Deamidated Human Triosephosphate Isomerase is a Promising Druggable Target


 , ,
, , 
Abstract
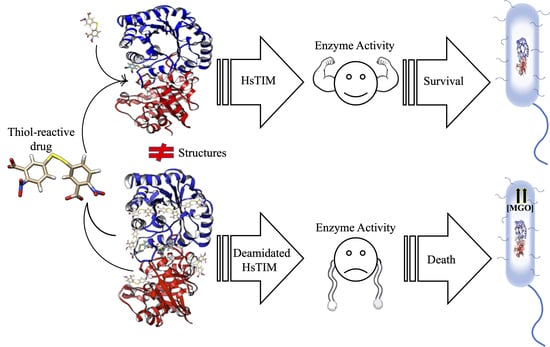
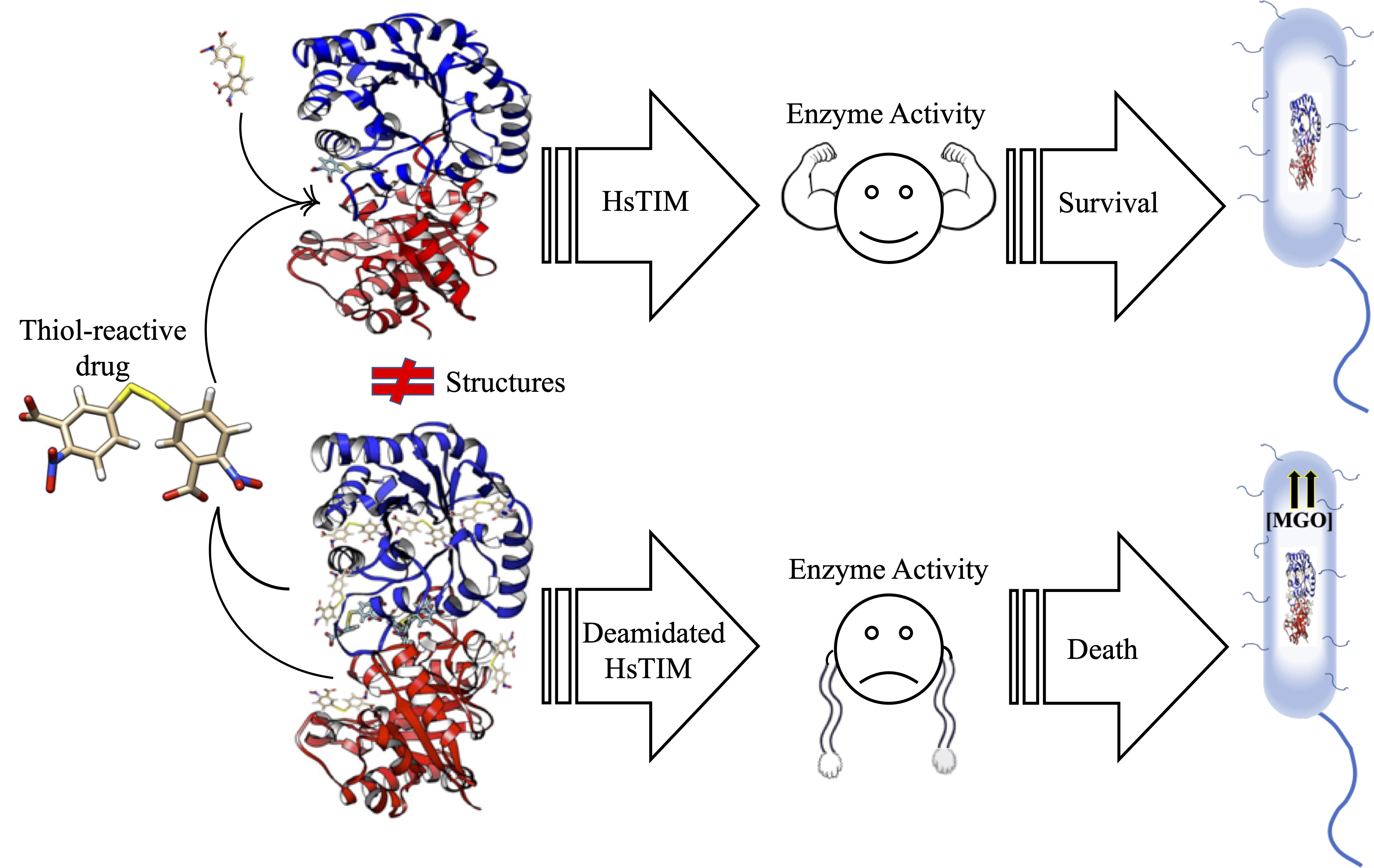
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. In Silico Analysis of the WT and N16D HsTIM Crystallographic Structures
2.3. Expression and Purification of Recombinant Enzymes
2.4. Inactivation Assays of WT and N16D HsTIM with MMTS, MTSES, and DTNB
2.5. Quantification of Derivatized Cys in WT and N16D HsTIM Treated with Sulfhydryl Reagents
2.6. Growth and Inhibition Curves of E. coli Δtim-BL21-Gold(DE3) Cells Complemented with WT and N16D Hstim Genes
2.7. Methylglyoxal and AGE Quantification in E. coli Δtim-BL21-Gold(DE3) Cells Complemented with WT and N16D Hstim Genes
2.8. Cellular Assays with E. coli BL21-CodonPlus (DE3)-RIL in the Presence of Omeprazole
2.9. Statistical Analysis
3. Results
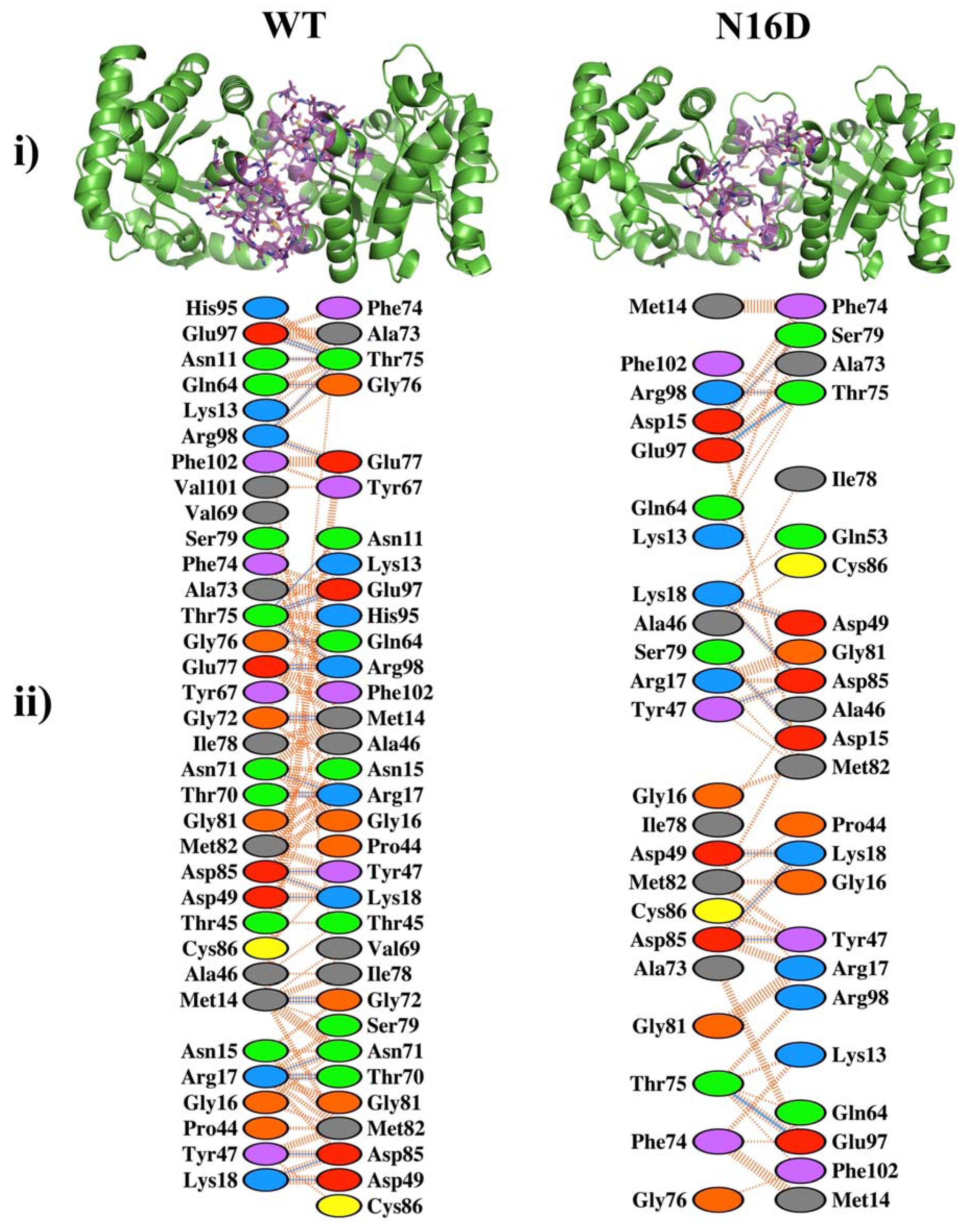
3.1. Deamidation Alters the Interatomic Interacting Network in HsTIM
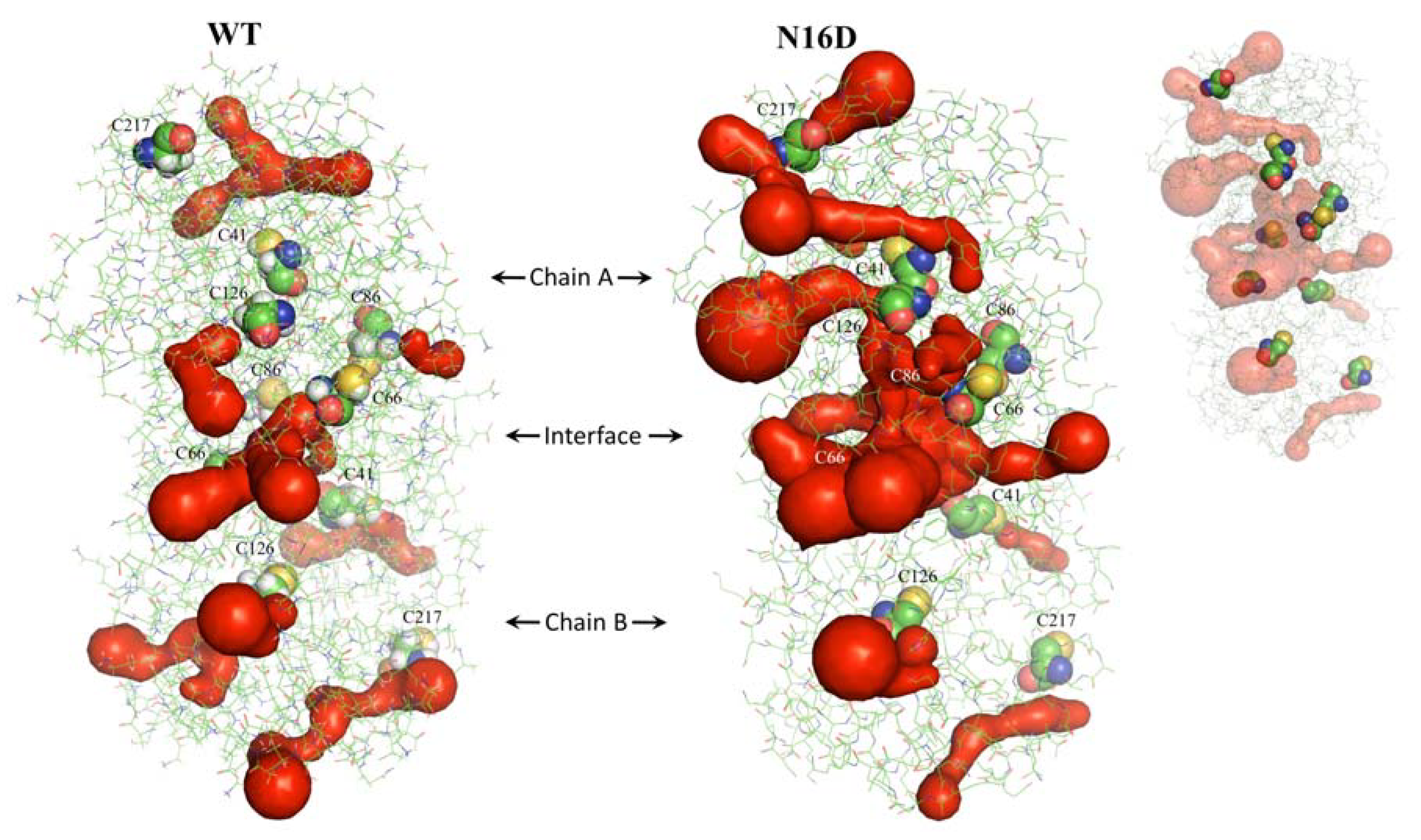
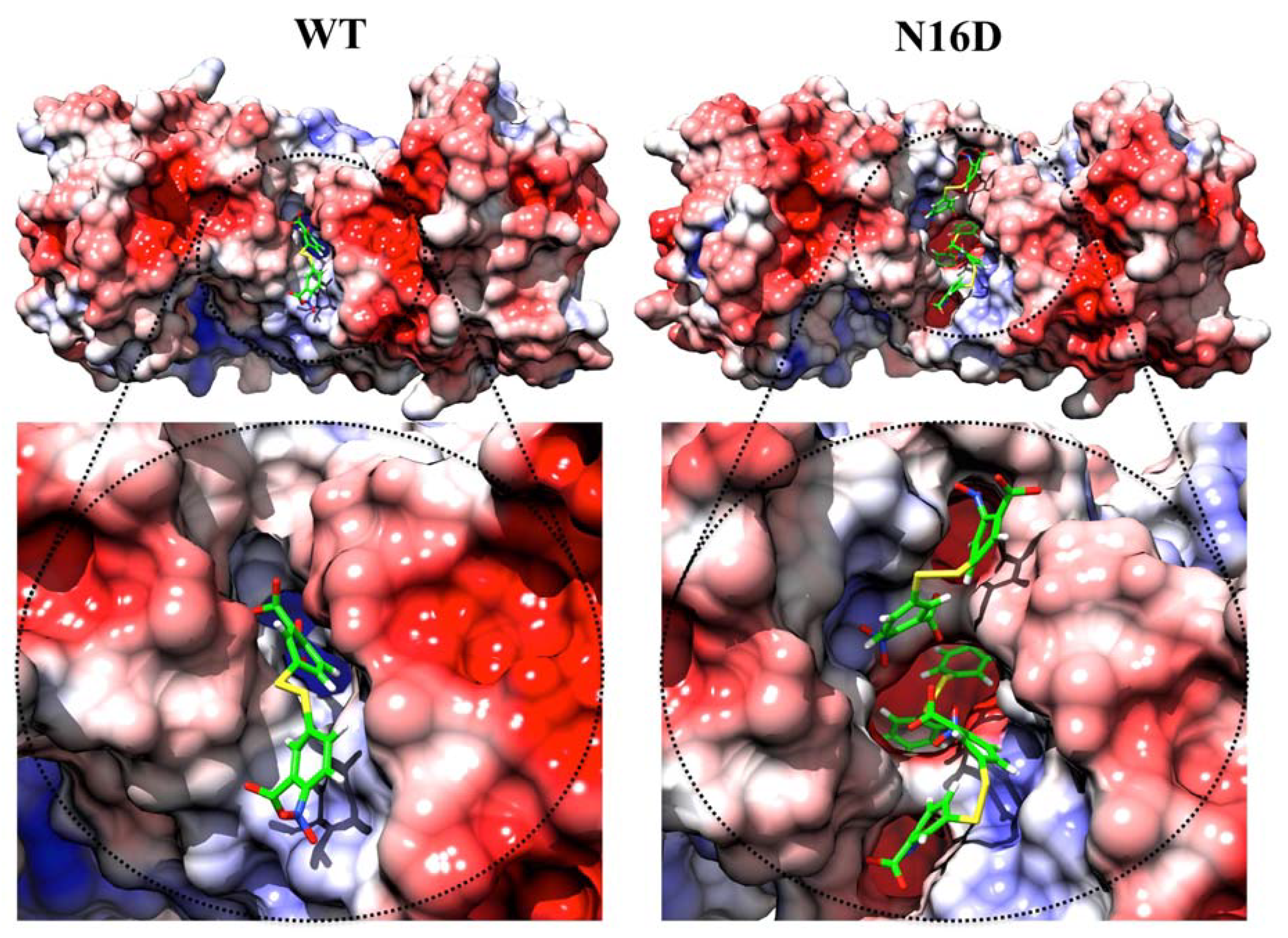
3.2. Binding Sites for Thiol-Reactive Compounds Are Increased into the Interface of N16D HsTIM
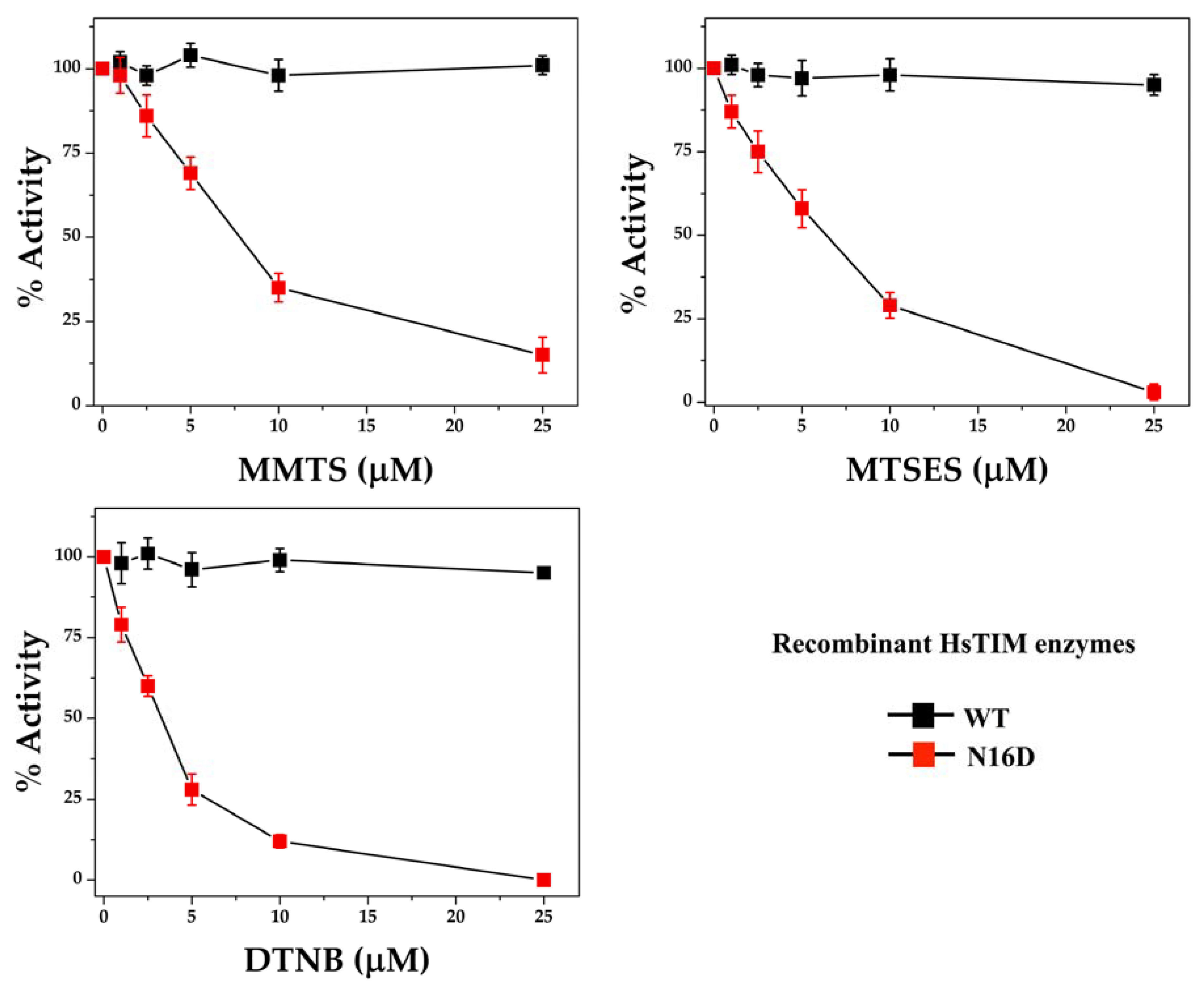
3.3. The N16D HsTIM Enzyme Is Totally and Selectively Inactivated with Thiol-Reactive Compounds
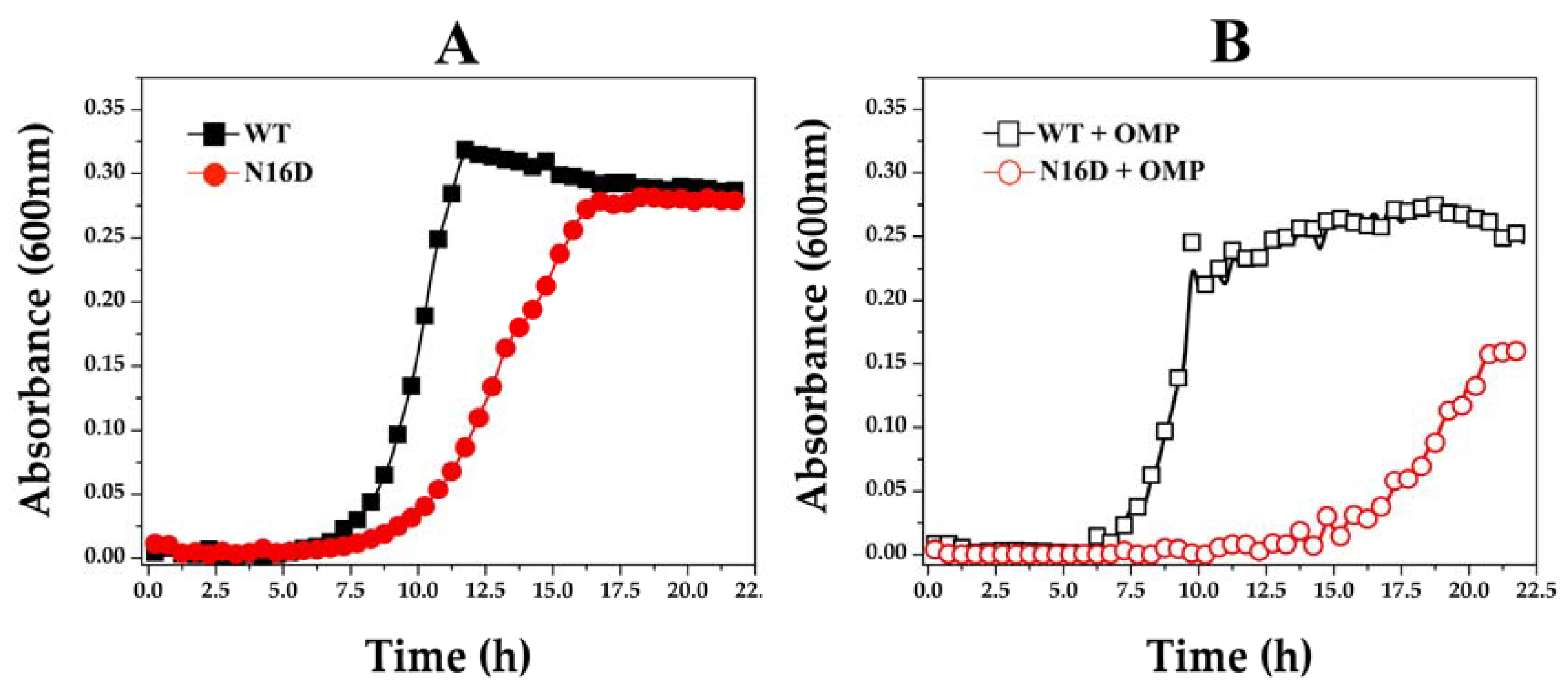
3.4. E. coli Δtim Cells Complemented with the WT and N16D Genes Are a Good Model to Study the Effects of N16D HsTIM at the Cellular Level
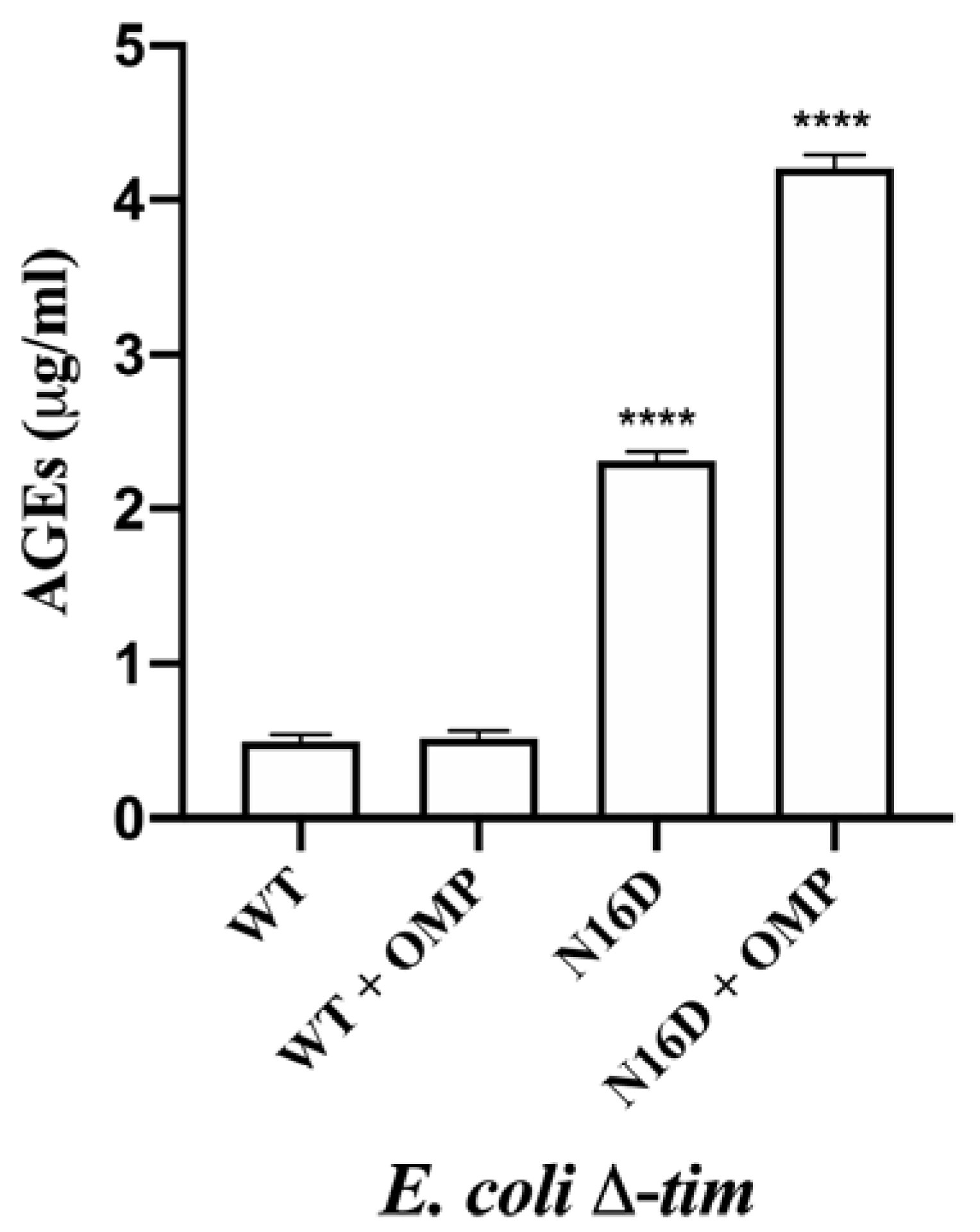
3.5. Omeprazole Induces Increasing Levels of MGO and AGEs in E. coli Δtim Cells Complemented with N16D HsTIM
4. Discussion
4.1. The Structural Differences between Deamidated and Nondeamidated HsTIM Are the Keystone to Being a Targetable Molecule
4.2. Deamidated TIM Is More Permeable to Thiol-Reactive Compounds than Its Nondeamidated Counterpart
4.3. Thiol-Reactive Compounds Selectively Affect N16D HsTIM
4.4. N16D HsTIM Is the Intracellular Druggable Target
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Park, J.S.; Lee, J.Y.; Nguyen, Y.; Kang, N.W.; Oh, E.K.; Jang, D.M.; Kim, H.J.; Kim, D.D.; Han, B.W. Structural Analyses on the Deamidation of N-Terminal Asn in the Human N-Degron Pathway. Biomolecules 2020, 10, 163. [Google Scholar] [CrossRef] [Green Version]
- Curnis, F.; Longhi, R.; Crippa, L.; Cattaneo, A.; Dondossola, E.; Bachi, A.; Corti, A. Spontaneous formation of L-isoaspartate and gain of function in fibronectin. J. Biol. Chem. 2006, 281, 36466–36476. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.E.; Robinson, A.B. Deamidation of human proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 12409–12413. [Google Scholar] [CrossRef] [Green Version]
- Sadakane, Y.; Kawahara, M. Implications of Metal Binding and Asparagine Deamidation for Amyloid Formation. Int. J. Mol. Sci. 2018, 19, 2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipkiss, A.R. Aging, Alzheimer’s Disease and Dysfunctional Glycolysis; Similar Effects of Too Much and Too Little. Aging Dis. 2019, 10, 1328–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar]
- Sinha, S.; Zhang, L.; Duan, S.; Williams, T.D.; Vlasak, J.; Ionescu, R.; Topp, E.M. Effect of protein structure on deamidation rate in the Fc fragment of an IgG1 monoclonal antibody. Protein Sci. 2009, 18, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumatin, F.; El Dhaybi, M.; Bobo, C.; Verdier, M.; Priault, M. Bcl-xL deamidation and cancer: Charting the fame trajectories of legitimate child and hidden siblings. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Magami, K.; Kim, I.; Fujii, N. A single Asp isomer substitution in an αA-crystallin-derived peptide induces a large change in peptide properties. Exp. Eye Res. 2020, 192, 107930. [Google Scholar] [CrossRef]
- De la Mora-de la Mora, I.; Torres-Larios, A.; Enríquez-Flores, S.; Méndez, S.T.; Castillo-Villanueva, A.; Gómez-Manzo, S.; López-Velázquez, G.; Marcial-Quino, J.; Torres-Arroyo, A.; García-Torres, I.; et al. Structural effects of protein aging: Terminal marking by deamidation in human triosephosphate isomerase. PLoS ONE 2015, 10, e0123379. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.Q.; Yüksel, K.U.; Gracy, R.W. Relationship between the catalytic center and the primary degradation site of triosephosphate isomerase: Effects of active site modification and deamidation. Arch. Biochem. Biophys. 1992, 293, 382–390. [Google Scholar] [CrossRef]
- Decker, R.S.; Mohrenweiser, H.W. Cell proliferation-associated expression of a recently evolved isozyme of triosephosphate isomerase. Biochem. Genet. 1985, 23, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matafome, P.; Rodrigues, T.; Sena, C.; Seiça, R. Methylglyoxal in Metabolic Disorders: Facts, Myths, and Promises. Med. Res. Rev. 2017, 37, 368–403. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Flores, S.; Rodriguez-Romero, A.; Hernandez-Alcantara, G.; De la Mora-De la Mora, I.; Gutierrez-Castrellon, P.; Carvajal, K.; Lopez-Velazquez, G.; Reyes-Vivas, H. Species-specific inhibition of Giardia lamblia triosephosphate isomerase by localized perturbation of the homodimer. Mol. Biochem. Parasitol. 2008, 157, 179–186. [Google Scholar] [CrossRef]
- Yuan, P.M.; Talent, J.M.; Gracy, R.W. Molecular basis for the accumulation of acidic isozymes of triosephosphate isomerase on aging. Mech. Ageing Dev. 1981, 17, 151–162. [Google Scholar] [CrossRef]
- Tollefsbol, T.O.; Cohen, H.J. The effect of age on the accumulation of labile triosephosphate isomerase and thymidine incorporation in pokeweed mitogen stimulated human lymphocytes. J. Gerontol. 1984, 39, 398–405. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; Souza, G.; Muraro, S.; Carregari, V.; Biagi, C.; Crunfli, F.; Restrepo, J.; Vendramini, P.; et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis dependent axis. Cell Metab. 2020. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Plaut, B.; Knowles, J.R. pH-dependence of the triose phosphate isomerase reaction. Biochem. J. 1972, 129, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellman, G.L. A colorimetric method for determining low concentrations of mercaptans. Arch. Biochem. Biophys. 1958, 74, 443–450. [Google Scholar] [CrossRef]
- Cabrera, N.; Torres-Larios, A.; García-Torres, I.; Enríquez-Flores, S.; Perez-Montfort, R. Differential effects on enzyme stability and kinetic parameters of mutants related to human triosephosphate isomerase deficiency. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1401–1409. [Google Scholar] [CrossRef]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Guix, F.X.; Clarimón, J.; Miscione, G.P.; Boada, M.; et al. Methylglyoxal produced by amyloid-β peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.P.; Brandt, R.B. Spectrophotometric determination of methyl glyoxal with 2,4-dinitrophenylhydrazine. Anal. Chem. 1975, 47, 2418–2422. [Google Scholar] [CrossRef]
- López-Velázquez, G.; Fernández-Lainez, C.; de la Mora-de la Mora, J.I.; Caudillo de la Portilla, D.; Reynoso-Robles, R.; González-Maciel, A.; Ridaura, C.; García-Torres, I.; Gutiérrez-Castrellón, P.; Olivos-García, A.; et al. On the molecular and cellular effects of omeprazole to further support its effectiveness as an antigiardial drug. Sci Rep. 2019, 9, 8922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalapos, M.P. Methylglyoxal in living organisms: Chemistry, biochemistry, toxicology and biological implications. Toxicol. Lett. 1999, 110, 145–175. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Pravda, L.; Sehnal, D.; Toušek, D.; Navrátilová, V.; Bazgier, V.; Berka, K.; Svobodová Vareková, R.; Koca, J.; Otyepka, M. MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef] [Green Version]
- Gkogkolou, P.; Böhm, M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinol 2012, 4, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kold-Christensen, R.; Johannsen, M. Methylglyoxal Metabolism and Aging-Related Disease: Moving from Correlation toward Causation. Trends Endocrinol. Metab. 2020, 31, 81–92. [Google Scholar] [CrossRef]
- Reyes-Vivas, H.; de la Mora-de la Mora, I.; Castillo-Villanueva, A.; Yépez-Mulia, L.; Hernández-Alcántara, G.; Figueroa-Salazar, R.; García-Torres, I.; Gómez-Manzo, S.; Méndez, S.T.; Vanoye-Carlo, A.; et al. Giardial triosephosphate isomerase as possible target of the cytotoxic effect of omeprazole in Giardia lamblia. Antimicrob. Agents Chemother. 2014, 58, 7072–7082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainfroid, V.; Terpstra, P.; Beauregard, M.; Frère, J.M.; Mande, S.C.; Hol, W.G.; Martial, J.A.; Goraj, K. Three hTIM mutants that provide new insights on why TIM is a dimer. J. Mol. Biol. 1996, 257, 441–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Almazán, C.; Arreola, R.; Rodríguez-Larrea, D.; Aguirre-López, B.; de Gómez-Puyou, M.T.; Pérez-Montfort, R.; Costas, M.; Gómez-Puyou, A.; Torres-Larios, A. Structural basis of human triosephosphate isomerase deficiency: Mutation E104D is related to alterations of a conserved water network at the dimer interface. J. Biol. Chem. 2008, 283, 23254–23263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Mora-De La Mora, I.; Torres-Larios, A.; Mendoza-Hernández, G.; Enriquez-Flores, S.; Castillo-Villanueva, A.; Mendez, S.T.; Garcia-Torres, I.; Torres-Arroyo, A.; Gómez-Manzo, S.; Marcial-Quino, J.; et al. The E104D mutation increases the susceptibility of human triosephosphate isomerase to proteolysis. Asymmetric cleavage of the two monomers of the homodimeric enzyme. Biochim. Biophys. Acta 2013, 1834, 2702–2711. [Google Scholar] [CrossRef]
- Maithal, K.; Ravindra, G.; Balaram, H.; Balaram, P. Inhibition of plasmodium falciparum triose-phosphate isomerase by chemical modification of an interface cysteine. Electrospray ionization mass spectrometric analysis of differential cysteine reactivities. J. Biol. Chem. 2002, 277, 25106–25114. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Romero, A.; Hernández-Santoyo, A.; del Pozo Yauner, L.; Kornhauser, A.; Fernández-Velasco, D.A. Structure and inactivation of triosephosphate isomerase from Entamoeba histolytica. J. Mol. Biol. 2002, 322, 669–675. [Google Scholar] [CrossRef]
- Enríquez-Flores, S.; Rodríguez-Romero, A.; Hernández-Alcántara, G.; Oria-Hernández, J.; Gutiérrez-Castrellón, P.; Pérez-Hernández, G.; de la Mora-de la Mora, I.; Castillo-Villanueva, A.; García-Torres, I.; Méndez, S.T.; et al. Determining the molecular mechanism of inactivation by chemical modification of triosephosphate isomerase from the human parasite Giardia lamblia: A study for antiparasitic drug design. Proteins 2011, 79, 2711–2724. [Google Scholar] [CrossRef]
- Pérez-Montfort, R.; Garza-Ramos, G.; Alcántara, G.H.; Reyes-Vivas, H.; Gao, X.G.; Maldonado, E.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Derivatization of the interface cysteine of triosephosphate isomerase from Trypanosoma brucei and Trypanosoma cruzi as probe of the interrelationship between the catalytic sites and the dimer interface. Biochemistry 1999, 38, 4114–4120. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcántara, G.; Oria-Hernández, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulby, A.J.; Heal, J.W.; Barrow, M.P.; Roemer, R.A.; O’Connor, P.B. Does deamidation cause protein unfolding? A top-down tandem mass spectrometry study. Protein Sci. 2015, 24, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romo-Mancillas, A.; Téllez-Valencia, A.; Yépez-Mulia, L.; Hernández-Luis, F.; Hernández-Campos, A.; Castillo, R. The design and inhibitory profile of new benzimidazole derivatives against triosephosphate isomerase from Trypanosoma cruzi: A problem of residue motility. J. Mol. Graph. Model. 2011, 30, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Cheng, L.S.; Amaro, R.E.; Xu, D.; Li, W.W.; Arzberger, P.W.; McCammon, J.A. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J. Med. Chem. 2008, 51, 3878–3894. [Google Scholar] [CrossRef] [Green Version]
- Le, L.; Lee, E.H.; Hardy, D.J.; Truong, T.N.; Schulten, K. Molecular dynamics simulations suggest that electrostatic funnel directs binding of Tamiflu to influenza N1 neuraminidases. PLoS Comput. Biol. 2010, 6, e1000939. [Google Scholar] [CrossRef] [Green Version]
- Vrouenraets, M.; Wierenga, J.; Meijberg, W.; Miedema, H. Chemical modification of the bacterial porin OmpF: Gain of selectivity by volume reduction. Biophys. J. 2006, 90, 1202–1211. [Google Scholar] [CrossRef] [Green Version]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat Commun. 2014, 5, 4740. [Google Scholar] [CrossRef] [Green Version]
- Garza-Ramos, G.; Cabrera, N.; Saavedra-Lira, E.; Tuena de Gómez-Puyou, M.; Ostoa-Saloma, P.; Pérez-Montfort, R.; Gómez-Puyou, A. Sulfhydryl reagent susceptibility in proteins with high sequence similarity–triosephosphate isomerase from Trypanosoma brucei, Trypanosoma cruzi and Leishmania mexicana. Eur. J. Biochem. 1998, 253, 684–691. [Google Scholar] [CrossRef]
- Moraes, J.; Arreola, R.; Cabrera, N.; Saramago, L.; Freitas, D.; Masuda, A.; da Silva Vaz, I., Jr.; Tuena de Gomez-Puyou, M.; Perez-Montfort, R.; Gomez-Puyou, A.; et al. Structural and biochemical characterization of a recombinant triosephosphate isomerase from Rhipicephalus (Boophilus) microplus. Insect. Biochem. Mol. Biol. 2011, 41, 400–409. [Google Scholar] [CrossRef]
- García-Torres, I.; De la Mora-De la Mora, I.; Hernández-Alcántara, G.; Molina-Ortiz, D.; Caballero-Salazar, S.; Olivos-García, A.; Nava, G.; López-Velázquez, G.; Enríquez-Flores, S. First characterization of a microsporidial triosephosphate isomerase and the biochemical mechanisms of its inactivation to propose a new druggable target. Sci. Rep. 2018, 8, 8591. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.J.; Durani, V.; Magliery, T.J. Triosephosphate isomerase by consensus design: Dramatic differences in physical properties and activity of related variants. J. Mol. Biol. 2011, 413, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Orosz, F.; Oláh, J.; Ovádi, J. Triosephosphate isomerase deficiency: New insights into an enigmatic disease. Biochim. Biophys. Acta 2009, 1792, 1168–1174. [Google Scholar] [CrossRef] [Green Version]
- Al-Motawa, M.; Abbas, H.; Wijten, P.; de la Fuente, A.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 virus to proteotoxicity—opportunity for repurposed chemotherapy of COVID-19 infection. bioRxiv 2020. [Google Scholar] [CrossRef]


 ) and +5.0 (
) and +5.0 (  ). Figures were modeled with the molecular graphics images produced with the UCSF Chimera package [18].
) and +5.0 ( ). Figures were modeled with the molecular graphics images produced with the UCSF Chimera package [18].
). Figures were modeled with the molecular graphics images produced with the UCSF Chimera package [18].
) and +5.0 ( ). Figures were modeled with the molecular graphics images produced with the UCSF Chimera package [18].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Thiol-Reactive Compound | Free Cys/Subunit | Derivatized Cys/Subunit |
|---|---|---|---|
| WT | Control * | 5.1 ± 0.2 | 0 |
| + MMTS | 3.9 ± 0.3 | 1 | |
| + MTSES | 4.1 ± 0.2 | 1 | |
| + DTNB | 3.8 ± 0.3 | 1 | |
| N16D | Control * | 4.8 ± 0.4 | 0 |
| + MMTS | 0.9 ± 0.3 | 4 | |
| + MTSES | 1.1 ± 0.4 | 4 | |
| + DTNB | 0.8 ± 0.3 | 4 |
| E. coli Δtim Cells Complemented with HsTIM | Condition | Enzyme Activity (%) | Enzyme Activity (µmol/min mg) |
|---|---|---|---|
| WT | Control * | 100 | 165 ± 11 |
| + Omeprazole | 96 ± 4 | 158 ± 4 | |
| N16D | Control * | 100 | 3.13 ± 0.045 |
| + Omeprazole | 1.95 ± 0.7 | 0.061 ± 0.023 |
| E. coli Δtim Cells Complemented with HsTIM | Condition | MGO (nmol/mL) | MGO (%) |
|---|---|---|---|
| WT | Control * | 580 | 100 |
| + Omeprazole | 694 ± 42 | 119 ± 6 ** | |
| N16D | Control * | 1506 ± 76 | 259 ± 5 ** |
| + Omeprazole | 2617 ± 183 | 451 ± 7 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enríquez-Flores, S.; Flores-López, L.A.; García-Torres, I.; de la Mora-de la Mora, I.; Cabrera, N.; Gutiérrez-Castrellón, P.; Martínez-Pérez, Y.; López-Velázquez, G. Deamidated Human Triosephosphate Isomerase is a Promising Druggable Target. Biomolecules 2020, 10, 1050. https://doi.org/10.3390/biom10071050
Enríquez-Flores S, Flores-López LA, García-Torres I, de la Mora-de la Mora I, Cabrera N, Gutiérrez-Castrellón P, Martínez-Pérez Y, López-Velázquez G. Deamidated Human Triosephosphate Isomerase is a Promising Druggable Target. Biomolecules. 2020; 10(7):1050. https://doi.org/10.3390/biom10071050
Chicago/Turabian StyleEnríquez-Flores, Sergio, Luis Antonio Flores-López, Itzhel García-Torres, Ignacio de la Mora-de la Mora, Nallely Cabrera, Pedro Gutiérrez-Castrellón, Yoalli Martínez-Pérez, and Gabriel López-Velázquez. 2020. "Deamidated Human Triosephosphate Isomerase is a Promising Druggable Target" Biomolecules 10, no. 7: 1050. https://doi.org/10.3390/biom10071050