Gene Expression Analysis of Astrocyte and Microglia Endocannabinoid Signaling during Autoimmune Demyelination


Abstract
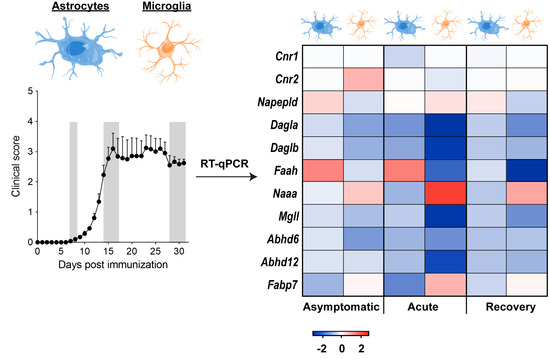
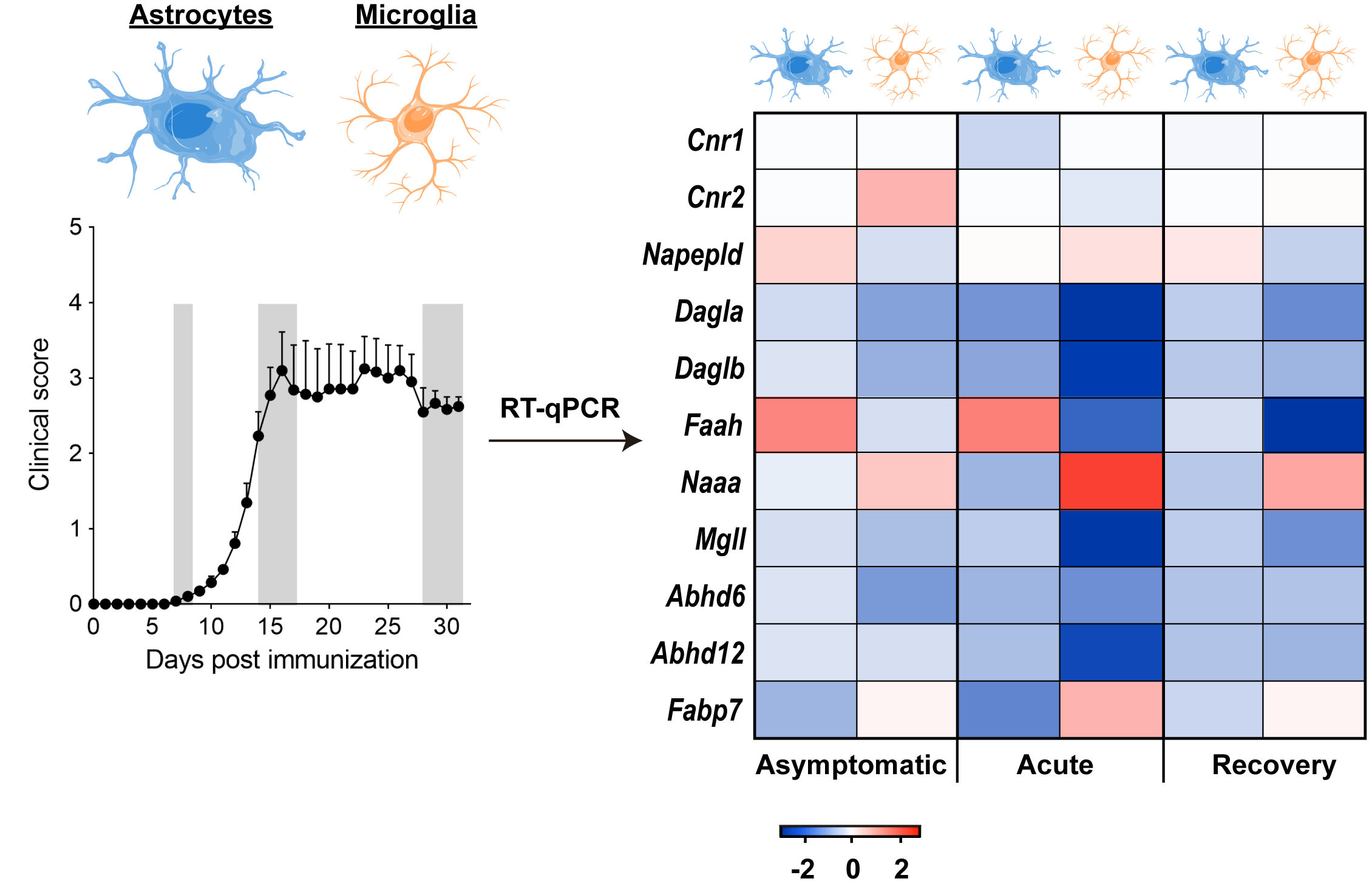
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Induction of EAE
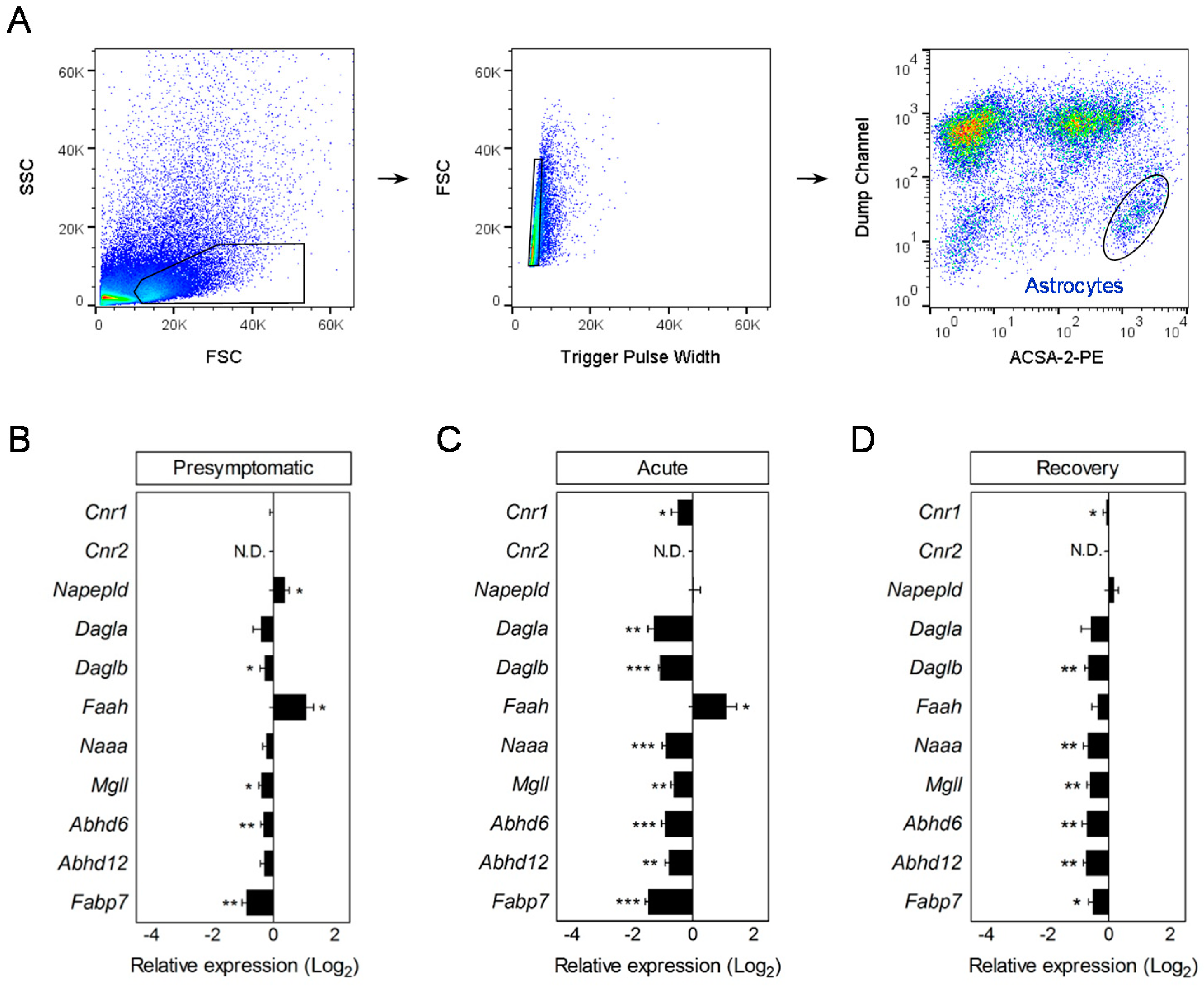
2.3. Flow Cytometry
2.4. Nanofluidic RT-qPCR in Purified Astrocytes and Microglia
2.5. Immunohistochemistry
2.6. Data Analysis
3. Results
3.1. Early Deregulation of Endocannabinoid System Genes in Astrocytes during EAE
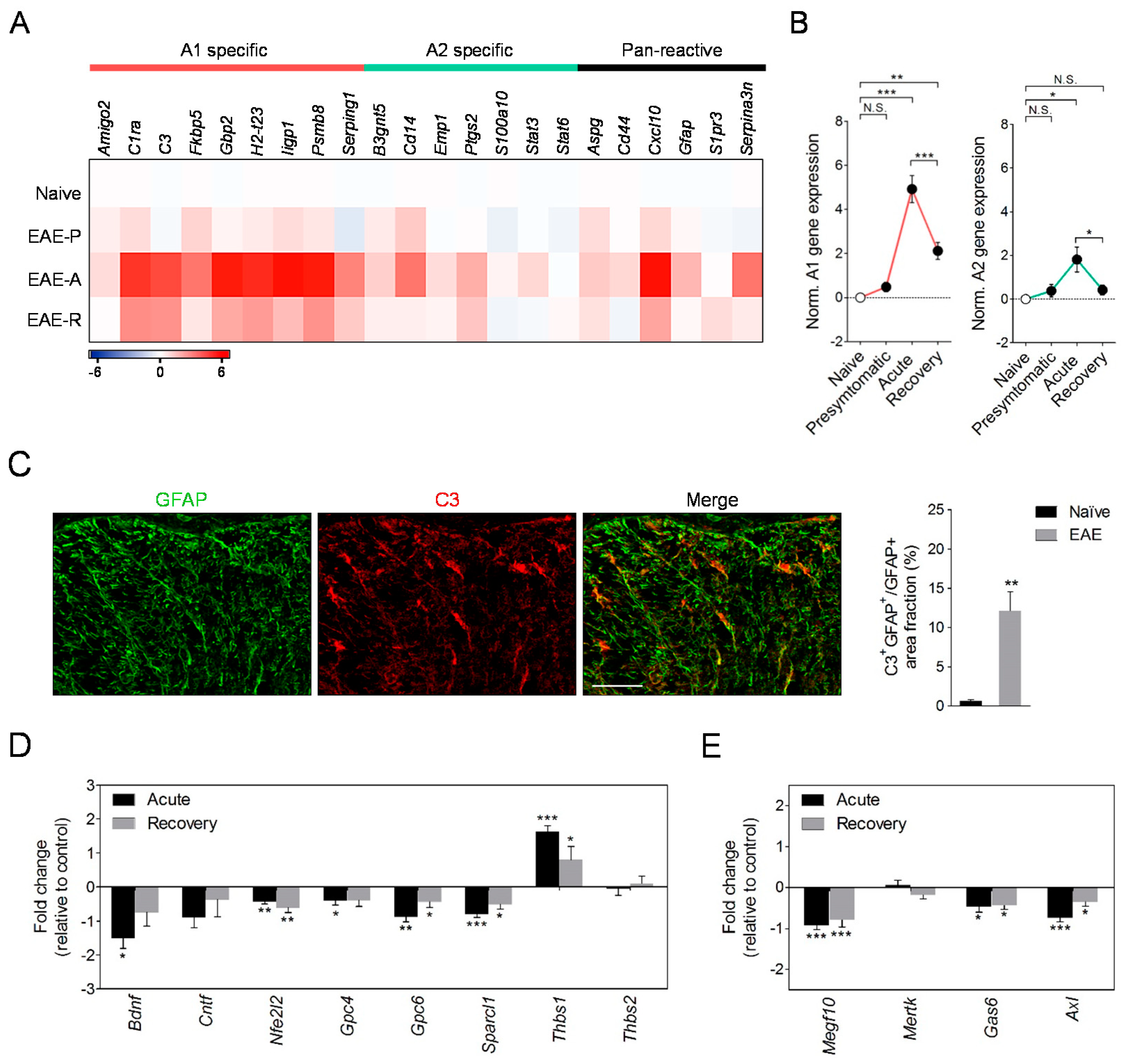
3.2. EAE Induction Promotes Astrocyte Conversion to a Neurotoxic A1 Phenotype
3.3. Modulation of Endocannabinoid Signaling-Related Genes in Microglia during EAE
3.4. Expression Patterns of Pro-Inflammatory and Protective Genes in Microglia during EAE
3.5. EAE Induces the Expression of Astrocyte Pan-Reactive and A1-Like Genes in Microglia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nylander, A.; Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2012, 122, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, L.; Quintana, F.J.; Weiner, H.L. The innate immune system in demyelinating disease. Immunol. Rev. 2012, 248, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2018. [Google Scholar] [CrossRef]
- Miron, V.E.; Franklin, R.J. Macrophages and CNS remyelination. J. Neurochem. 2014, 130, 165–171. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Tassoni, A.; Farkhondeh, V.; Itoh, Y.; Itoh, N.; Sofroniew, M.V.; Voskuhl, R.R. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci. Rep. 2019, 9, 10010. [Google Scholar] [CrossRef]
- Hou, B.; Zhang, Y.; Liang, P.; He, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczocha, M.; Glaser, S.T.; Deutsch, D.G. Identification of intracellular carriers for the endocannabinoid anandamide. Proc. Natl. Acad. Sci. USA 2009, 106, 6375–6380. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [Green Version]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Kaczocha, M.; Glaser, S.T.; Maher, T.; Clavin, B.; Hamilton, J.; O’Rourke, J.; Rebecchi, M.; Puopolo, M.; Owada, Y.; Thanos, P.K. Fatty acid binding protein deletion suppresses inflammatory pain through endocannabinoid/N-acylethanolamine-dependent mechanisms. Mol. Pain 2015, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Gutiérrez, S.; Molina-Holgado, E.; Arévalo-Martín, A.; Correa, F.; Viso, A.; López-Rodríguez, M.L.; Di Marzo, V.; Guaza, C. Activation of the endocannabinoid system as therapeutic approach in a murine model of multiple sclerosis. FASEB J. 2005, 19, 1338–1340. [Google Scholar] [CrossRef]
- Bernal-Chico, A.; Canedo, M.; Manterola, A.; Victoria Sánchez-Gómez, M.; Pérez-Samartín, A.; Rodríguez-Puertas, R.; Matute, C.; Mato, S. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo. Glia 2015, 63, 163–176. [Google Scholar] [CrossRef]
- Manterola, A.; Bernal-Chico, A.; Cipriani, R.; Canedo-Antelo, M.; Moreno-García, Á.; Martín-Fontecha, M.; Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Ortega-Gutiérrez, S.; Brown, J.M.; et al. Deregulation of the endocannabinoid system and therapeutic potential of ABHD6 blockade in the cuprizone model of demyelination. Biochem. Pharmacol. 2018, 157, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Martín, A.; Vela, J.M.; Molina-Holgado, E.; Borrell, J.; Guaza, C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J. Neurosci. 2003, 23, 2511–2516. [Google Scholar]
- Palazuelos, J.; Davoust, N.; Julien, B.; Hatterer, E.; Aguado, T.; Mechoulam, R.; Benito, C.; Romero, J.; Silva, A.; Guzmán, M.; et al. The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: Involvement in the pathogenesis of an animal model of multiple sclerosis. J. Biol. Chem. 2008, 283, 13320–13329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresz, K.; Pryce, G.; Ponomarev, E.D.; Marsicano, G.; Croxford, J.L.; Shriver, L.P.; Ledent, C.; Cheng, X.; Carrier, E.J.; Mann, M.K.; et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat. Med. 2007, 13, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Berrendero, F.; Sánchez, A.; Cabranes, A.; Puerta, C.; Ramos, J.A.; García-Merino, A.; Fernández-Ruiz, J. Changes in cannabinoid CB(1) receptors in striatal and cortical regions of rats with experimental allergic encephalomyelitis, an animal model of multiple sclerosis. Synapse 2001, 41, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Cabranes, A.; Pryce, G.; Baker, D.; Fernández-Ruiz, J. Changes in CB1 receptors in motor-related brain structures of chronic relapsing experimental allergic encephalomyelitis mice. Brain Res. 2006, 1107, 199–205. [Google Scholar] [CrossRef]
- Loría, F.; Petrosino, S.; Mestre, L.; Spagnolo, A.; Correa, F.; Hernangómez, M.; Guaza, C.; Di Marzo, V.; Docagne, F. Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur. J. Neurosci. 2008, 28, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Jean-Gilles, L.; Feng, S.; Tench, C.R.; Chapman, V.; Kendall, D.A.; Barrett, D.A.; Constantinescu, C.S. Plasma endocannabinoid levels in multiple sclerosis. J. Neurol. Sci. 2009, 287, 212–215. [Google Scholar] [CrossRef]
- Di Filippo, M.; Pini, L.A.; Pelliccioli, G.P.; Calabresi, P.; Sarchielli, P. Abnormalities in the cerebrospinal fluid levels of endocannabinoids in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry. 2008, 79, 1224–1229. [Google Scholar] [CrossRef]
- Centonze, D.; Bari, M.; Rossi, S.; Prosperetti, C.; Furlan, R.; Fezza, F.; De Chiara, V.; Battistini, L.; Bernardi, G.; Bernardini, S.; et al. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain 2007, 130, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 2006, 49, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.; Pryce, G.; Croxford, J.L.; Brown, P.; Pertwee, R.G.; Makriyannis, A.; Khanolkar, A.; Layward, L.; Fezza, F.; Bisogno, T.; et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001, 15, 300–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, B.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705–718. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; De Vin, F.; Duqué, S.I.; Li, C.; Saito, T.; Saido, T.; Fiers, M.; Belgard, T.G.; Holt, M.G. An immunoaffinity-based method for isolating ultrapure adult astrocytes based on ATP1B2 targeting by the ACSA-2 antibody. J. Biol. Chem. 2017, 292, 8874–8891. [Google Scholar] [CrossRef] [Green Version]
- Kantzer, C.G.; Boutin, C.; Herzig, I.D.; Wittwer, C.; Reiß, S.; Tiveron, M.C.; Drewes, J.; Rockel, T.D.; Ohlig, S.; Ninkovic, J.; et al. Anti-ACSA-2 defines a novel monoclonal antibody for prospective isolation of living neonatal and adult astrocytes. Glia 2017, 65, 990–1004. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Jang, J.S.; Simon, V.A.; Feddersen, R.M.; Rakhshan, F.; Schultz, D.A.; Zschunke, M.A.; Lingle, W.L.; Kolbert, C.P.; Jen, J. Quantitative miRNA expression analysis using fluidigm microfluidics dynamic arrays. BMC Genomics 2011, 12, 144. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Reisenberg, M.; Singh, P.K.; Williams, G.; Doherty, P. The diacylglycerol lipases: Structure, regulation and roles in and beyond endocannabinoid signaling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yamasaki, R.; Yamaguchi, H.; Nagata, S.; Une, H.; Cui, Y.; Masaki, K.; Nakamuta, Y.; Iinuma, K.; Watanabe, M.; et al. Oligodendroglial connexin 47 regulates neuroinflammation upon autoimmune demyelination in a novel mouse model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 2160–2169. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, L.; Da Cunha, A.P.; Madi, A.; Beynon, V.; Yang, Z.; Alvarez, J.I.; Prat, A.; Sobel, R.A.; Kobzik, L.; Lassmann, H.; et al. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 2016, 139, 1939–1957. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.-A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Lewis, N.D.; Hill, J.D.; Juchem, K.W.; Stefanopoulos, D.E.; Modis, L.K. RNA sequencing of microglia and monocyte-derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J. Neuroimmunol. 2014, 277, 26–38. [Google Scholar] [CrossRef]
- Zabala, A.; Vazquez-Villoldo, N.; Rissiek, B.; Gejo, J.; Martin, A.; Palomino, A.; Perez-Samartín, A.; Pulagam, K.R.; Lukowiak, M.; Capetillo-Zarate, E.; et al. P2X4 receptor controls microglia activation and favors remyelination in autoimmune encephalitis. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflammation 2013, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noristani, H.N.; Gerber, Y.N.; Sabourin, J.C.; Le Corre, M.; Lonjon, N.; Mestre-Frances, N.; Hirbec, H.E.; Perrin, F.E. RNA-Seq Analysis of microglia reveals time-dependent activation of specific genetic programs following spinal cord injury. Front. Mol. Neurosci. 2017, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, I.M.; Morimoto, E.T.A.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito, C.; Romero, J.P.; Tolón, R.M.; Clemente, D.; Docagne, F.; Hillard, C.J.; Guaza, C.; Romero, J. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J. Neurosci. 2007, 27, 2396–2402. [Google Scholar] [CrossRef] [Green Version]
- Muccioli, G.G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; López Rodríguez, M.L.; Bajjalieh, S.; Stella, N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J. Neurosci. 2007, 27, 2883–2889. [Google Scholar] [CrossRef] [Green Version]
- Marrs, W.R.; Blankman, J.L.; Horne, E.A.; Thomazeau, A.; Lin, Y.H.; Coy, J.; Bodor, A.L.; Muccioli, G.G.; Hu, S.S.; Woodruff, G.; et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 2010, 13, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Hermes, D.J.; Xu, C.; Poklis, J.L.; Niphakis, M.J.; Cravatt, B.F.; Mackie, K.; Lichtman, A.H.; Ignatowska-Jankowska, B.M.; Fitting, S. Neuroprotective effects of fatty acid amide hydrolase catabolic enzyme inhibition in a HIV-1 Tat model of neuroAIDS. Neuropharmacology 2018, 141, 55–65. [Google Scholar] [CrossRef]
- Chicca, A.; Nicolussi, S.; Bartholomäus, R.; Blunder, M.; Aparisi Rey, A.; Petrucci, V.; Reynoso-Moreno, I.; del Carmen Reynoso-Moreno, I.; Viveros-Paredes, J.M.; Dalghi Gens, M.; et al. Chemical probes to potently and selectively inhibit endocannabinoid cellular reuptake. Proc. Natl. Acad. Sci. USA 2017, 114, E5006–E5015. [Google Scholar] [CrossRef] [Green Version]
- Metna-Laurent, M.; Marsicano, G. Rising stars: Modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia 2015, 63, 353–364. [Google Scholar] [CrossRef]
- Ashton, J.C. The use of knockout mice to test the specificity of antibodies for cannabinoid receptors. Hippocampus 2012, 22, 643–644. [Google Scholar] [CrossRef]
- Grimsey, N.L.; Goodfellow, C.E.; Scotter, E.L.; Dowie, M.J.; Glass, M.; Graham, E.S. Specific detection of CB1 receptors; cannabinoid CB1 receptor antibodies are not all created equal! J. Neurosci. Methods 2008, 171, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Vickstrom, C.R.; Liu, M.J.; Zhao, L.; Viader, A.; Cravatt, B.F.; Liu, Q.S. Neuronal and astrocytic monoacylglycerol lipase limit the spread of endocannabinoid signaling in the cerebellum. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Viader, A.; Blankman, J.L.; Zhong, P.; Liu, X.; Schlosburg, J.E.; Joslyn, C.M.; Liu, Q.S.; Tomarchio, A.J.; Lichtman, A.H.; Selley, D.E.; et al. Metabolic interplay between astrocytes and neurons regulates endocannabinoid action. Cell Rep. 2015, 12, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, Y.; Vickstrom, C.R.; Li, Y.; Viader, A.; Cravatt, B.F.; Liu, Q. Coordinated regulation of endocannabinoid-mediated retrograde synaptic suppression in the cerebellum by neuronal and astrocytic monoacylglycerol lipase. Sci. Rep. 2016, 6, 35829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.-C.; Gutiérrez-Vázquez, C.; Rothhammer, V.; Mayo, L.; Wheeler, M.A.; Tjon, E.C.; Zandee, S.E.J.; Blain, M.; de Lima, K.A.; Takenaka, M.C.; et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 2019, 179, 1483–1498.e22. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Smith, B.C.; Sinyuk, M.; Jenkins, J.E.; Psenicka, M.W.; Williams, J.L. The impact of regional astrocyte interferon-γ signaling during chronic autoimmunity: A novel role for the immunoproteasome. J. Neuroinf. 2020, 17, 184. [Google Scholar] [CrossRef]
- Hindinger, C.; Bergmann, C.C.; Hinton, D.R.; Phares, T.W.; Parra, G.I.; Hussain, S.; Savarin, C.; Atkinson, R.D.; Stohlman, S.A. IFN-γ signaling to astrocytes protects from autoimmune mediated neurological disability. PLoS ONE 2012, 7, e42088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalai, A.J.; Hu, X.; Adams, J.E.; Barnum, S.R. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol. Immunol. 2007, 44, 3132–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, A.; Kirchhoff, F. Endocannabinoids and heterogeneity of glial cells in brain function. Front. Integr. Neurosci. 2016, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Araque, A.; Castillo, P.E.; Manzoni, O.J.; Tonini, R. Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 2017, 124, 13–24. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-García, Á.; Bernal-Chico, A.; Colomer, T.; Rodríguez-Antigüedad, A.; Matute, C.; Mato, S. Gene Expression Analysis of Astrocyte and Microglia Endocannabinoid Signaling during Autoimmune Demyelination. Biomolecules 2020, 10, 1228. https://doi.org/10.3390/biom10091228
Moreno-García Á, Bernal-Chico A, Colomer T, Rodríguez-Antigüedad A, Matute C, Mato S. Gene Expression Analysis of Astrocyte and Microglia Endocannabinoid Signaling during Autoimmune Demyelination. Biomolecules. 2020; 10(9):1228. https://doi.org/10.3390/biom10091228
Chicago/Turabian StyleMoreno-García, Álvaro, Ana Bernal-Chico, Teresa Colomer, Alfredo Rodríguez-Antigüedad, Carlos Matute, and Susana Mato. 2020. "Gene Expression Analysis of Astrocyte and Microglia Endocannabinoid Signaling during Autoimmune Demyelination" Biomolecules 10, no. 9: 1228. https://doi.org/10.3390/biom10091228