tissue-restricted patterns. Gene Expr. Patterns 2011, 11, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Colazo, J.; Hu, J.; Dahir, K.; Simmons, J.H. Neurological symptoms in Hypophosphatasia. Osteoporos. Int. 2019, 30, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.L.; Mugnol, F.; Bica, I.; Fiori, R.M. Pyridoxine-dependent seizures associated with hypophosphatasia in a newborn. J. Child. Neurol. 2002, 17, 222–224. [Google Scholar] [CrossRef]
- Brun-Heath, I.; Ermonval, M.; Chabrol, E.; Xiao, J.; Palkovits, M.; Lyck, R.; Miller, F.; Couraud, P.-O.; Mornet, E.; Fonta, C. Differential expression of the bone and the liver tissue non-specific alkaline phosphatase isoforms in brain tissues. Cell Tissue Res. 2010, 343, 521–536. [Google Scholar] [CrossRef]
- Fonta, C.; Négyessy, L.; Renaud, L.; Barone, P. Areal and Subcellular Localization of the Ubiquitous Alkaline Phosphatase in the Primate Cerebral Cortex: Evidence for a Role in Neurotransmission. Cereb. Cortex 2004, 14, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Négyessy, L.; Xiao, J.; Kántor, O.; Kovacs, G.G.; Palkovits, M.; Doczi, T.; Renaud, L.; Baksa, G.; Glasz, T.; Ashaber, M.; et al. Layer-specific activity of tissue non-specific alkaline phosphatase in the human neocortex. Neurosciemce 2011, 172, 406–418. [Google Scholar] [CrossRef]
- Langer, D.; Hammer, K.; Koszałka, P.; Schrader, J.; Robson, S.; Zimmermann, H. Distribution of ectonucleotidases in the rodent brain revisited. Cell Tissue Res. 2008, 334, 199–217. [Google Scholar] [CrossRef]
- Wan, J.; Goldman, D. Retina regeneration in zebrafish. Curr. Opin. Genet. Dev. 2016, 40, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Sehring, I.M.; Jahn, C.; Weidinger, G. Zebrafish fin and heart: What’s special about regeneration? Curr. Opin. Genet. Dev. 2016, 40, 48–56. [Google Scholar] [CrossRef]
- Kizil, C.; Kaslin, J.; Kroehne, V.; Brand, M. Adult neurogenesis and brain regeneration in zebrafish. Dev. Neurobiol. 2012, 72, 429–461. [Google Scholar] [CrossRef] [PubMed]
- Bayés, À.; Collins, M.O.; Reig-Viader, R.; Gou, G.; Goulding, D.; Izquierdo, A.; Choudhary, J.S.; Emes, R.D.; Grant, S.G.N. Evolution of complexity in the zebrafish synapse proteome. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cruz, T.; Gleizes, M.; Balayssac, S.; Mornet, E.; Marsal, G.; Millán, J.L.; Malet-Martino, M.; Nowak, L.G.; Gilard, V.; Fonta, C. Identification of altered brain metabolites associated with TNAP activity in a mouse model of hypophosphatasia using untargeted NMR-based metabolomics analysis. J. Neurochem. 2017, 140, 919–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramaniam, S.; Bowling, F.; Carpenter, K.; Earl, J.; Chaitow, J.; Pitt, J.; Mornet, E.; Sillence, D.; Ellaway, C. Perinatal hypophosphatasia presenting as neonatal epileptic encephalopathy with abnormal neurotransmitter metabolism secondary to reduced co-factor pyridoxal-5′-phosphate availability. J. Inherit. Metab. Dis. 2010, 33, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Diez-Zaera, M.; Diaz-Hernandez, J.I.; Hernandez-Alvarez, E.; Zimmermann, H.; Díaz-Hernández, M.; Miras-Portugal, M.T. Tissue-nonspecific alkaline phosphatase promotes axonal growth of hippocampal neurons. Mol. Biol. Cell 2011, 22, 1014–1024. [Google Scholar] [CrossRef]
- Street, S.E.; Sowa, N.A. TNAP and Pain Control. Subcell. Biochem. 2015, 76, 283–305. [Google Scholar] [CrossRef]
- Salles, J.-P. Clinical Forms and Animal Models of Hypophosphatasia. Subcell. Biochem. 2015, 76, 3–24. [Google Scholar] [CrossRef]
- Seefried, L.; Dahir, K.; Petryk, A.; Hogler, W.; Linglart, A.; Martos-Moreno, G.A.; Ozono, K.; Fang, S.; Rockman-Greenberg, C.; Kishnani, P.S. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J. Bone Miner. Res. 2020, 35, 2171–2178. [Google Scholar] [CrossRef]
- Négyessy, L.; Győrffy, B.; Hanics, J.; Bányai, M.; Fonta, C.; Bazsó, F. Signal Transduction Pathways of TNAP: Molecular Network Analyses. Subcell. Biochem. 2015, 76, 185–205. [Google Scholar] [CrossRef]
- Sebastián-Serrano, Á.; Engel, T.; De Diego-García, L.; Olivos-Oré, L.A.; Arribas-Blázquez, M.; Martínez-Frailes, C.; Pérez-Díaz, C.; Millán, J.L.; Artalejo, A.R.; Miras-Portugal, M.T.; et al. Neurodevelopmental alterations and seizures developed by mouse model of infantile hypophosphatasia are associated with purinergic signalling deregulation. Hum. Mol. Genet. 2016, 25, 4143–4156. [Google Scholar] [CrossRef] [Green Version]
- Kántor, O.; Varga, A.; Kovács-Öller, T.; Énzsöly, A.; Balogh, L.; Baksa, G.; Szepessy, Z.; Fonta, C.; Roe, A.W.; Nitschke, R.; et al. TNAP activity is localized at critical sites of retinal neurotransmission across various vertebrate species. Cell Tissue Res. 2014, 358, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Kántor, O.; Varga, A.; Toth, R.; Énzsöly, A.; Pálfi, E.; Kovács-Öller, T.; Nitschke, R.; Szél, Á.; Székely, A.; Völgyi, B.; et al. Stratified organization and disorganization of inner plexiform layer revealed by TNAP activity in healthy and diabetic rat retina. Cell Tissue Res. 2014, 359, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Gasque, K.C.; Foster, B.L.; Kuss, P.; Yadav, M.C.; Liu, J.; Kiffer-Moreira, T.; Van Elsas, A.; Hatch, N.E.; Somerman, M.J.; Millán, J.L. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl−/− mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone 2015, 72, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, P.A.; Barthel, L.K.; Bernardos, R.; Perkowski, J.J. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev. Biol. 2006, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurm, A.; Pannicke, T.; Iandiev, I.; Francke, M.; Hollborn, M.; Wiedemann, P.; Reichenbach, A.; Osborne, N.N.; Bringmann, A. Purinergic signaling involved in Müller cell function in the mammalian retina. Prog. Retin. Eye Res. 2011, 30, 324–342. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Role of Purines in Müller Glia. J. Ocul. Pharmacol. Ther. 2016, 32, 518–533. [Google Scholar] [CrossRef]
- Iandiev, I.; Wurm, A.; Pannicke, T.; Wiedemann, P.; Reichenbach, A.; Robson, S.C.; Zimmermann, H.; Bringmann, A. Ectonucleotidases in Müller glial cells of the rodent retina: Involvement in inhibition of osmotic cell swelling. Purinergic Signal. 2007, 3, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Gospe, S.M.; Santiago-Turla, C.; DeArmey, S.M.; Cummings, T.J.; Kishnani, P.S.; Bhatti, M.T. Ectopic Ocular Surface Calcification in Patients With Hypophosphatasia Treated With Asfotase Alfa. Cornea 2019, 38, 896–900. [Google Scholar] [CrossRef]
- Rosemberg, D.B.; Rico, E.P.; Langoni, A.S.; Spinelli, J.T.; Pereira, T.C.B.; Dias, R.D.; De Souza, D.O.G.; Bonan, C.D.; Bogo, M.R. NTPDase family in zebrafish: Nucleotide hydrolysis, molecular identification and gene expression profiles in brain, liver and heart. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2010, 155, 230–240. [Google Scholar] [CrossRef]
- Chida-Naomiya, R.; Shimura, M.; Nagao, R.; Kumada, A.; Kawashima, H. Hearing impairment improved after treatment with asfotase alfa in a case of perinatal hypophosphatasia. Mol. Genet. Metab. Rep. 2020, 24. [Google Scholar] [CrossRef]
- Pinto-Teixeira, F.; Viader-Llargués, O.; Torres-Mejia, E.; Turan, M.; González-Gualda, E.; Pola-Morell, L.; López-Schier, H. Inexhaustible hair-cell regeneration in young and aged zebrafish. Biol. Open 2015, 4, 903–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viader-Llargués, O.; Lupperger, V.; Pola-Morell, L.; Marr, C.; López-Schier, H. Live cell-lineage tracing and machine learning reveal patterns of organ regeneration. eLife 2018, 7, e30823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakisaka, N.; Miyasaka, N.; Koide, T.; Masuda, M.; Hiraki-Kajiyama, T.; Yoshihara, Y. An Adenosine Receptor for Olfaction in Fish. Curr. Biol. 2017, 27, 1437–1447.e4. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Hernandez, M.; Hernández, F.; Miras-Portugal, M.T.; Avila, J. TNAP Plays a Key Role in Neural Differentiation as well as in Neurodegenerative Disorders. Subcell. Biochem. 2015, 76, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Salles, J.P. Hypophosphatasia: Biological and Clinical Aspects, Avenues for Therapy. Clin. Biochem. Rev. 2020, 41, 13–26. [Google Scholar] [CrossRef]
- Hahnel, A.C.; Rappolee, D.A.; Millan, J.L.; Manes, T.; Ziomek, C.A.; Theodosiou, N.G.; Werb, Z.; Pedersen, R.A.; Schultz, G.A. Two alkaline phosphatase genes are expressed during early development in the mouse embryo. Development 1990, 110, 555–564. [Google Scholar]
- Fraser, D. Hypophosphatasia. Am. J. Med. 1957, 22, 730–746. [Google Scholar] [CrossRef]
- Weiss, M.J.; Cole, D.E.; Ray, K.; Whyte, M.P.; Lafferty, M.A.; Mulivor, R.A.; Harris, H. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc. Natl. Acad. Sci. USA 1988, 85, 7666–7669. [Google Scholar] [CrossRef] [Green Version]
- Di Rocco, F.; Baujat, G.; Cormier-Daire, V.; Rothenbuhler, A.; Linglart, A. Craniosynostosis and hypophosphatasia. Archives de Pédiatrie 2017, 24, 5S89–5S92. [Google Scholar] [CrossRef]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological Role of Vascular Smooth Muscle Alkaline Phosphatase in Medial Artery Calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Vardy, E.R.; Kellett, K.A.; Cocklin, S.L.; Hooper, N.M. Alkaline Phosphatase Is Increased in both Brain and Plasma in Alzheimer’s Disease. Neurodegener. Dis. 2012, 9, 31–37. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | Mouse and Rat | Zebrafish and Other Vertebrates | |
|---|---|---|---|
| ALPL/Alpl/alpl Expression | |||
| Expression database links | The Human Protein Atlas (https://www.proteinatlas.org/ENSG00000162551-ALPL) | Mouse Genome Informatics (http://www.informatics.jax.org/marker/MGI:87983) Rat Genome Database (https://rgd.mcw.edu/rgdweb/report/gene/main.html?id=2100) | Zebrafish Information Network (https://zfin.org/ZDB-GENE-040420-1) Xenopus/Xenbase (http://www.xenbase.org/gene/expression.do?tabId=1&method=displayGenePageExpression&geneId=1010301&objId=1010301) Chicken/Geisha (http://geisha.arizona.edu/geisha/search.jsp?search=NCBI+ID&text=396317) |
| Embryonic development | Expression of Alpl/alpl has been intensively studied in mice and to a smaller extent in zebrafish, Xenopus and chicken embryos. It has been detected in developing brain regions (telencephalon and diencephalon), bone structures, genital organs, muscle/myotome, kidney, and heart. | ||
| N/A | [197] | [7] | |
| Bone and mineralizing structures | Expression in skeletal bone and other mineralizing structures, e.g., teeth, has been reported for humans and rodents. In addition, expression in limbs/fins has been reported in mice and zebrafish. | ||
| [101] | [112] | Expression database | |
| Neurons and brain | Expression in distinct brain regions is common to all vertebrates. Most prominently detected in the forebrain/telencephalon, epiphyses, and layers within the human neocortex. | ||
| [168] | [158] | [166,167] | |
| Eye | Expression in the retina is reported for a wide number of vertebrates and is evolutionary highly conserved. | ||
| [183] | [183] | [183] | |
| Consequences of TNAP/Tnap Loss of Function | |||
| Hypophosphatasia | A large number of disease-causing mutations have been described and result in HPP in humans. Similar disease phenotypes have been reported in knockout and mutated mice, including decreased circulating TNAP levels in blood. Variable severity grades can be observed and result in different pathological disease classes in human patients and murine models. | ||
| [32,33] | [68,70,129] | N/A | |
| Defects in bone and mineralizing structures | Changed levels of TNAP activity result in prominent skeletal and mineralizing defects in humans and mice, e.g., decreased bone density, lack of dentin mineralization, and increased bone resorption. Blocking of Tnap results in a delay of mineralization in zebrafish embryos. | ||
| [198,199] | [70,129] | [7] | |
| Defects in teeth | Lack of mineralization results in abnormal tooth development, morphological deformations, and short tooth roots in humans and mice. Observed changes are ALPL/Alpl mutation-dependent. | ||
| [97,102,103] | [73,98] | N/A | |
| Defects in neurons and Brain | Most prominently, neurological symptoms of HPP patients are seizures, anxiety disorders, and depression. In murine knockout models and heavily affected patients, seizures can be lethal. Although, neuronal tube defects, demonstrated by differences in spine nerve morphology and lumbar nerve roots development, can be abnormal in knockout mice. | ||
| [147,164] | [69,146,155] | N/A | |
| Craniosynostosis | Fusion of sutures has been reported in human HPP patients and in knockout mice. However, the phenotype is mutation-dependent and highly variable. | ||
| [200] | [72] | N/A | |
| Vascular calcification | HPP patients do not develop vascular calcification, although transgenic mouse models can show pathological arterial calcification. | ||
| N/A | [201] | N/A | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liedtke, D.; Hofmann, C.; Jakob, F.; Klopocki, E.; Graser, S. Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease. Biomolecules 2020, 10, 1648. https://doi.org/10.3390/biom10121648
Liedtke D, Hofmann C, Jakob F, Klopocki E, Graser S. Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease. Biomolecules. 2020; 10(12):1648. https://doi.org/10.3390/biom10121648
Chicago/Turabian StyleLiedtke, Daniel, Christine Hofmann, Franz Jakob, Eva Klopocki, and Stephanie Graser. 2020. "Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease" Biomolecules 10, no. 12: 1648. https://doi.org/10.3390/biom10121648