
Characterization of Tumor-Associated Macrophages and the Immune Microenvironment in Limited-Stage Neuroendocrine-High and -Low Small Cell Lung Cancer

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Tissue Processing
2.3. Immunohistochemistry and Immunofluorescence
2.4. Cell Counting and Morphometry
2.5. Cut-Off Values and Tumor Classification
2.6. Molecular Analysis
2.7. Data Pre-Processing and fGSEA Pathway Analysis
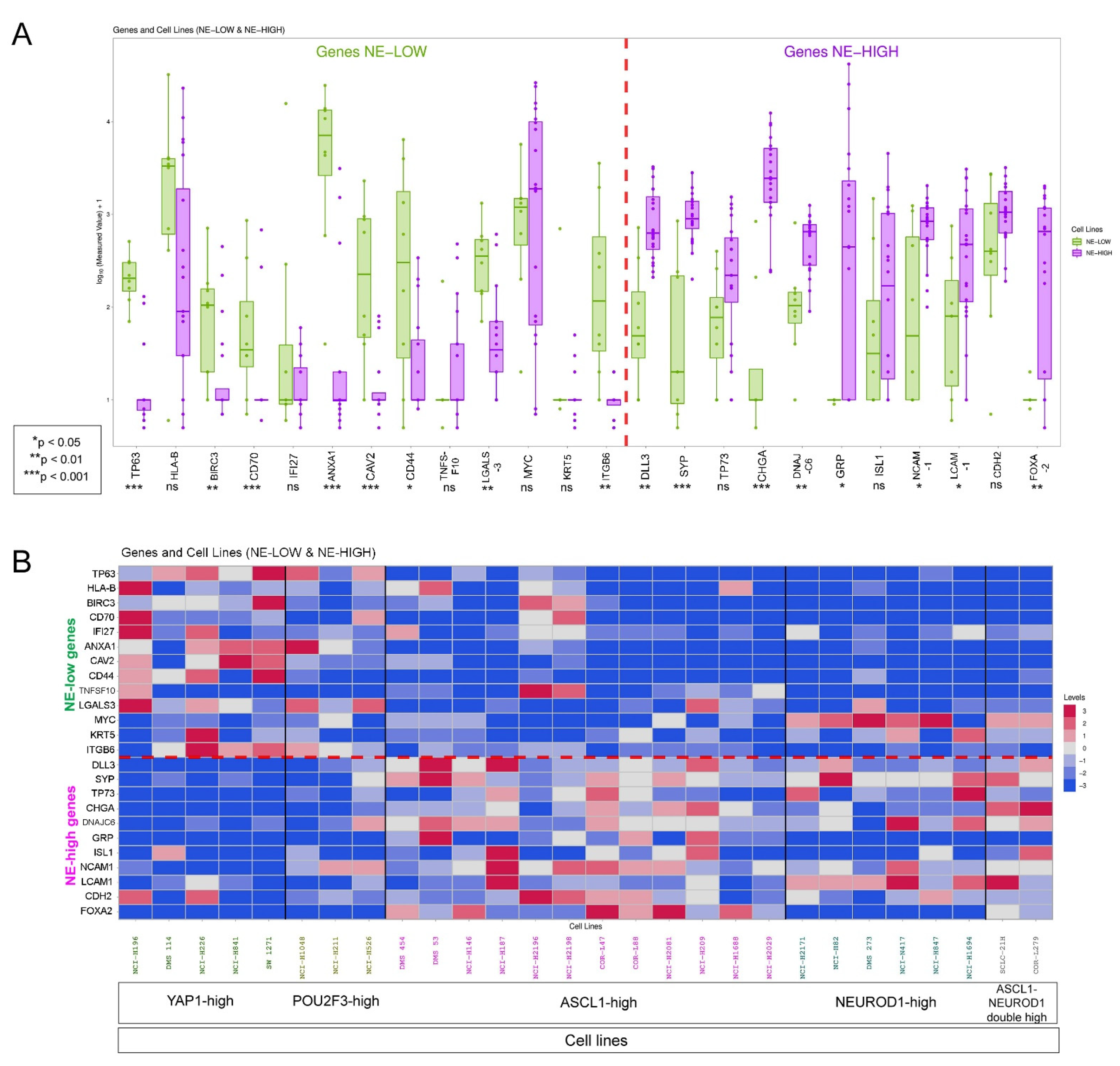
2.8. Cell Line Data
2.9. Statistical Methods
3. Results
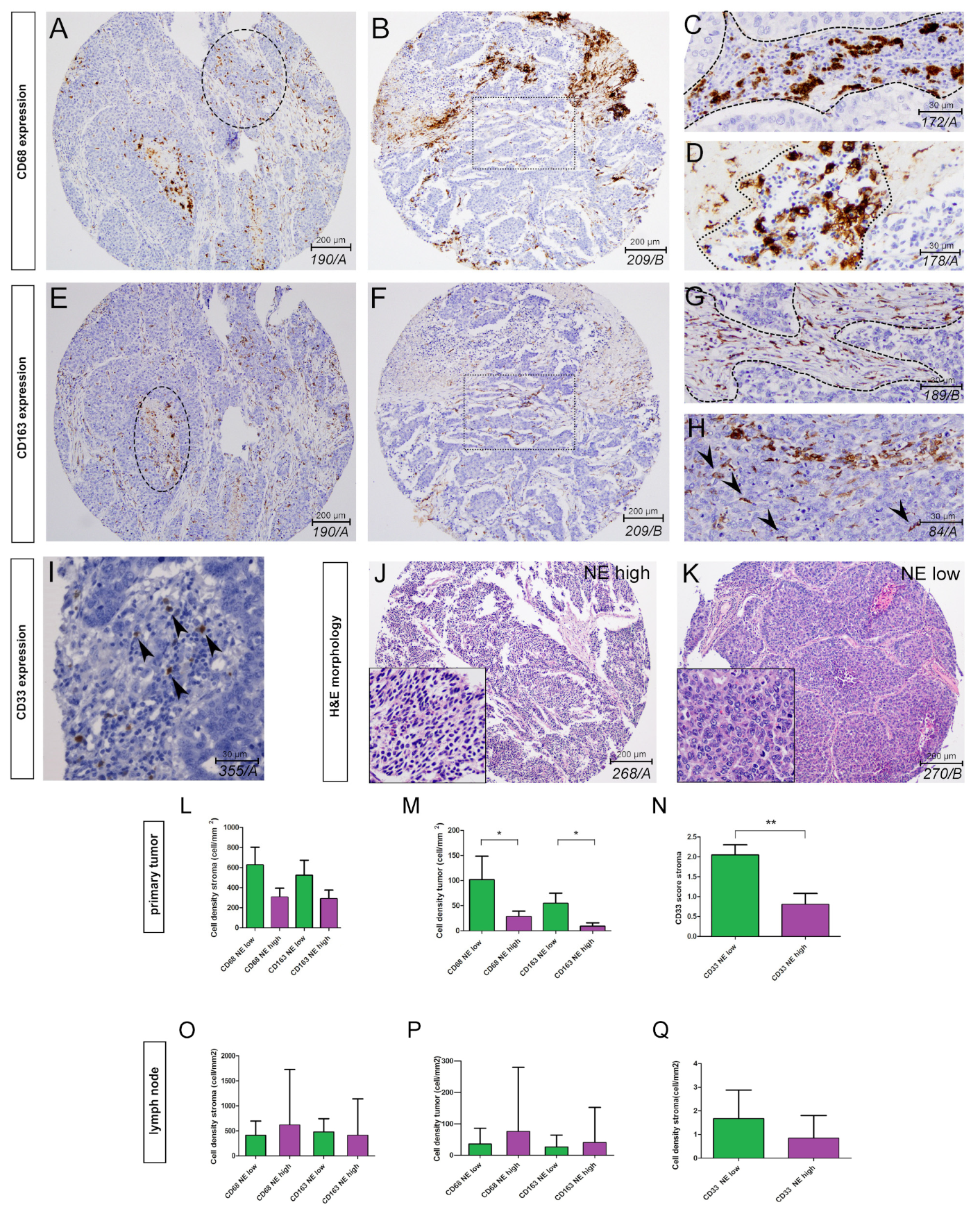
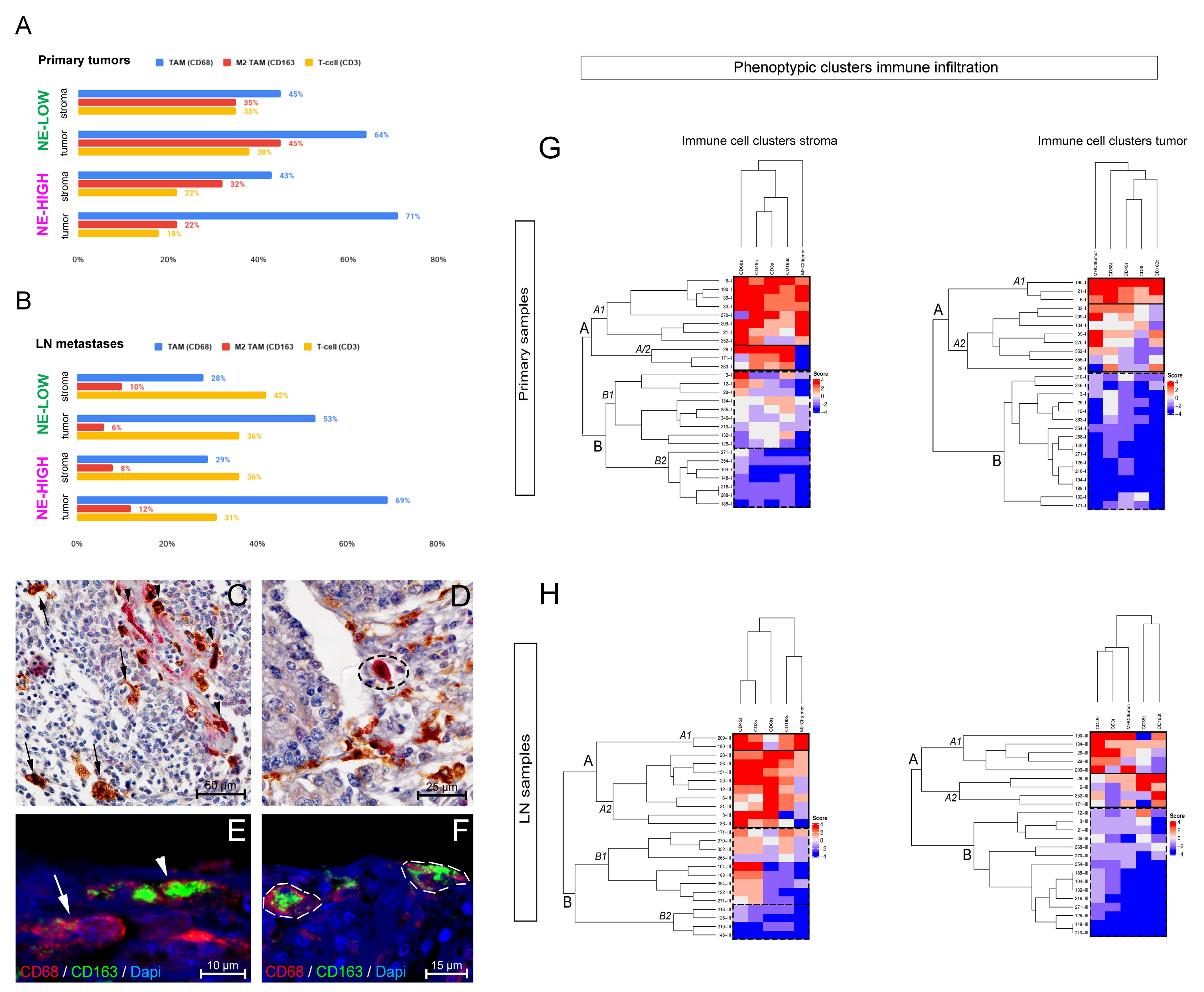
3.1. The Phenotype of Tumor-Associated Macrophages and Their Tissue Distribution According to SCLC NE Subtypes
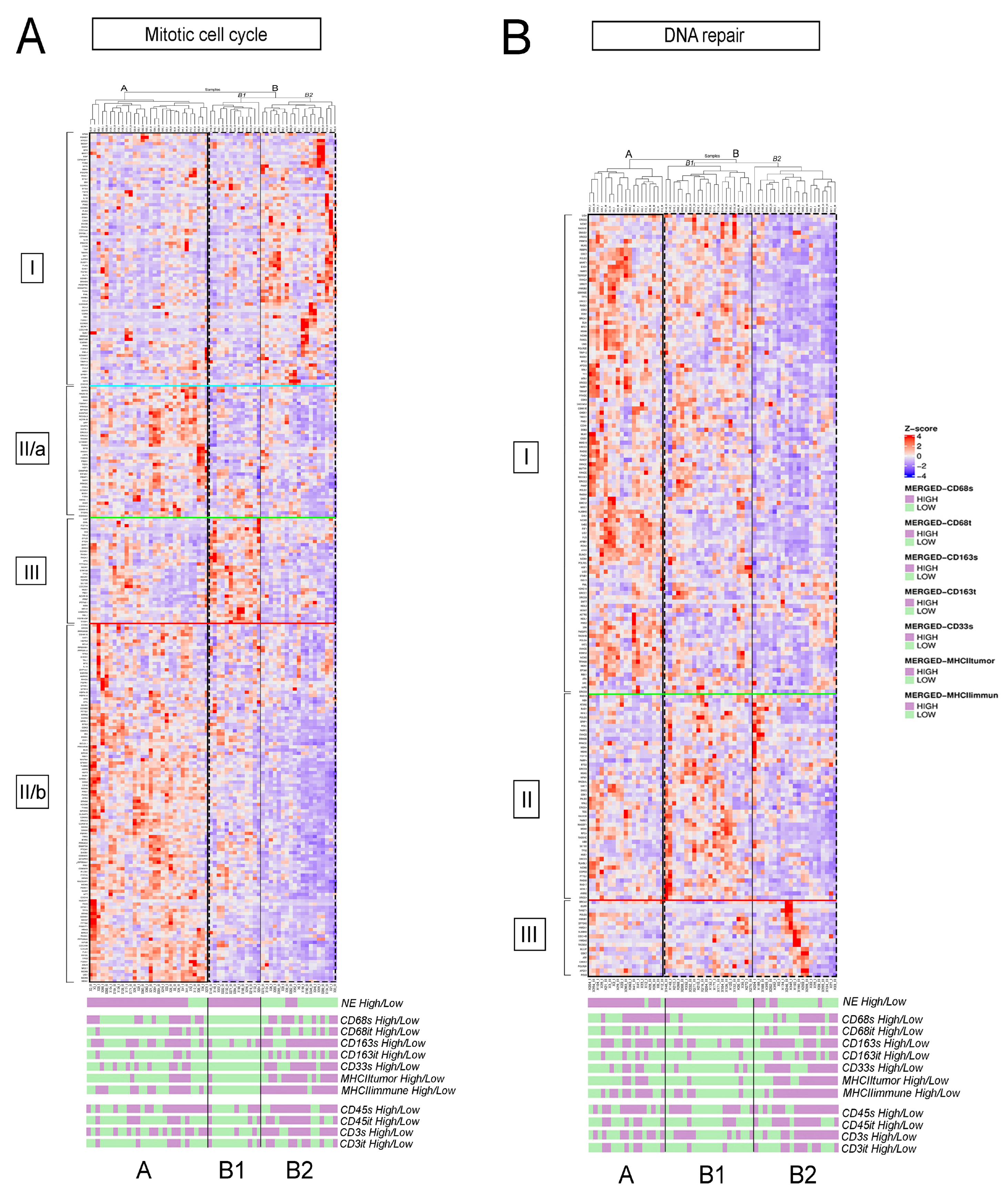
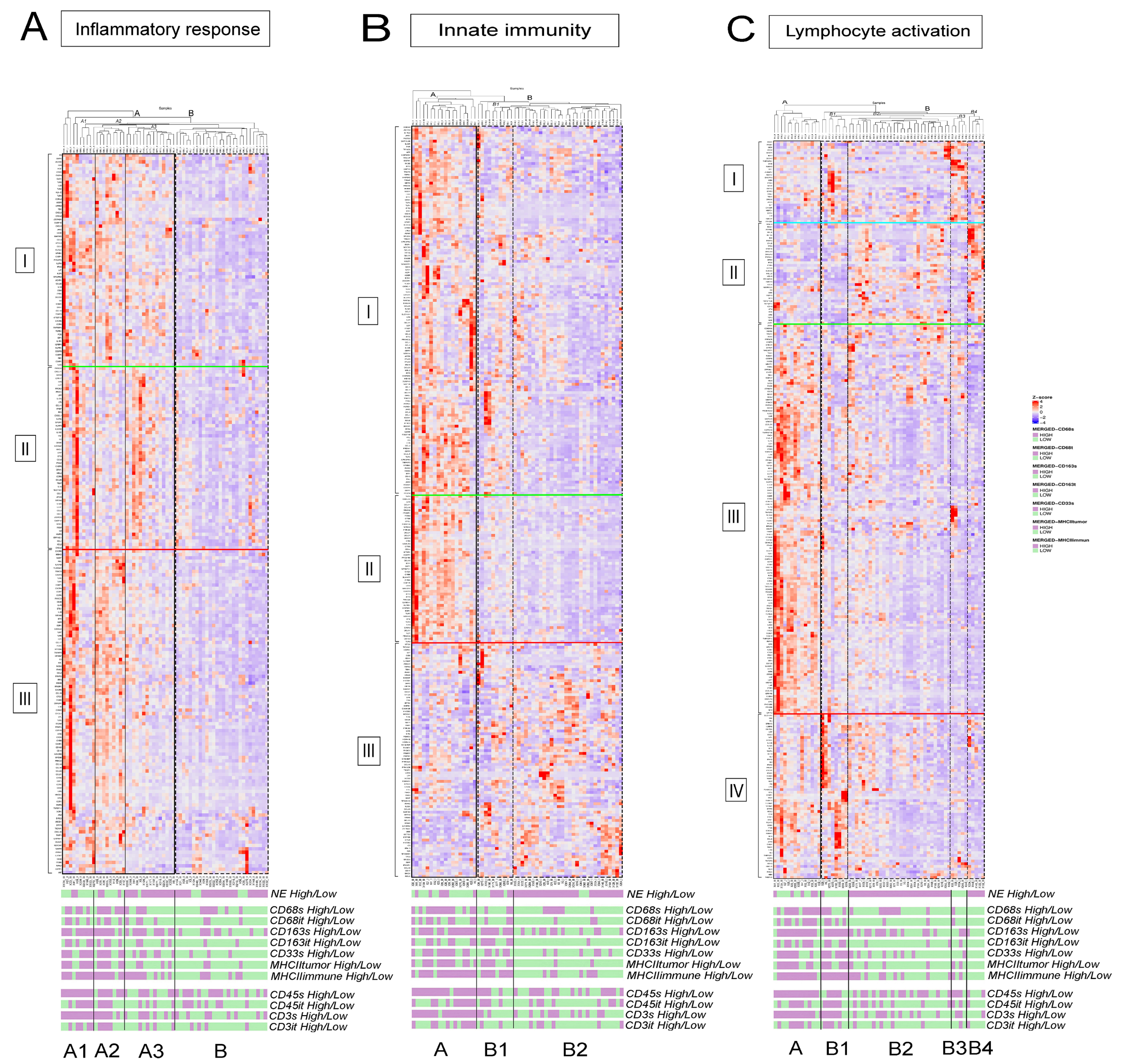
3.2. fGSEA Pathway Analysis in Different Subsets of Tumors According to NE Phenotype and Macrophage-Infiltration
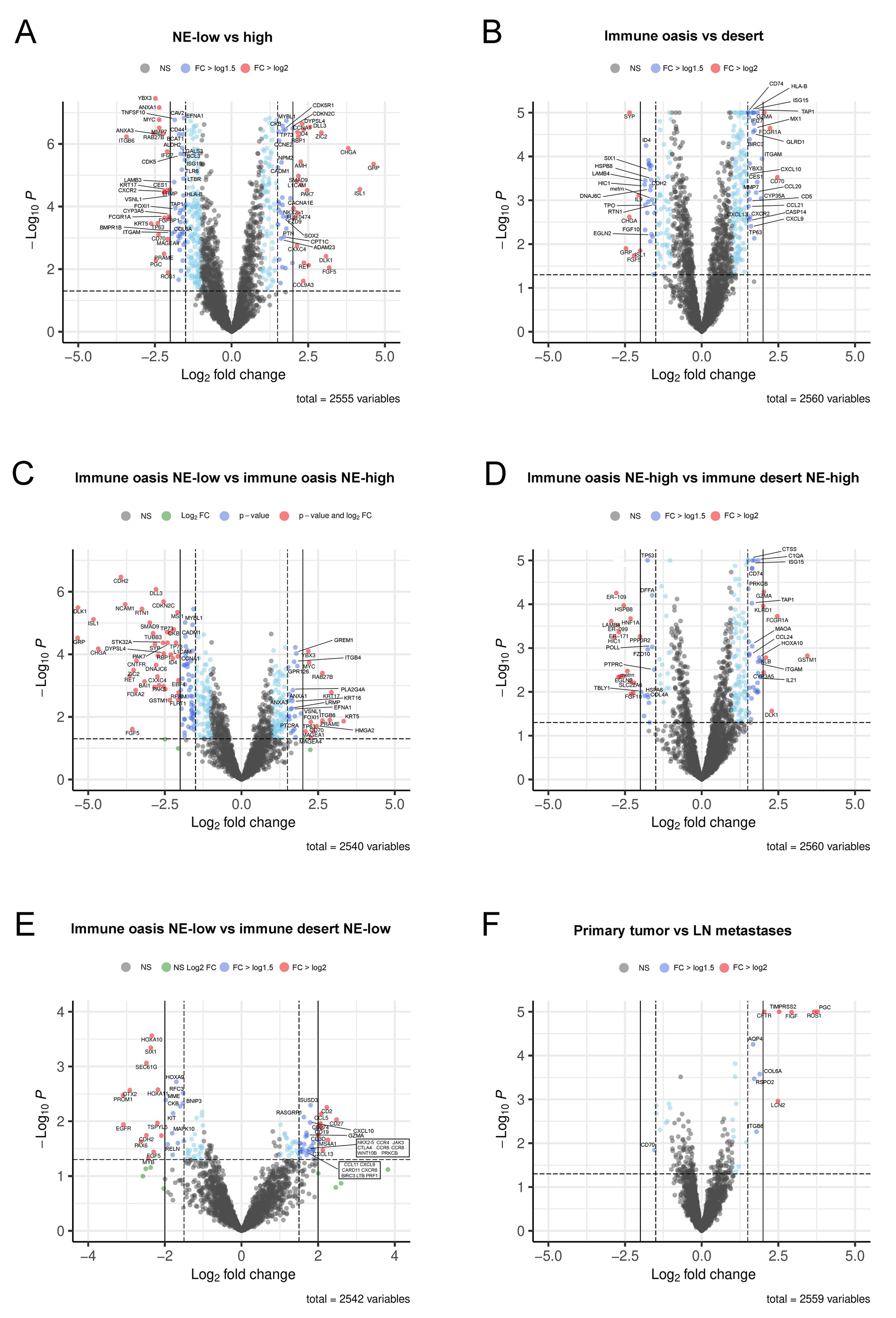
3.3. Detailed Analysis of Representative “Oncogenic” and “Immune Response” Pathways Related to the Gene Ontology (GO) System
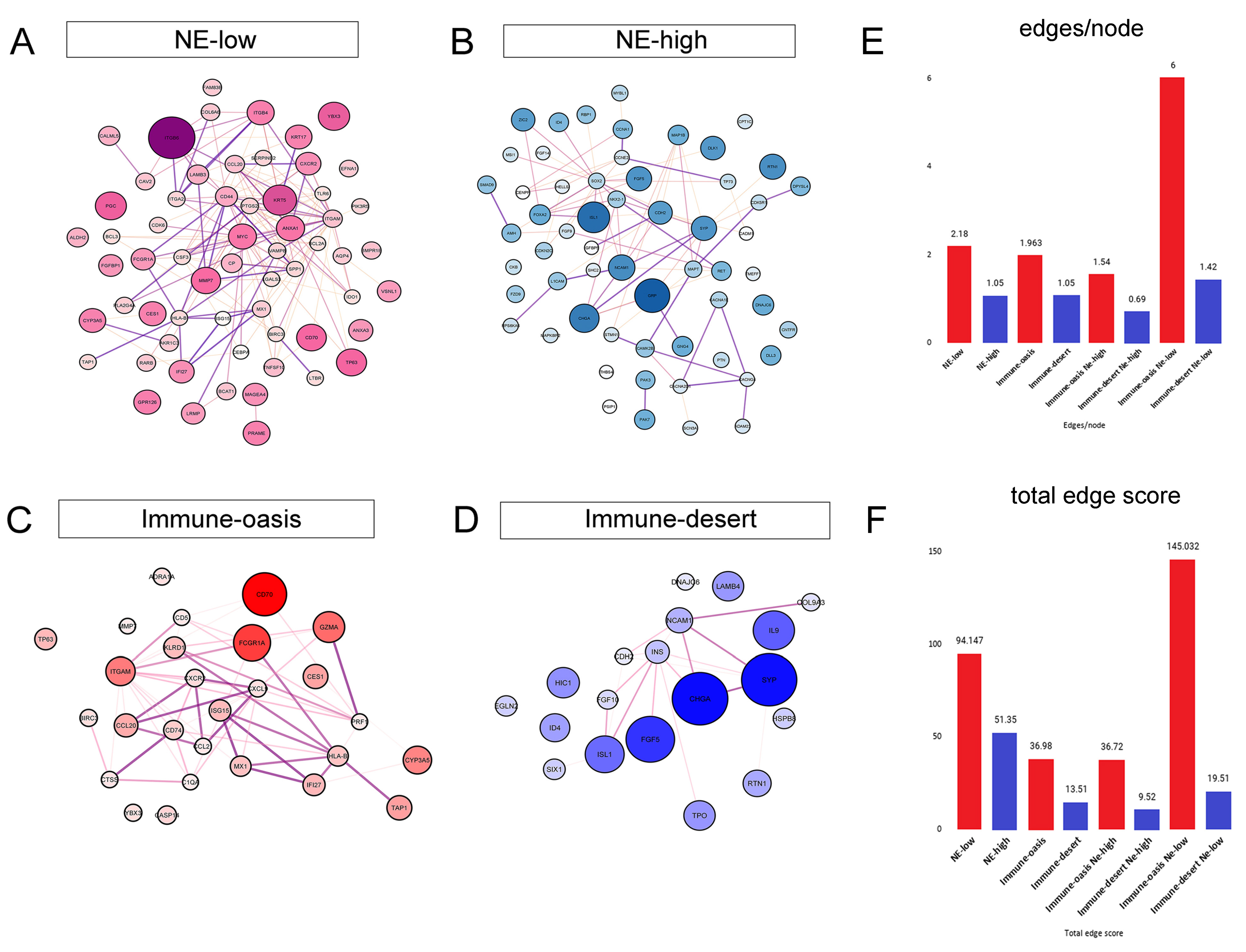
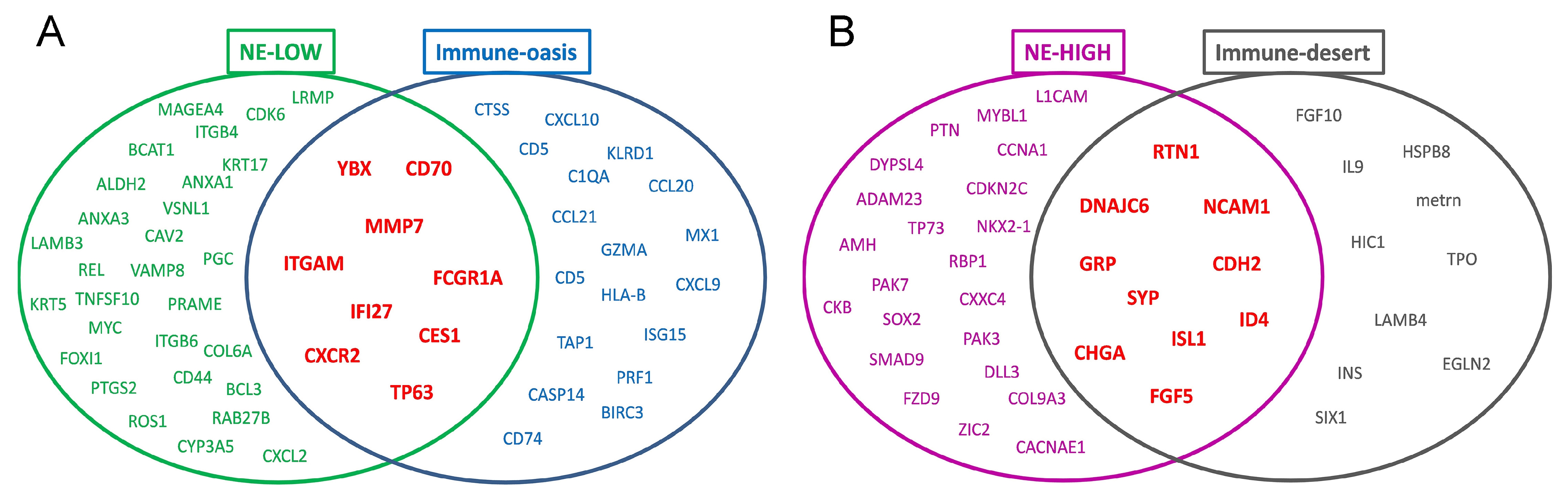
3.4. Molecular Targets and Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Horn, L.; Mansfield, A.S.; Szczesna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Sørensen, M.; Pijls-Johannesma, M.; Felip, E. ESMO Guidelines Working Group. Small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21, v120–v125. [Google Scholar] [CrossRef]
- Lohinai, Z.; Megyesfalvi, Z.; Suda, K.; Harko, T.; Ren, S.; Moldvay, J.; Laszlo, V.; Rivard, C.; Dome, B.; Hirsch, F.R. Comparative expression analysis in small cell lung carcinoma reveals neuroendocrine pattern change in primary tumor versus lymph node metastases. Transl. Lung Cancer Res. 2020, 8, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Molecular Phenotypes of SCLC. In Proceedings of the International Association for the Study of Lung Cancer-19th World Conference on Lung Cancer, Toronto, ON, Canada, 23–26 September 2018. [Google Scholar]
- Saito, M.; Shiraishi, K.; Goto, A.; Suzuki, H.; Kohno, T.; Kono, K. Development of targeted therapy and immunotherapy for the treatment of small cell lung cancer. Jpn. J. Clin. Oncol. 2018, 48, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef] [Green Version]
- Dora, D.; Rivard, C.; Yu, H.; Bunn, P.; Suda, K.; Ren, S.; Pickard, S.L.; Laszlo, V.; Harko, T.; Megyesfalvi, Z.; et al. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol. Oncol. 2020, 14, 1947–1965. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Knelson, E.H.; Wolff, J.O.; Vajdi, A.; Saigi, M.; Campisi, M.; Hong, D.; Thai, T.C.; Piel, B.; Han, S.; et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov. 2021, candisc.0913.2020. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N.; Chanmee, T. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Binmadi, N.O.; Yang, Y.-H.; Proia, P.; Basile, J.R. Semaphorin 4D cooperates with VEGF to promote angiogenesis and tumor progression. Angiogenesis 2012, 15, 391–407. [Google Scholar] [CrossRef]
- Astekar, M.; Metgud, R.; Sharma, A.; Soni, A. Hidden keys in stroma: Unlocking the tumor progression. J. Oral Maxillofac. Pathol. 2013, 17, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.L.; Rios, E.; Durães, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.M.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’Ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Barbera-Guillem, E.; Nyhus, J.K.; Wolford, C.C.; Friece, C.R.; Sampsel, J.W. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res. 2002, 62, 7042–7049. [Google Scholar] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neuro Oncol. 2015, 122, 293–301. [Google Scholar] [CrossRef]
- Porembka, M.; Mitchem, J.; Belt, B.A.; Hsieh, C.-S.; Lee, H.-M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Safi, S.; Blattner, C.; Rathinasamy, A.; Umansky, L.; Juenger, S.; Warth, A.; Eichhorn, M.; Muley, T.; Herth, F.J.F.; et al. Circulating and Tumor Myeloid-derived Suppressor Cells in Resectable Non-Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 777–787. [Google Scholar] [CrossRef]
- Battifora, H. Methods in laboratory investigation. The multitumor (sausage) tissue block: Novel method for immunohistochemical antibody testing. Lab. Investig. 1986, 55, 244–248. [Google Scholar]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; Dezonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Evans, J.D. Straightforward Statistics for the Behavioral Sciences; Thomson Brooks/Cole Publishing, Co.: Three Lakes, WI, USA, 1996. [Google Scholar]
- Lin, A.; Schildknecht, A.; Nguyen, L.T.; Ohashi, P.S. Dendritic cells integrate signals from the tumor microenvironment to modulate immunity and tumor growth. Immunol. Lett. 2010, 127, 77–84. [Google Scholar] [CrossRef]
- Chen, J.J.; Lin, Y.-C.; Yao, P.-L.; Yuan, A.; Chen, H.-Y.; Shun, C.-T.; Tsai, M.-F.; Chen, C.-H.; Yang, P.-C. Tumor-associated macrophages: The double-edged sword in cancer progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Dhabekar, G.S.; Dandekar, R.C.; Kingaonkar, A.V. Role of macrophages in malignancy. Ann. Maxillofac. Surg. 2011, 1, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dötzer, K.; Schlüter, F.; Schoenberg, M.B.; Bazhin, A.V.; Von Koch, F.E.; Schnelzer, A.; Anthuber, S.; Grab, D.; Czogalla, B.; Burges, A.; et al. Immune Heterogeneity Between Primary Tumors and Corresponding Metastatic Lesions and Response to Platinum Therapy in Primary Ovarian Cancer. Cancers 2019, 11, 1250. [Google Scholar] [CrossRef] [Green Version]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.; Patwardhan, G.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Park, I.A.; Hwang, S.-H.; Song, I.H.; Heo, S.-H.; Kim, Y.-A.; Bang, W.S.; Park, H.S.; Lee, M.; Gong, G.; Lee, H.J. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. PLoS ONE 2017, 12, e0182786. [Google Scholar] [CrossRef]
- Mortara, L.; Castellani, P.; Meazza, R.; Tosi, G.; Barbaro, A.D.L.; Procopio, F.A.; Comes, A.; Zardi, L.; Ferrini, S.; Accolla, R. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin. Cancer Res. 2006, 12, 3435–3443. [Google Scholar] [CrossRef] [Green Version]
- Leoni, G.; Neumann, P.-A.; Kamaly, N.; Quiros, M.; Nishio, H.; Jones, H.R.; Sumagin, R.; Hilgarth, R.S.; Alam, A.; Fredman, G.; et al. Annexin A1′containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J. Clin. Investig. 2015, 125, 1215–1227. [Google Scholar] [CrossRef] [Green Version]
- Boudhraa, Z.; Rondepierre, F.; Ouchchane, L.; Kintossou, R.; Trzeciakiewicz, A.; Franck, F.; Kanitakis, J.; Labeille, B.; Joubert-Zakeyh, J.; Bouchon, B.; et al. Annexin A1 in primary tumors promotes melanoma dissemination. Clin. Exp. Metastasis 2014, 31, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lin, G.; Fang, W.; Zhu, H.; Chu, K. Increased expression of annexin A1 predicts poor prognosis in human hepatocellular carcinoma and enhances cell malignant phenotype. Med. Oncol. 2014, 31, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-Y.; Wu, M.-S.; Lin, J.-T.; Lin, M.-T.; Shun, C.-T.; Huang, H.-Y.; Hua, K.-T.; Kuo, M.-L. Annexin A1 is associated with gastric cancer survival and promotes gastric cancer cell invasiveness through the formyl peptide receptor/extracellular signal-regulated kinase/integrin beta-1-binding protein 1 pathway. Cancer 2012, 118, 5757–5767. [Google Scholar] [CrossRef] [PubMed]
- Biaoxue, R.; Xiling, J.; Shuanying, Y.; Wei, Z.; Xiguang, C.; Jinsui, W.; Min, Z. Upregulation of Hsp90-beta and annexin A1 correlates with poor survival and lymphatic metastasis in lung cancer patients. J. Exp. Clin. Cancer Res. 2012, 31, 70. [Google Scholar] [CrossRef] [Green Version]
- Sheu, M.-J.; Li, C.-F.; Lin, C.-Y.; Lee, S.-W.; Lin, L.-C.; Chen, T.-J.; Ma, L.-J. Overexpression of ANXA1 confers independent negative prognostic impact in rectal cancers receiving concurrent chemoradiotherapy. Tumor Biol. 2014, 35, 7755–7763. [Google Scholar] [CrossRef]
- De Marchi, T.; Timmermans, A.M.; Smid, M.; Look, M.P.; Stingl, C.; Opdam, M.; Linn, S.C.; Sweep, F.; Span, P.N.; Kliffen, M.; et al. Annexin-A1 and caldesmon are associated with resistance to tamoxifen in estrogen receptor positive recurrent breast cancer. Oncotarget 2015, 7, 3098–3110. [Google Scholar] [CrossRef] [Green Version]
- de Graauw, M.; van Miltenburg, M.H.; Schmidt, M.; Pont, C.; Lalai, R.; Kartopawiro, J.; Pardali, E.; Le Devedec, S.E.; Smit, V.T.; van der Wal, A.; et al. Annexin A1 regulates TGF-beta signaling and promotes metastasis formation of basal-like breast cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6340–6345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bist, P.; Leow, S.C.; Phua, Q.H.; Shu, S.; Zhuang, Q.; Loh, W.T.; Nguyen, T.H.; Zhou, J.B.; Hooi, S.C.; Lim, L. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-B: Implication in breast cancer metastasis. Oncogene 2011, 30, 3174–3185. [Google Scholar] [CrossRef] [Green Version]
- Duggleby, R.C.; Shaw, T.N.F.; Jarvis, L.B.; Kaur, G.; Gaston, J.S.H. CD27 expression discriminates between regulatory and non-regulatory cells after expansion of human peripheral blood CD4+ CD25+ cells. Immunology 2007, 121, 129–139. [Google Scholar] [CrossRef]
- Nolte, A.; van Olffen, R.W.; van Gisbergen, K.; van Lier, R. Timing and tuning of CD27–CD70 interactions: The impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol. Rev. 2009, 229, 15. [Google Scholar] [CrossRef] [PubMed]
- Law, C.L.; Gordon, K.A.; Toki, B.E.; Yamane, A.K.; Hering, M.A.; Cerveny, C.G.; Petroziello, J.M.; Ryan, M.C.; Smith, L.; Simon, R.; et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. 2006, 66, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Z.; Novak, A.J.; Ziesmer, S.C.; Witzig, T.E.; Ansell, S.M. CD70+ non-Hodgkin lymphoma B cells induce Foxp3 expression and regulatory function in intratumoral CD4+CD25 T cells. Blood 2007, 110, 2537–2544. [Google Scholar] [CrossRef] [Green Version]
- Claus, C.; Riether, C.; Schürch, C.; Matter, M.; Hilmenyuk, T.; Ochsenbein, A.F. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 2012, 72, 3664–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.; Zwaenepoel, K.; Rolfo, C.; Bossche, J.V.D.; Deben, C.; Silence, K.; Hermans, C.; Smits, E.; Van Schil, P.; Lardon, F.; et al. Unlocking the potential of CD70 as a novel immunotherapeutic target for non-small cell lung cancer. Oncotarget 2015, 6, 13462–13475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Groom, J.R.; Richmond, J.; Murooka, T.T.; Sorensen, E.W.; Sung, J.H.; Bankert, K.; von Andrian, U.H.; Moon, J.J.; Mempel, T.R.; Luster, A.D. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 2012, 37, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, A.; Gómez, L.; Lustgarten, J.; Mira, E.; Mañes, S.; Rechsteiner, M.P.; Von Teichman, A.; Nowicka, A.; Sulser, T.; Schraml, P.; et al. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4+ and CD8+ T cells. Cancer Res. 2011, 71, 5455–5466. [Google Scholar] [CrossRef] [Green Version]
- Hoves, S.; Sutton, V.R.; Trapani, J.A. A novel role for granzymes in anti-tumor immunity. OncoImmunology 2012, 1, 219–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, T.; Li, X.; Huang, J.; Wu, B.; Huang, X.; Zhou, Y.; Zhu, J.; Hou, J. Predictive value of MMP-7 expression for response to chemotherapy and survival in patients with non-small cell lung cancer. Cancer Sci. 2008, 99, 2185–2192. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Keppler, M.D.; Jazayeri, M.; Thomas, G.J.; Parsons, M.; Violette, S.; Weinreb, P.; Hart, I.R.; Marshall, J.F. HS1-associated protein X-1 regulates carcinoma cell migration and invasion via clathrin-mediated endocytosis of integrin alphavbeta6. Cancer Res. 2007, 67, 5275–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A.; Schwarzl, T.; Huppertz, I.; Kramer, G.; Mantas, P.; Alleaume, A.-M.; Huber, W.; Krijgsveld, J.; Hentze, M.W. The RNA-Binding Protein YBX3 Controls Amino Acid Levels by Regulating SLC mRNA Abundance. Cell Rep. 2019, 27, 3097–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Lu, X.; Chen, X.; Zou, Y.; Liu, A.; Li, W.; He, B.; He, S.; Chen, Q. Eight potential biomarkers for distinguishing between lung adenocarcinoma and squamous cell carcinoma. Oncotarget 2017, 8, 71759–71771. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.-H.; Chang, Y.-Y.; Lai, L.-C.; Tsai, M.-H.; Hsiao, C.K.; Chang, K.-J.; Chuang, E.Y. Molecular Characteristics and Metastasis Predictor Genes of Triple-Negative Breast Cancer: A Clinical Study of Triple-Negative Breast Carcinomas. PLoS ONE 2012, 7, e45831. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Xie, Y. Elevated nuclear CDK6 is associated with an unfavorable prognosis in lung adenocarcinoma patients. Int. J. Clin. Exp. Pathol. 2017, 10, 9614–9620. [Google Scholar]
- Massion, P.P.; Taflan, P.M.; Rahman, S.M.J.; Yildiz, P.; Shyr, Y.; Edgerton, M.E.; Westfall, M.D.; Roberts, J.R.; Pietenpol, J.A.; Carbone, D.P.; et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res. 2003, 63, 14612504. [Google Scholar]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [Green Version]
- van der Poel, C.E.; Spaapen, R.M.; van de Winkel, J.G.J.; Leusen, J.H.W. Functional characteristics of the high affinity IgG receptor, FcγRI. J. Immunol. 2011, 186, 2699–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dora, D.; Rivard, C.; Yu, H.; Pickard, S.L.; Laszlo, V.; Harko, T.; Megyesfalvi, Z.; Dinya, E.; Gerdan, C.; Szegvari, G.; et al. Characterization of Tumor-Associated Macrophages and the Immune Microenvironment in Limited-Stage Neuroendocrine-High and -Low Small Cell Lung Cancer. Biology 2021, 10, 502. https://doi.org/10.3390/biology10060502
Dora D, Rivard C, Yu H, Pickard SL, Laszlo V, Harko T, Megyesfalvi Z, Dinya E, Gerdan C, Szegvari G, et al. Characterization of Tumor-Associated Macrophages and the Immune Microenvironment in Limited-Stage Neuroendocrine-High and -Low Small Cell Lung Cancer. Biology. 2021; 10(6):502. https://doi.org/10.3390/biology10060502
Chicago/Turabian StyleDora, David, Christopher Rivard, Hui Yu, Shivaun Lueke Pickard, Viktoria Laszlo, Tunde Harko, Zsolt Megyesfalvi, Elek Dinya, Csongor Gerdan, Gabor Szegvari, and et al. 2021. "Characterization of Tumor-Associated Macrophages and the Immune Microenvironment in Limited-Stage Neuroendocrine-High and -Low Small Cell Lung Cancer" Biology 10, no. 6: 502. https://doi.org/10.3390/biology10060502