
Massive Parallel Alignment of RNA-seq Reads in Serverless Computing


1
Department of Health Sciences, Magna Graecia University of Catanzaro, 88100 Catanzaro, Italy
2
Department of Computer Architecture and Automation, Universidad Complutense de Madrid, 28040 Madrid, Spain
3
Data Analytics Research Center, Department of Medical and Surgical Sciences, Magna Graecia University of Catanzaro, 88100 Catanzaro, Italy
*
Author to whom correspondence should be addressed.
Big Data Cogn. Comput. 2023, 7(2), 98; https://doi.org/10.3390/bdcc7020098
Submission received: 19 April 2023
/
Revised: 3 May 2023
/
Accepted: 12 May 2023
/
Published: 15 May 2023
(This article belongs to the Special Issue Data-Based Bioinformatics and Applications)
Abstract
:In recent years, the use of Cloud infrastructures for data processing has proven useful, with a computing potential that is not affected by the limitations of a local infrastructure. In this context, Serverless computing is the fastest-growing Cloud service model due to its auto-scaling methodologies, reliability, and fault tolerance. We present a solution based on in-house Serverless infrastructure, which is able to perform large-scale RNA-seq data analysis focused on the mapping of sequencing reads to a reference genome. The main contribution was bringing the computation of genomic data into serverless computing, focusing on RNA-seq read-mapping to a reference genome, as this is the most time-consuming task for some pipelines. The proposed solution handles massive parallel instances to maximize the efficiency in terms of running time. We evaluated the performance of our solution by performing two main tests, both based on the mapping of RNA-seq reads to Human GRCh38. Our experiments demonstrated a reduction of , , and , compared to the local environments with 16, 8, and 4 virtual cores, respectively. Furthermore, serverless limitations were investigated.
1. Introduction
In recent years, the use of high-throughput Next-Generation Sequencing (NGS) technologies has improved diagnostic effectiveness by increasing the need for novel bioinformatics solutions that can allow for the processing and analysis of genomic data, e.g., via data-mining, machine learning, and data-integration techniques [1], as well as via network analysis [2,3,4].
RNA-sequencing (RNA-seq) offers a sensitive and accurate methodology for gene expression [5]. For instance, it for the genes that are expressed in the synthesis of a functional product to be identified, and can evaluate the differentially expressed transcripts between two molecular conditions. RNA-seq has become the de facto standard for the analysis of the transcriptome. Generally, the processing of large-scale genomic data requires many computational resources, resulting in a highly expansive process in terms of computational power and running time. Traditional approaches are not fully efficient from this perspective; therefore, large set of samples are generally analyzed using the ad-hoc pipeline to reduce the downtimes [6].
The use of Cloud infrastructures for data processing can be even more useful, obtaining computing potential that is not affected by the limitations of a local infrastructure.
In the era of Big Data and Artificial Intelligence (AI), the need for increasing computing capacities is growing rapidly. Infrastructures based on self-managed servers or local workstations are not convenient from an economic point of view due to management costs, the need for specialized personnel, and purely evolutionary issues related to scalability and obsolescence. Specifically, Cloud computing allows for users to reduce management costs, as well as to improve the management in terms of simplicity and practicality. Other non-trivial advantages concern the reliability, scalability, and fault tolerance of such infrastructures. Outside the enterprise environment, fewer Cloud models are based on virtual machines (or virtual servers), to support integrated security paradigms, elastic storage, or computer storage to preserve the state of an execution at a specific time point (i.e., snapshot). These approaches are known as Infrastructure as a Service (IaaS), or, in meanings with greater abstraction, as Platform as a Service (PaaS). Other well-known execution models include Cloud as a Service (CaaS), Software as a Service (SaaS), and Function-as-a-Service (FaaS). At a high level, the terms FaaS and Serverless Computing (or just Serverless) are used interchangeably, even by Cloud Providers (CPs). Therefore, the infrastructure of both orchestration is managed by the CP. However, FaaS only provides an event-driven execution model on a Cloud architecture, which is limited to the development of an in-house own function, while Serverless enables the development of this function by extending the runtime environment to other services and resources, e.g., by including a storage space or an entire support architecture (to be designed). In addition, Serverless also supports the auto-scaling, leading to (theoretical) unlimited expansion potential, and uses a container-based technology to allocate the resources on-demand.
Serverless is supported by several CPs, such as Amazon Web Services (AWS), Google Cloud, and Microsoft Azure [7].
In this paper, we present a solution based on an in-house Serverless infrastructure, which is able to perform large-scale RNA-seq data analysis focused on mapping the sequencing reads to a reference genome.
Our main contribution was to bring the computation of genomic data into serverless computing, focusing on RNA-seq reads mapping to a reference genome, which is the most time-consuming task for some pipelines. Furthermore, we summarize this as follows:
- Deploying the methodologies for RNA-seq data analysis in a serverless environment;
- Maximize the parallelization of data processing via serverless computing;
- Modeling a cloud architecture that is capable of overcoming the typical limitations of serverless technology;
- Designing a ready-to-use solution for the massive parallel alignment of RNA-seq reads in serverless computing;
- Evaluating the limits (e.g., timeout) of using serverless computing in our case study.
The rest of the paper is organized as follows. Section 2 presents the design of our own architecture based on Serverless, as well as the method applied to the massive parallel mapping of RNA-seq reads to a reference genome. Section 3 describes a set of experiments for performance evaluation in real use-cases. Section 4 discusses the results on the basis of the evidence reported by our experimentation. Finally, Section 5 concludes the paper.
Related Works
In this section, we investigated the scientific literature using the PubMed search engine (https://pubmed.ncbi.nlm.nih.gov, accessed on 3 May 2023), maintained by the National Center for Biotechnology Information (NCBI). We focused on keywords related to the term “serverless”. We applied the following inclusion and exclusion criteria:
- we reported articles concerning the bioinformatics solution based on serveless computing and focused on specific tasks;
- we preferred articles that reported detailed information about the development and prototyping of in-house solutions;
- we excluded articles older than 5 years, and/or ones with topics outside the bioinformatics context;
- we excluded articles already mentioned in the introduction;
- we excluded articles purely dedicated to presenting the state-of-the-art.
In recent years, serverless computing has become a relevant solution for data processing, allowing for the analysis of big data in several fields, including biomedical information [8]. Its main advantage is its propensity for scalability and massive parallel computation, as well as the related flexible services allowing for data storage and on-demand access to applications and resources. As there are not many Serverless-based solutions in bioinformatics, and this technology is still being studied in depth, we report the studies that are most relevant to our research below.
Mrozek et al. [9] adapted the concept of Data Lake for storing and processing NGS data. In this study, the authors proposed a large-scale and serverless computational approach that is able to improve the quality of NGS data by bringing together the benefits of the Data Lake Analytics and the serverless computing paradigm. Their approach used U-SQL query language to allow for the extraction, processing, and storing of data from NGS-based multi-omics data analyses.
In a similar context, Ansari et al. [10] focused their attention on removing the dependence on cloud providers via serverless computing. This study proposed a deep learning sequence-based prediction model for peptide properties. This objective was addressed by developing a specific architecture supporting the bidirectional recurrent neural networks, based on edge machine learning, to remove the dependence on cloud providers. Similarly, Benjamin D. Lee et al. [11] presented a Serverless web tool for DNA sequence visualization based on AWS Lambda that, in its free tier, allows for one million function invocations, totaling 3.2 million seconds of computing time each month.
Other studies have focused on the serverless paradigm, with the aim of maximizing parallelism and reducing execution times. For instance, Niu et al. [12] applied Serverless computing to improve sequence comparison in terms of execution time. They considered several Cloud services, such as AWS Lambda and Google Cloud Functions. According to their experimentation, Google Cloud Functions require more latencies for the invocation of numerous functions (in the order of hundreds) compared to AWS Lambda, which was found to be more efficient. The latter reported a speed-up that was 400× faster than the local workstation used in the tests, while the benefits offered by the Serverless service managed by Google were also significant, even if limited to 47× compared to the same workstation. This issue was investigated in Aji John et al. [13], who also reported an advantage to using AWS Lambda compared to other Serverless services.
Soohyun Lee et al. [14] used AWS Lambda to deploy their own application for the scalable execution of portable pipelines on the Cloud. The authors were able to process terabytes of information from the 4D Nucleome (4DN) portal [15] related to the organization and evolution of curated nucleomics data.
Other contexts were studied to evaluate the benefits provided by Serverless technology. At the conclusion of our overview, and to better present the effectiveness of serverless computing, we report on the study proposed by Bebortta et al. [16], which, due to the type of analysis, is strongly related to this context, even if in a different area. Here, the authors focused on geospatial data analysis, considering two main issues: the former focused on an analysis of the worldwide density of mineral resources, while the latter focused on 30-year household forecasts at a parcel level. They demonstrated that their own serverless framework was able to reduce timing constraints by also improving the analysis of geospatial data in terms of performance. In addition, they observed that AWS Lambda has much higher efficiency compared to other solutions (i.e., Google Cloud Functions and Microsoft Azure Functions).
In accordance with the mentioned studies, AWS Lambda proved to be the most advantageous choice in terms of execution times and costs among all Serverless services. For this reason, we decided to exploit it for the design and implementation of the solution that we propose in this paper.
Note that we did not find any studies focusing on the mapping of RNA-seq reads to a reference genome via serverless computing.
2. Materials and Methods
In this section, we describe the materials and methods involved in the design and development of the proposed solution.
Our solution was designed as a set of AWS Lambda functions. Each one is dedicated to a specific task, of which the environment handles massive parallel instances in order to maximize the efficiency in terms of running time. The functions were run in a Serverless environment based on our own architecture, consisting of (i) a custom Serverless architecture, and (ii) a set of functions dedicated to specific tasks.
We report the main features and limitations related to AWS Lambda, as follows:
- Maximum timeout: 900 s (15 min);
- Maximum virtual CPUs (vCPUs): 6.
- Maximum memory: 10,240 MB;
- Maximum ephemeral storage: 10,240 MB;
- Maximum concurrent executions: 1024.
According to the official AWS Lambda’s documentation (https://docs.aws.amazon.com/lambda/latest/dg/gettingstarted-limits.html, accessed on 3 May 2023), at 1769 MB of memory, a function has the equivalent of 1 virtual CPU (vCPU); at 10,240 MB of memory, 6 vCPUs are allocated. Furthermore, Lambda does not support any accelerated computing device (e.g., Graphics Processing Units). Special needs may be granted to a specific account on demand and at the discretion of AWS; in any case, there would be additional costs. However, requests forwarded by new or basic accounts almost certainly could not be satisfied.
The mentioned issues were overcome by designing a custom architecture that extended the core capabilities provided by AWS Lambda. The latter was, therefore, performed in a more elastic and performing environment, especially in terms of data management.
2.1. Serverless Architecture
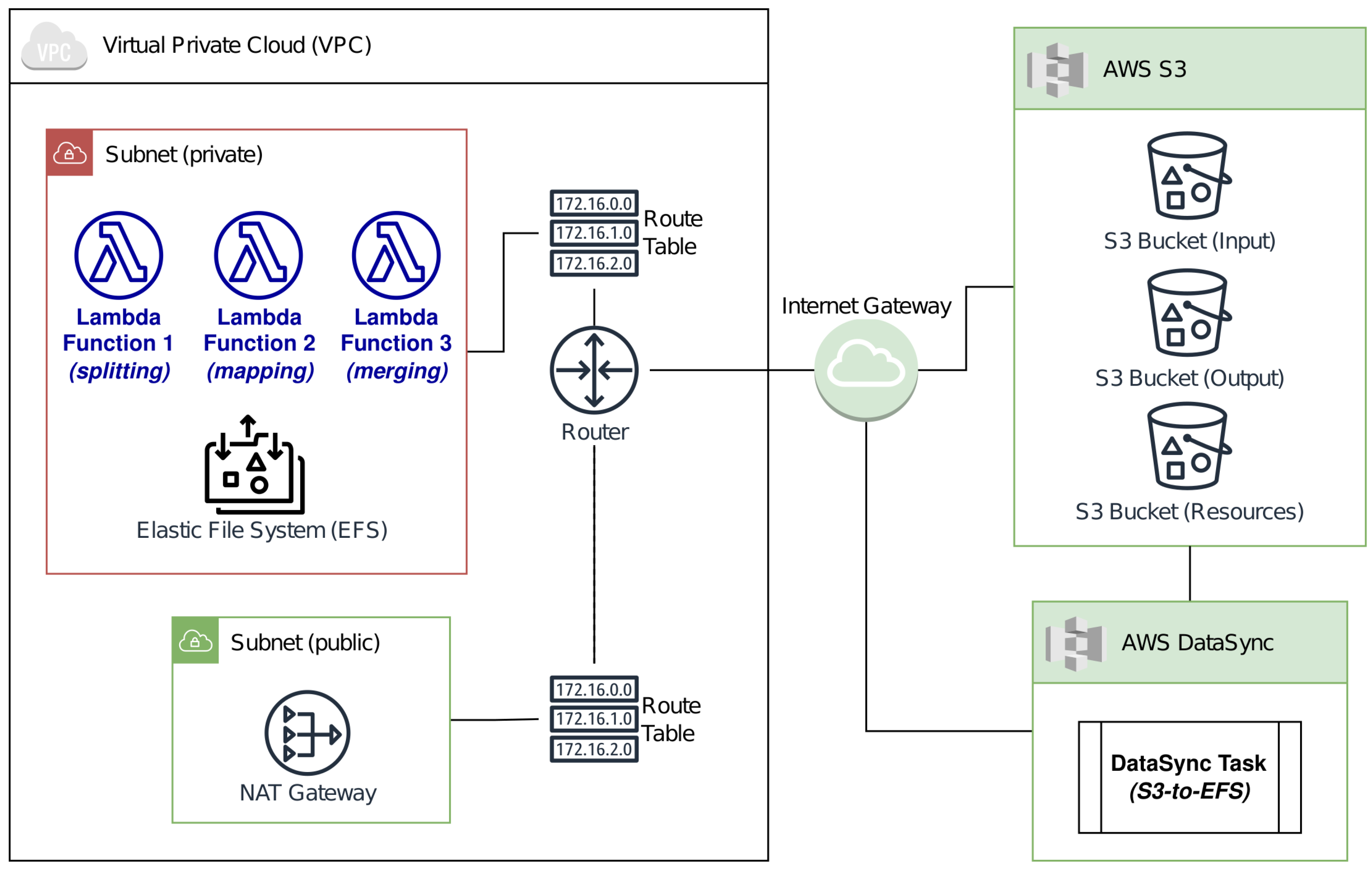
The basic environment offered by AWS Lambda is not a good choice for our purpose, due to several issues. The main ones are briefly listed as follows: the times and means of data exchange (e.g, upload and download over HTTP/s from/to an AWS storage service), the limited space allocated for the ephemeral storage, and the destruction of allocated data when an instance terminates. Therefore, we designed our own architecture using the features made available by AWS in order to support a large amount of data and ensure a persistent storage that survives the whole processing and can be shared among all instances allocated during the runtime, remaining available even after timeout.
We used two types of persistent storage, obtained via well-known AWS services, to overcome the discussed limitations to AWS Lambda.
The first one is an Elastic File System (EFS), a scalable set-and-forget elastic file system that supports auto-shrinking properties. Its main advantage is the possibility of sharing this storage among all instances, even when different functions or applications are used. This issue allows for persistence and data exchange, overcoming the ephemeral storage of a function.
The second one is based on AWS Simple Storage Service (S3). AWS S3 is an object-oriented storage service based on bucket for data organization. It is affected by the cross-/same-region replication and data exchange over HyperText Transfer Protocol Secure (HTTPS), which introduces some latencies compared to other solutions that manage exchanges via file system access (e.g., EFS or Lambda Ephemeral Storage). However, it allows external clients to download/upload data, which would be impossible when using file system access.
According to this issue, we stored the input in a dedicated bucket by moving data from S3 to the EFS using AWS DataSync; the latter works like the producer/consumer model by moving data from bucket to EFS based on availability.
Each instance of a Lambda function retrieves only the fragment of interest from S3 by moving it into the function’s ephemeral storage. Instead, the reference genome is moved from S3 to the EFS using AWS DataSync during the initialization phase only; this is a non-recurring process that consequently reduces any latency, as each instance mounts the EFS by directly accessing the data.
To relate the mentioned services to our application, an Ad-Hoc architecture was modeled using a Virtual Private Cloud (VPC). This allowed for efficient communication among all services used by our solution.
We modeled the VPC by configuring both a private and a public subnetwork (i.e., subnet). The former hosted the Lambda functions and the EPS, while the latter consists of a virtual Network Address Translation (NAT) Gateway that enables data to be retrieved from external sources over HTTPS. In addition, we defined two main routing tables to route traffic between internal (i.e., Lambda) and external (i.e., S3) services according to the access grants handled by a virtual router.
Our own architecture is shown in Figure 1.
2.2. Serverless Application
Our solution performs the computation using a Lambda application consisting of the following layers:
- Three Lambda functions (∼5 KB each);
- Third-party dependencies (∼75 MB).
We report the main implementation details as follows.
Lambda functions were implemented in Python 3 as three Lambda functions within the same environment. We integrated SeqKit in Lambda Function 1 to split the FASTQ file into subsequences, while SamTools was integrated in Lambda Function 3 to merge the aligned subsequences in the final output. Furthermore, we included HISAT2 in Lambda Function 2 for RNA-seq read mapping.
Our implementation uses Subprocess and Zipfile, two modules available into the Python Standard Library. The former handles several compressed file formats. The latter can invoke third-party software tools by also handling the related input, output, and error pipes. In addition, we used Botocore and Boto3 (https://boto3.amazonaws.com/v1/documentation/api/latest/reference/services/s3.html, accessed on 3 May 2023) provided by AWS for the low-level management of the objects stored on an AWS S3 bucket.
The third-party software tool invocations were handled via CLI. The main commands and the main specification are reported below for each function; more information can be found within the official documentation related to the software tool of interest.
Lambda Function 1 was based on the command split2 of SeqKit. It splits sequences by size/parts (paired- or single-end FASTQ) as follows (only the minimum flags are reported):
- > seqkit split2 [flags]
- Flags (others are available, but not used):
- -1 # read 1, file path
- -2 # read 2, file path
- -s # number of subsequences
Lambda Function 2 was based on HISAT2. It aligns RNA-Seq reads to a reference genome as follows (only the minimum flags are reported):
- > hisat2 [flags]
- Flags (others are available, but not used):
- -x # path of the reference genome
- -1 # read 1, file path
- -2 # read 2, file path
- -S # output, file path
Lambda Function 3 was based on the command merge of SamTools. It merges multiple sorted alignment files, maintaining the existing sort order, as follows (only the minimum parameters are reported):
- > samtools merge [output] [subsequences]
- Parameters (others are available, but not used):
- [ourput] # output, file path
- [subsequences] # list of subsequences, or * for all ones
Note that if the fragment is not split, only Lambda Function 2 is executed, while Lambda Function 1 and Lambda Function 3 are skipped. The threshold at which to split the fragments is chosen by the user and implemented in the configuration, and should take into account the maximum time that the function can process before being stopped due to timeout. We suggest choosing the threshold based on the specifications of the samples of interest, and in accordance with the total number of reads to be analysed, in order to minimize latencies.
2.3. Massive Parallel Alignment in Serverless
An RNA-seq data analysis may be conducted using several optimal approaches based on specific best practices [19]. According to Pertea et al. [20], we performed the mapping using a well-known software tool named Hierarchical Indexing for Spliced Alignment of Transcripts 2 (HISAT2) [21], as this is able to guarantee an optimal performance for read mapping. Therefore, the proposed methodology integrates the advantages of HISAT2 in terms of alignment quality with the efficient parallelization of an environment based on Serverless.
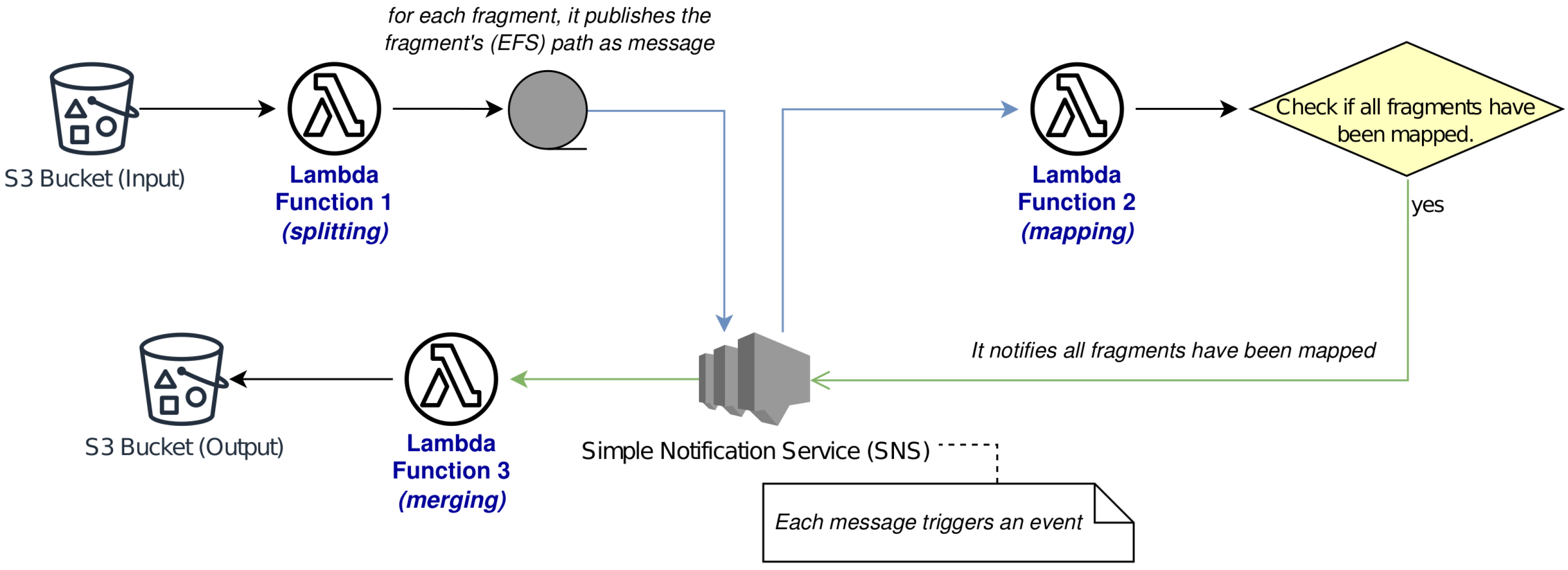
In addition, this was based on the divide-et-impera approach to maximize the parallelization: the original data are divided in several fragments based on a defined number of reads for each resulting file in order to obtain a mass of input data that can be processed within the times imposed by the Lambda execution environment. After all raw fragments were processed, the resulting mapped ones were merged; this process is not particularly expensive in terms of running time. Note that the proposed solution may be extended to other alignment methods, provided that the latter support a command line execution.
We used the AWS Simple Notification Service (SNS) to monitor the success of fragment processing. If of these processes fails, it is re-processed by a novel instance. Note that the failure rate was zero in our tests, but we retained the ability to handle these situations should they occur.
Figure 2 reports a non-exhaustive workflow for illustrative purposes only.
3. Results
This section reports information about the dataset used for testing and the results from the performance evaluation, as well as the hardware configuration used for comparative tests.
We studied the computational optimization offered by our solution in terms of execution time, and compared this to the local environment based on the following hardware: Intel Xeon GHz, 8 cores or 16 virtual cores (vcores), and 16 GB of memory. It was declined in the following configurations:
- Two cores (4 vcores);
- Four cores (8 vcores);
- Eight cores (16 vcores).
Furthermore, the AWS environment that hosted the proposed solution had the following configuration:
- Ephemeral storage: 512 MB;
- Memory (i.e., RAM): 10,240 MB;
- Function timeout: 900 s.
As discussion, the vCPU allocation is handled by AWS Lambda; at 10,240 MB of memory, 6 vCPUs are allocated, while up to 1769 MB, only one vCPU is allocated. For the other ranges, please refer to the official documentation of AWS’s Lambda. The architectural information related to our solution is discussed in Section 2.1.
3.1. Datasets
We used a set of 10 random RNA-seq reads for Chromosome X, related to the RNA-sequencing of “465 lymphoblastoid cell lines from the 1000 Genomes” (ftp://ftp.sra.ebi.ac.uk/vol1/fastq/, accessed on 3 May 2023; uncompressed data size: ∼2.8 GB).
The samples are listed as follows:
- ERR188337;
- ERR188401;
- ERR188257;
- ERR188383;
- ERR188044;
- ERR204651;
- ERR204713;
- ERR204822;
- ERR204904;
- ERR204916.
Each paired-end FastQ sample consists of two compressed files containing forward and reverse reads, respectively. To provide an example, the sample ERR204916 consists of ERR204916_1.fq and ERR204916_2.fq.
The samples were mapped on the Ensemble Genome Reference Consortium Human Build 38 (Human GRCh38) pre-indexed for HISAT2 (ftp.ccb.jhu.edu/pub/infphilo/HISAT2/data/hg38_tran.tar.gz, accessed on 3 May 2023; uncompressed data size: GB). Note that HISAT2 needs the genome to be indexed in its own format (.ht2) before it can be used for mapping.
3.2. Performance Evaluation
We evaluated the performance of our solution by performing two main tests, both based on the mapping of an input data to a Human GRCh38 pre-indexed for HISAT2; we will simply refer to the latter as “Human GRCh38”. The samples chosen for testing only refer to the Chromosome X, and these can be aligned without the need to apply the divide-et-impera approach. This allowed for us to fully evaluate the abilities of the infrastructure; the main purpose was to saturate the resources made available by AWS in our case studies.
Test 1 was performed in parallel on different instances that were dynamically allocated by AWS Lambda, while Test 2 was performed over a single instance. As discussed, AWS Lambda provides a concurrency limit of 1000 across all functions in a region. In all tests, our account was entirely dedicated to the test of interest, and no other functions were instantiated or running.
3.2.1. Test 1
Test 1 concerned the mapping of RNA-Seq reads to Human GRCh38, which are both presented in the previous subsection. This test was repeated as the number of samples increased, and a comparison with a set of local environments was also reported, in accordance with the configurations presented above.
In addition, we compared the performance of a single core configuration (1 vCPU, 1769 MB of memory) and multicore configurations with the maximum allocable resources for our basic account (6 vCPUs, 10,240 MB of memory) to relate the single and the multiple cores execution in the same Serverless environment, as well as to evaluate the computational performance of a single virtual CPU offered by the AWS Lambda environment.
3.2.2. Test 2
Similarly to the previous one, Test 2 evaluated the performance in terms of running time by focusing on the maximum number of reads that can be processed by only one instance before the environment interrupts the execution for timeout.
The mapping of fragments consisted of 1,321,477 reads, each one. The test was repeated as the number of reads increased by processing a total of 64 fragments to Human GRCh38.
The fragment and the related number of reads were randomly chosen in our samples. The multiplicative factor (from 1 to 64) was obtained by replicating the same fragment to obtain a set of results that may indicate the proportionality or degradation of the allocated resources, as time and space increase during computation.
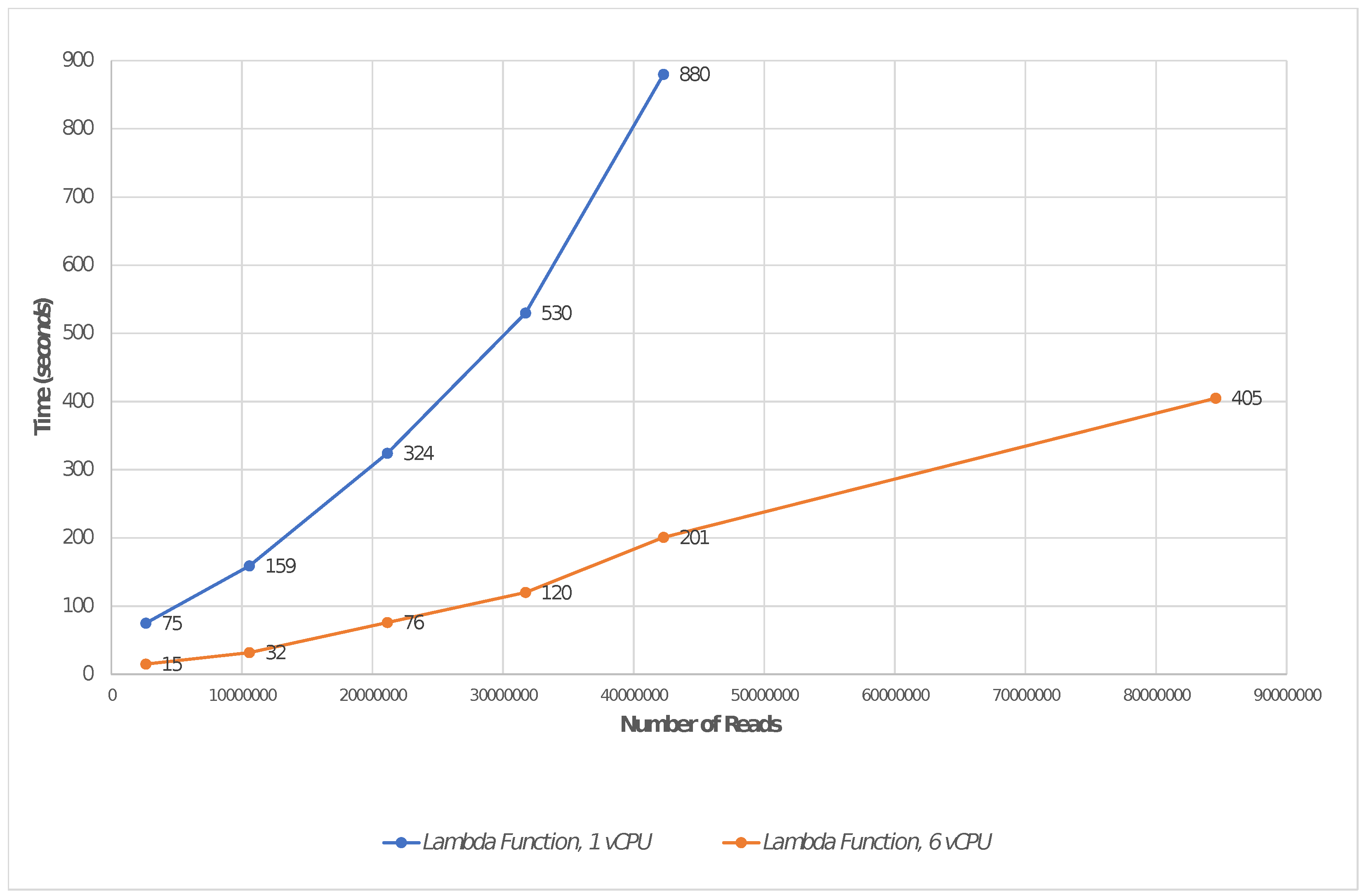
Table 2 reports the running times (in seconds) by highlighting where timeout occurs. These are also shown in Figure 4. This represents the maximum number of reads that were processed before the environment interrupts the execution of at least one instance due to timeout. This test was repeated as the number of samples increased, and a comparison with a set of local environments was also carried out, in accordance with the configurations presented above.
4. Discussion
In our tests, we evaluated the performance of our solution by mapping a set of samples to Human GRCh38, pre-indexed for HISAT2, to study the computational optimization provided by Serverless computing when applied to genomic data analysis.
Test 1 showed that our solution allows for a reduction of up to of the running time required by the processing of 10 samples compared to a local environment configured with four vcores in several configurations. In details, a reduction of , , and was obtained compared to a local environment with 16, 8, and 4 vcores, respectively. The running time of the local environment with a configuration based on 16 vcores reports results comparable with our solution, when deployed on a Serverless environment with only 1 vCPU and our own architecture.
This test demonstrated the significant scalability and parallelism advantage that Serverless computing can offer. However, the latter has some memory and vCPU usage limitations when used on the basic accounts in AWS. We were unable to test the configurations reserved for company accounts, for which a form of confidential negotiation is foreseen in terms of configuration and, above all, in terms of costs; however, let us assume that the advantage must be even more tangible. Note that each instance of a Lambda function is handled by the environment based on the same configuration, and its execution does not afflict the other ones; it is atomic and independent. Consequently, 1024 computations on 1024 instances (one instance for one sample) will take as much as 1 sample out of 1 instance; as reported, 1024 is the maximum number of concurrent executions. However, the costs will be calculated for all allocated resources.
In addition, we performed a second test (i.e., Test 2) to evaluate the performance in terms of maximum number of reads that can be processed by only one instance before the environment interrupts the execution for timeout.
We performed the mapping of a growing set of fragments consisting of 1,321,477 reads each. The fragment was randomly chosen from our sample, and replicated to conform to the scale of the analyses as the amount of data increases. In each experiment, the fragments were mapped to Human GRCh38. The test shows that a configuration based on the maximum specification provided by AWS Lambda (6 vCPU, 10,240 MB of memory) is able to process up to 64 fragments, or 84,574,528 reads, before being interrupted by the provided for timeout. As discussed, the latter can be extended to a maximum of 900 s.
This test shows a relevant limitation referring to the maximum timeout defined for each instance, as well as more obvious limitations referring to the maximum number of memory (and related vCPU) that can be allocated. However, this limit can be circumvented by defining an ad hoc contract with AWS; the latter case is reserved for very large corporate contexts, for which a private negotiation with the provider is necessary, and at least possible.
Finally, our solution was more suitable for highly parallel computations, and is not recommended for a single atomic operation that requires a lot of resources. Definitely, it was found to be more suited to large numbers of computations, rather than small groups. Therefore, the performances of a single computation cannot compete with a local workstation (even of a medium–low level), but these solutions are clearly better for numerous samples, due to both the high scalability and parallelism offered by Serverless computing, rather than the large number of computational resources made available by Cloud services.
5. Conclusions
In this paper, we presented a solution based on an in-house Serverless infrastructure, which is able to perform large-scale RNA-seq data analysis by improving the execution times required for the mapping of NGS reads. This work showed the potential of Serverless in complex genomic analysis.
Our tests demonstrated a reduction of , , and , compared to a local environment with 16, 8, and 4 vcores, respectively.
However, some limitations have emerged regarding the maximum number of reads that a solution of this type is able to process in the same instance. Specifically, we managed to process a maximum of 64 fragments, consisting of a total of 84,574,528 reads. This limitation is due to the maximum timeout that is allowed for each instance. Other limitations refer to the maximum number of memory that can be allocated; even if used in an enterprise context, this limit should not be valid as AWS allows for you to agree on ad-hoc contracts for specific needs. However, the costs and details are not disclosed.
Finally, our solution (and, more generally, other Serverless ones) is clearly better on numerous samples, both due to the high scalability and parallelism offered by Serverless computing compared to the large number of computational resources made available by Cloud services.
Future studies may concern the extension of this methodology to more complex tasks, such as the porting of a whole pipeline, e.g., for the transcript-level quantification or gene expression from RNA-seq data. In the latter case, several adjustments will be necessary, such that a constant monitoring of the progress can be performed, with the management of a possible rollback in case of failure. The serverless paradigm may not be suitable for this activity, but the proposed architecture may be able to overcome most of the limitations in this case as well; only a future in-depth study will be able to confirm or deny the possibility of carrying out what has been described.
Author Contributions
Data curation, P.C.; Formal analysis, P.C.; Investigation, P.C. and M.C.; Writing—Original Draft Preparation, P.C.; Writing—Review and Editing, P.C., M.C. and J.L.V.-P.; Methodology, P.C.; Supervision, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
HISAT2 is available on https://daehwankimlab.github.io/HISAT2 (accessed on 3 May 2023). SeqKit is available on https://github.com/shenwei356/seqkit (accessed on 3 May 2023). Samtools is available on https://www.htslib.org/download (accessed on 3 May 2023). Samples are available on ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ (accessed on 3 May 2023). Human GRCh38 (pre-indexed for HISAT2) is available on ftp.ccb.jhu.edu/pub/infphilo/HISAT2/data/hg38_tran.tar.gz (accessed on 3 May 2023). A package consisting of the Lambda functions and the resources related to the proposed solution is available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cinaglia, P.; Guzzi, P.H.; Veltri, P. INTEGRO: An algorithm for data-integration and disease-gene association. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018; pp. 2076–2081. [Google Scholar] [CrossRef]
- Cinaglia, P.; Cannataro, M. Network alignment and motif discovery in dynamic networks. Netw. Model. Anal. Health Inform. Bioinform. 2022, 11, 38. [Google Scholar] [CrossRef]
- Cinaglia, P.; Cannataro, M. A Method Based on Temporal Embedding for the Pairwise Alignment of Dynamic Networks. Entropy 2023, 25, 665. [Google Scholar] [CrossRef]
- Elhesha, R.; Sarkar, A.; Cinaglia, P.; Boucher, C.; Kahveci, T. Co-evolving Patterns in Temporal Networks of Varying Evolution. In Proceedings of the BCB ’19: 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, New York, NY, USA, 7–10 September 2019; pp. 494–503. [Google Scholar] [CrossRef]
- Ji, F.; Sadreyev, R.I. RNA-seq: Basic Bioinformatics Analysis. Curr. Protoc. Mol. Biol. 2018, 124, e68. [Google Scholar] [CrossRef] [PubMed]
- Cinaglia, P.; Cannataro, M. A Flexible Automated Pipeline Engine for Transcript-Level Quantification from RNA-seq. In Proceedings of the Advances in Conceptual Modeling, Hyderabad, India, 17–20 October 2022; Guizzardi, R., Neumayr, B., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2022; pp. 45–54. [Google Scholar]
- Grzesik, P.; Augustyn, D.R.; Lik, L.; Mrozek, D. Serverless computing in omics data analysis and integration. Briefings Bioinform. 2022, 23, bbab349. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Cepeda, R.; Agapito, G.; Vazquez-Poletti, J.L.; Cannataro, M. Challenges and Opportunities of Amazon Serverless Lambda Services in Bioinformatics. In Proceedings of the BCB ’19: 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics. Association for Computing Machinery, Niagara Falls, NY, USA, 7–10 September 2019; pp. 663–668. [Google Scholar] [CrossRef]
- Mrozek, D.; Stępień, K.; Grzesik, P.; Małysiak Mrozek, B. A Large-Scale and Serverless Computational Approach for Improving Quality of NGS Data Supporting Big Multi-Omics Data Analyses. Front. Genet. 2021, 12, 699280. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; White, A.D. Serverless Prediction of Peptide Properties with Recurrent Neural Networks. J. Chem. Inf. Model. 2023, 63, 2546–2553. [Google Scholar] [CrossRef]
- Lee, B.D.; Timony, M.A.; Ruiz, P. DNAvisualization.org: A serverless web tool for DNA sequence visualization. Nucleic Acids Res. 2019, 47, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Kumanov, D.; Hung, L.H.; Lloyd, W.; Yeung, K.Y. Leveraging Serverless Computing to Improve Performance for Sequence Comparison. In Proceedings of the BCB ’19: 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, New York, NY, USA, 7–10 September 2019; pp. 683–687. [Google Scholar] [CrossRef]
- John, A.; Muenzen, K.; Ausmees, K. Evaluation of serverless computing for scalable execution of a joint variant calling workflow. PLoS ONE 2021, 16, e0254363. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Johnson, J.; Vitzthum, C.; Kırlı, K.; Alver, B.H.; Park, P.J. Tibanna: Software for scalable execution of portable pipelines on the cloud. Bioinformatics 2019, 35, 4424–4426. [Google Scholar] [CrossRef] [PubMed]
- Reiff, S.B.; Schroeder, A.J.; Kırlı, K.; Cosolo, A.; Bakker, C.; Mercado, L.; Lee, S.; Veit, A.D.; Balashov, A.K.; Vitzthum, C.; et al. The 4D Nucleome Data Portal as a resource for searching and visualizing curated nucleomics data. Nat. Commun. 2022, 13, 2365. [Google Scholar] [CrossRef] [PubMed]
- Bebortta, S.; Das, S.K.; Kandpal, M.; Barik, D.R.; Dubey, H. Geospatial Serverless Computing: Architectures, Tools and Future Directions. ISPRS Int. J. Geo-Inf. 2020, 9, 311. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
Figure 1.
The figure shows our own architecture supporting AWS Lambda.

Figure 2.
The divide-et-impera approach was performed by our solution, in accordance with the reported workflow.
Figure 2.
The divide-et-impera approach was performed by our solution, in accordance with the reported workflow.

Figure 3.
The figure shows the plot related to Table 1: it reports the running times estimated for each set of samples based on the processing performed by our Lambda function (i.e., Lambda Function) on two environments with 1 vCPU and 6 vCPUs, respectively, and by a set of local environments (i.e., Local) configured with 2 cores (4 vcores), 4 cores (8 vcores), and 8 cores (16 vcores). The time is reported in seconds.
Figure 3.
The figure shows the plot related to Table 1: it reports the running times estimated for each set of samples based on the processing performed by our Lambda function (i.e., Lambda Function) on two environments with 1 vCPU and 6 vCPUs, respectively, and by a set of local environments (i.e., Local) configured with 2 cores (4 vcores), 4 cores (8 vcores), and 8 cores (16 vcores). The time is reported in seconds.

Figure 4.
The figure shows the plot related to Table 2: it reports the running times by focusing on the maximum number of reads that can be processed before the environment interrupts the execution of at least one instance due to timeout. The time is reported in seconds. Note that the last point in each curve is the last acquisition before timeout occurs.
Figure 4.
The figure shows the plot related to Table 2: it reports the running times by focusing on the maximum number of reads that can be processed before the environment interrupts the execution of at least one instance due to timeout. The time is reported in seconds. Note that the last point in each curve is the last acquisition before timeout occurs.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The table reports the running times estimated for each set of samples based on the processing of our Lambda function (i.e., Lambda Function) in two environments with 1 vCPU and 6 vCPUs, respectively, and by a set of local environments (i.e., Local) configured with 2 cores (4 vcores), 4 cores (8 vcores), and 8 cores (16 vcores). The time is reported in seconds. For each experiment, the best result is shown in bold.
Table 1.
The table reports the running times estimated for each set of samples based on the processing of our Lambda function (i.e., Lambda Function) in two environments with 1 vCPU and 6 vCPUs, respectively, and by a set of local environments (i.e., Local) configured with 2 cores (4 vcores), 4 cores (8 vcores), and 8 cores (16 vcores). The time is reported in seconds. For each experiment, the best result is shown in bold.
| Samples | Lambda Function, 1 vCPU | Lambda Function, 6 vCPU | Local, 4 Vcores | Local, 8 Vcores | Local, 16 Vcores |
|---|---|---|---|---|---|
| 1 | 124 | 25 | 69 | 24 | 12 |
| 2 | 124 | 25 | 136 | 47 | 25 |
| 4 | 124 | 25 | 271 | 96 | 50 |
| 6 | 124 | 25 | 410 | 142 | 73 |
| 8 | 124 | 25 | 551 | 202 | 99 |
| 10 | 124 | 25 | 691 | 252 | 124 |
Table 2.
The table reports the running times by focusing on the maximum number of reads that can be processed before the environment interrupts the execution of at least one instance due to timeout. The time is reported in seconds.
Table 2.
The table reports the running times by focusing on the maximum number of reads that can be processed before the environment interrupts the execution of at least one instance due to timeout. The time is reported in seconds.
| Fragments | Reads | Lambda Function, 1 vCPU | Lambda Function, 6 vCPU |
|---|---|---|---|
| 1 | 1321,477 | 75 | 15 |
| 2 | 2,642,954 | 159 | 32 |
| 8 | 10,571,816 | 324 | 76 |
| 16 | 21,143,632 | 530 | 120 |
| 24 | 31,715,448 | 880 * | 201 |
| 32 | 42,287,264 | Timeout | 405 |
| 64 | 84,574,528 | Timeout | 826 * |
* This is the maximum limit experienced before timeout occurs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cinaglia, P.; Vázquez-Poletti, J.L.; Cannataro, M. Massive Parallel Alignment of RNA-seq Reads in Serverless Computing. Big Data Cogn. Comput. 2023, 7, 98. https://doi.org/10.3390/bdcc7020098
AMA Style
Cinaglia P, Vázquez-Poletti JL, Cannataro M. Massive Parallel Alignment of RNA-seq Reads in Serverless Computing. Big Data and Cognitive Computing. 2023; 7(2):98. https://doi.org/10.3390/bdcc7020098
Chicago/Turabian StyleCinaglia, Pietro, José Luis Vázquez-Poletti, and Mario Cannataro. 2023. "Massive Parallel Alignment of RNA-seq Reads in Serverless Computing" Big Data and Cognitive Computing 7, no. 2: 98. https://doi.org/10.3390/bdcc7020098