
Mechanisms of Electron-Induced Chemistry in Molecular Ices
by
 , , , and
, , , and
Fabian Schmidt
 ,
,
Tobias Borrmann
,
Martin Philipp Mues
,
Sanna Benter
,
Petra Swiderek
and
Jan Hendrik Bredehöft
* Institute for Applied and Physical Chemistry, University of Bremen, Leobener Straße 5, 28359 Bremen, Germany
*
Author to whom correspondence should be addressed.
Atoms 2022, 10(1), 25; https://doi.org/10.3390/atoms10010025
Submission received: 20 December 2021
/
Revised: 9 February 2022
/
Accepted: 17 February 2022
/
Published: 21 February 2022
(This article belongs to the Special Issue Interaction of Electrons with Atoms, Molecules and Surfaces)
Abstract
:Electron-induced chemistry is relevant to many processes that occur when ionizing radiation interacts with matter. This includes radiation damage, curing of polymers, and nanofabrication processes but also the formation of complex molecules in molecular ices grown on dust particles in space. High-energy radiation liberates from such materials an abundance of secondary electrons of which most have energies below 20 eV. These electrons efficiently trigger reactions when they attach to molecules or induce electronic excitation and further ionization. This review focuses on the present state of insight regarding the mechanisms of reactions induced by electrons with energies between 0 and 20 eV that lead to formation of larger products in binary ice layers consisting of small molecules (H2O, CO, CH3OH, NH3, CH4, C2H4, CH3CN, C2H6) or some derivatives thereof (C2H5NH2 and (C2H5)2NH, CH2=CHCH3). It summarizes our approach to identify products and quantify their amounts based on thermal desorption spectrometry (TDS) and electron-stimulated desorption (ESD) experiments performed in ultrahigh vacuum (UHV). The overview of the results demonstrates that, although the initial electron-molecule interaction is a non-thermal process, product formation from the resulting reactive species is often governed by subsequent reactions that follow well-known thermal and radical-driven mechanisms of organic chemistry.
1. Introduction
The interaction of free electrons with molecules can trigger chemical reactions that are relevant to a wide range of phenomena. This includes, for instance, radiation damage of living tissue [1,2], the curing of polymeric materials [3,4], nanofabrication processes such as focused electron beam induced deposition (FEBID) or extreme ultraviolet lithography (EUVL) [1,5,6,7] and also the chemistry in molecular ices that form on dust particles in space as studied in the field of astrochemistry [1,8,9,10,11]. In these processes, ionizing particulate or electromagnetic radiation incident on the material induces the chemical reactions. Common to all kinds of ionizing radiation is that it releases low-energy electrons when interacting with matter. High energy electrons or ions produce an abundance of low-energy secondary electrons (LESEs) with energies ranging from near 0 eV up to 50 eV [1,5,10] while electromagnetic radiation with energy above the ionization threshold of the irradiated material releases photoelectrons (PEs) that, if energetic enough, again produce LESEs [12]. As an example, it is now widely accepted that the chemistry inherent in FEBID is in fact driven by LESEs [1,13,14]. In EUVL, PEs and LESEs can have a similar efficiency with respect to the chemical conversion of a resist as the initial EUV photon [15]. In the context of astrochemistry, it is also nowadays proposed that LESEs released under the effect of ionizing cosmic radiation contribute to the chemistry that occurs in interstellar molecular ices [8,10,11].
Considering only the initial electron-molecule encounter, electron-induced chemistry may appear predominantly destructive in nature. In fact, interaction with an electron often leads to dissociative states of a molecule and consequent bond rupture, processes that are particularly efficient within the typical energy range of LESEs [1,16,17]. More specifically, after electron attachment (EA) to a molecule, the resulting radical anion can decay to an anion and at least one neutral fragment. This process is called dissociative electron attachment (DEA). Neutral dissociation (ND) refers to the fragmentation of a dissociative state formed by electronic excitation (EE) resulting in neutral fragments. As a third pathway, dissociative ionization (DI) leads to fragmentation of a radical cation formed by electron ionization (EI) yielding a cation and again at least one neutral product. Such fragmentation is, for instance, desired in FEBID which produces even freestanding 3D nanostructures by scanning a surface with a keV electron beam to control the fragmentation of adsorbed metal organic precursor molecules and thus the formation of metal deposits [5,6]. However, the reactive fragments produced upon electron-molecule interaction can also cause unwanted effects. As an example, radiation-driven chemistry of adsorbates is relevant to materials used in space. Even the most thoroughly cleaned instruments for trace-analysis [18] have water and other organics adsorbed on their surfaces so that desorption and possibly out-gassing are still measurable years after leaving Earth’s atmosphere [19]. Radiation-induced chemical reactions of the adsorbate or ice layer can then lead to corrosion of the underlying surface [20] and thus degradation of materials [21].
In contrast to the destructive effects mentioned so far, radiation processing of ice layers that accrete over time on cold interstellar dust grains is considered as the most likely scenario for formation of complex molecules in space [8,9]. This is based on the observation that electron interactions with small molecules can lead to the synthesis of larger and structurally more complex products. Such electron-induced syntheses have been summarized before [8,10,16,22] and we refer the reader to these earlier reviews for an overview of the developments in the field. Herein, we aim at providing an overarching perspective of work on the chemistry driven by low energy electrons in binary mixed ices that has been performed in our laboratory for more than ten years [16,23,24,25,26,27,28,29,30,31,32]. In particular, the goal of this review is to demonstrate that after the initial non-thermal electron-molecule interaction, the fate of the thus produced reactive species is not simply governed by random encounters with other molecules. In fact, these species often rather involve in thermal reactions which can be understood based on established reaction mechanisms of synthetic chemistry.
The reactants that were included in the studies reviewed herein comprise the majority of those molecules that are considered to be most abundant in interstellar icy grain mantles [33], namely water (H2O), carbon monoxide (CO), methanol (CH3OH), ammonia (NH3), and methane (CH4). Ethylene (C2H4) is also an important reactant because it is particularly involved in reaction mechanisms that lead to the synthesis of larger products and can be considered as the simplest model compound for unsaturated hydrocarbon species. In these studies, the ice constituents were systematically varied as summarized in Table 1 which shows target products that incorporate all atoms of the reactants via a minimal amount of bond reorganization. Such systematic screening of ice systems is an important strategy of our approach. By exchanging one of the two constituents of the binary ice, it is possible to identify which of the reactants yields a key intermediate that leads to a particular synthesis. Thereby, deeper insight into the reaction mechanisms is obtained as will be discussed herein. Because of the large number of different products that are formed upon electron irradiation [8,11,34], CH3OH stands out among the reactants of the binary mixed ices summarized in Table 1 and has therefore also been investigated as pure ice [31]. In addition, earlier work has studied electron-induced reactions in CD3CN/C2H6 mixed ices [23] as well as in pure CH3CN [25]. Furthermore, the electron-induced reactions of different amines (C2H5NH2 and (C2H5)2NH) with C2H4 and of NH3 and propene (CH2=CHCH3) were also investigated [29]. Based on these combined results, this review:
- (i)
- provides a summary of our experimental approach which relies on a sufficient resolution in electron energy to identify resonant enhancements and energetic thresholds in the energy dependence of product yields and thus the electron-molecule interactions that initiate product formation,
- (ii)
- shows how information on the mechanisms of electron-driven chemistry in ices can be obtained by investigating the energy dependence of several products in the same sample or by comparing different ice systems, and
- (iii)
- discusses that the chemistry following the initial electron-molecule encounter can be understood based on established reaction mechanisms known from organic chemistry combined with quantum chemical calculations.
In line with the aims outlined above, this review is organized as follows. The experimental approach and methods are presented in Section 2. This is followed by a summary of the fundamental electron-molecule interactions that have been observed to initiate further reactions in the systems discussed herein (Section 3). Section 4 then discusses the principles of chemical reactions that are initiated by electron-molecule encounters and that lead to the synthesis of new stable products. Section 5 addresses some open questions and gives an outlook on potential topics for further study. Thereby, this review will hopefully serve future studies as reference to the chemical reaction mechanisms that underly the electron-induced chemistry in ices and multilayer molecular adsorbates.
2. Experimental Approach
The general approach of the work reviewed herein is to unravel the mechanisms of electron-induced chemical reactions in molecular ices by post-irradiation thermal desorption spectrometry (TDS) that can be combined, if appropriate, with experiments that directly monitor the electron-stimulated desorption (ESD) of products during irradiation. Both techniques rely on an ultrahigh vacuum (UHV) setup equipped with a variable temperature metal foil onto which ice layers are condensed, a defocused electron gun for irradiation, and a mass spectrometer for the detection of neutral desorbing species. In a TDS experiment, the surface with the ice layer is warmed up at a constant heating rate (typically 1 K/s) and the desorbing species are detected as function of temperature. ESD is studied at constant temperature. In both cases, mass scans can be performed to assess the range of products. In experiments that monitor the desorption rate of specific products as function of time, a maximum of four selected characteristic m/z values is measured in parallel. Based on this approach described in detail in our previous original publications [23,24,25,26,27,28,29,30,31,32], the following sequence of experiments is performed:
- (i)
- Products that are formed after electron irradiation are identified from TDS data for characteristic m/z values. In the ideal case, the desorption of a particular species can be seen in TDS curves for several m/z values and the intensity of the desorption peaks for these different m/z reflects the intensities in the mass spectra (MS) of this compound recorded on the same instrument when leaking the pure vapor of the product into the UHV chamber.
- (ii)
- Product formation is quantified from integrated desorption signals for a characteristic m/z ratio. The absolute product yields can be derived by comparing these integrated desorption signals to those obtained from ices that have been prepared with defined composition and surface coverage, as can be deduced from TDS, so that the amount of the respective product is known [31,35,36]. Relative amounts of different products can also be obtained without such a standard sample by using partial ionization cross sections for the characteristic m/z ratios used to monitor the products. The partial ionization cross sections for a given m/z ratio with intensity of a specific product is defined by , where are the intensities of all m/z ratios of that product and is its total ionization cross section. These cross sections, that refer to the electron energy applied for ionization in the mass spectrometer, define the intensities with which specific m/z ratios appear in the mass spectrum. If known, they can be used as correction factor that is applied to compare the intensities for specific m/z ratios and thus also the desorption peak areas of two different products and to convert them to a relative yield [24,29,31].
- (iii)
- The thus obtained product yields are determined as function of electron exposure, which is defined as the transmitted charge per surface area as measured on the metal foil, to establish over which range they increase linearly with exposure. In this linear regime, the decay of the initial reactants is still negligible and the product is not yet consumed by electron irradiation [24,26,29,30,31,32,35,36]. Under this condition, the rate with which a product is formed is directly proportional to the applied electron exposure. This is the basis on which the rates of formation can be compared between different products.
- (iv)
- Product yields are then measured as function of electron energy (E0) for an exposure that is within the linear regime for all investigated electron energies. The energy dependence of the product yields gives information on the electron-molecule interactions that initiate product formation. This includes thresholds for electronic excitation or ionization as well as maxima within specific energy ranges that point to a contribution of electron attachment. These processes are summarized in Section 3. When monitoring product formation in mixed ices, it is not obvious from the energy dependence alone which constituent undergoes the initiating electron-molecule interaction if both show similar DI or ND thresholds or energy ranges for DEA. In these cases, more detailed mechanistic information can be derived by comparing the energy dependent yields of several products [24,26] as will be exemplified in Section 4. In particular cases, a unique assignment can be obtained by exchanging one component of the ice for another and checking whether product formation still occurs [37].
Figure 1 illustrates the individual steps of the data analysis performed to obtain electron energy dependent product yields from TDS experiments using the example of CO/NH3 mixed ices. In the example presented here, m/z ratios of 43 and 45 have been monitored to reveal the formation of the stable neutral products formamide (H2NCHO) and isocyanic acid (HNCO). In this particular case, the desorption peaks of the two compounds overlap (Figure 1a). While m/z 45 is representative of formamide alone, m/z 43 represents both the molecular ion of isocyanic acid and a cationic fragment obtained from formamide, both being produced in the ion source of the mass spectrometer. The desorption signal obtained for m/z 43 must therefore be deconvoluted into contributions of the two products. This is achieved by scaling the m/z 45 desorption signal of formamide with the relative intensity of the m/z 43 fragment which was deduced from the mass spectrum of formamide (Figure 1b). Subsequently, the area of this scaled peak is subtracted from the integrated m/z 43 desorption signal leaving only the contributions of HNCO to the desorption peak (Figure 1c). These integrated desorption signals are then converted to relative product yields using partial ionization cross sections (Figure 1d). An appropriate electron exposure within the linear regime is then selected based on the dependence of relative yields on exposure (Figure 1e) after which the energy dependence is obtained by measuring the relative product yields at this selected electron exposure for a range of different electron energies (Figure 1f).
For reference, we also summarize the technical details of the experiments. Briefly, the experiment is housed in a UHV chamber (Figure 2) with a base pressure of 10−10 mbar maintained by turbomolecular pumps. The UHV chamber is connected to a gas handling manifold which consists of two storage and one mixing reservoir (see Figure 3). Reactants can be leaked from the storage reservoirs into the mixing reservoir by precision leak valves. The increase of partial pressures in the mixing reservoir which is typically chosen as a few tens of mTorr is monitored with a MKS Baratron type 622B capacitance manometer which allows one to establish a defined gas composition. In between two consecutive experiments, the mixing reservoir is evacuated by a turbomolecular pump.
Molecular ices were deposited by leaking gases or vapors from the mixing reservoir into the main chamber via a stainless-steel tube whose end is located in front of the metal foil substrate (Au [23,25,27,28,29,30,35,36] or Ta [24,26,31,32]) held at 30–35 K by a closed-cycle helium cryostat (Leybold RGD210 or C/P 2-10). During this deposition step, the pressure in the main chamber typically increases by 1–2 orders of magnitude. This ensures that there is sufficient partial pressure of the gas from the manifold in the chamber to form a film. The only major background gas under UHV conditions is H2, which does not co-condense. Prior to each deposition, the substrate is resistively heated. This ensures that adsorbates which may have accumulated on the surface are removed.
The thickness of the investigated ice layers is estimated from TDS data for individual compounds by observing at which pressure drop the transition from monolayer to multilayer desorption signal occurs [23]. TDS data are acquired by resistively heating the substrate with heating rates of ~1 K/s while monitoring characteristic m/z ratios of the investigated neutral desorbing species using a quadrupole mass spectrometer (QMS, Stanford Research Systems RGA200 or RGA300) residual gas analyzer with electron-impact ion source operating at an electron energy of 70 eV. Typically, ice layers are prepared with mean thickness in the range of 10 to 20 monolayers. This minimizes the contribution of the interface between the ice and the underlying substrate to the chemistry that is monitored. On the other hand, these layers are thin enough for the electrons to efficiently penetrate the ice. In the case of new results, the actual layer thickness is stated in the figure caption. We refer to the original publications for more detailed information on data from previous work.
A targeted mixing ratio of the two ice components of, for instance, 1:1 can be established in the simplest case by leaking a gas mixture with a molar fraction of 1:1 onto the substrate [28,29,32]. The actual stochiometric ratio of the two reactants in the ice can be deduced from the area under the TDS curves and considering the partial cross sections for electron impact ionization of the characteristic fragments that are monitored in the QMS [32]. The thus determined ice composition can deviate from the mixing ratio of the gases if one of the compounds adsorbs more strongly than the other. In this case, the gas mixture can be adjusted to produce a 1:1 composition of the ice as deduced from TDS [32]. Alternatively, the two compounds can be leaked sequentially. This is advantageous when one of them tends to form a porous ice such as H2O. The pores can then accommodate the molecules of the second gas and thus enhance its sticking probability. Better control over layer thickness is achieved, when the two gases are leaked sequentially.
Commercial non-monochromatized electron sources (Specs FG15/40 or STAIB NEK-150-1) with energies between 1 eV and 500 eV with a typical energy resolution of 0.5–1 eV are used for electron irradiation [24,25,26,28,29,31,32]. The electron exposure was determined by integrating the transmitted current over time and dividing by the sample area. Before and after irradiation, the substrate is biased with a negative potential which exceeds the range of investigated electron energies, while the electron source is left on. This allows for precise control over start and end of the irradiation, while at the same time preventing any electrons from reaching the sample during deposition or TDS.
Data analysis and visualization is performed with Python [38] using the Numpy [39], SciPy [40], and Matplotlib [41] packages. Baseline correction of the ESD and TDS data was performed, in the simplest case, by subtracting a constant value from the raw data. This value can be calculated by averaging the background signal. Alternatively, the baseline is fitted by a line or polynomial or, in the case of ESD experiments, with an exponential function to correct for any temporal drifts of the background signal. In the case of low signal intensities, the noise level is reduced by applying a second-order Savitzky-Golay filter [42,43] with a window length of 9 points. In order to compare signals from different experiments quantitatively, the area under the desorption signal is numerically integrated by the trapezoidal method. An experimental error of the integrated desorption signals is established by repeating selected experiments 3–5 times to calculate a standard deviation.
3. How Do Reactive Species Form under Electron Irradiation?
3.1. Basics of Electron Molecule Interactions
Upon interaction with a free electron, a molecule can be promoted to different states that are often more reactive than the molecular ground state. In electron ionization (EI), the impinging electron liberates another electron from the target to leave a radical cation behind. Electronic excitation (EE) leads to a neutral excited state while electron attachment (EA) to the molecule yields a radical anion:
M + e− → M•+ + 2 e− electron ionization (EI)
M + e− → M* + e− electronic excitation (EE)
M + e− → M•− electron attachment (EA)
The dependence of the three processes on electron energy is different, with EE and EI showing a threshold-type behavior. When the energy of the impinging electron exceeds the threshold for electronic excitation (EE) or the ionization threshold (EI), the cross section for interaction steadily increases until it reaches a plateau between 30 to 70 eV (for EE) and 50 to 100 eV (for EI) [16]. After this plateau, the cross section decreases due to ever shorter interaction times owing to the increasing speed of the electron. In contrast, electrons can attach to a molecule only if there is an anion state available for the energy of the incoming electron. This leads to EA in well-defined energy ranges called resonances. At energies typically below 5 eV, this happens in so-called single particle resonances, where the incoming electron occupies a previously unoccupied orbital. At higher energies, electron attachment is usually accompanied by electronic excitation leading to core-excited resonances.
The three states that a molecule M can occupy after the encounter with the electron (radical cation, radical anion, excited) can be more chemically reactive than the ground state molecule M. If these reactive species are in contact with other molecules or atoms, further chemical synthesis can be triggered.
The described processes assume that the molecule itself stays intact. Yet, molecules do in fact often dissociate after interaction with an electron, if the energy of the electron is sufficiently high to overcome the dissociation energy of a chemical bond in the molecule. This energy can be offset to some degree by the formation of new chemical bonds [16]. There are thus a number of further processes that can lead to the formation of reactive species:
M + e− → A+ + B• + 2 e− dissociative ionization (DI)
M + e− → A• + B• + e− neutral dissociation (ND)
M + e− → A− + B• dissociative electron attachment (DEA)
For DI, the thresholds are the same as for EI increased by the required bond dissociation energy. The same can hold for ND as compared to EE. Often, however, the presence of dissociation is not governed by additional energy but by whether the primary excited state M* is dissociative or not. This also applies to anion states M•− formed by EA but here dissociation competes with autodetachment of the electron [44]. Furthermore, anion states M•− are preferentially formed at near-thermal electron energies where excess energy can be removed from a particular bond by redistribution of energy to different internal degrees of freedom of the molecule [1]. There is also an additional dissociative process following electronic excitation, namely dipolar dissociation (DD) of the excited state into a cation and an anion. However, DD is usually considered to only have significant cross sections at energies higher than the ones discussed in this review [17,45,46,47]. We note that ND can also result from EA to a high-lying anion state which subsequently decays via autodetachment of the electron to produce neutral fragments [48]. This type of reaction mechanism has also been brought forward as bond breaking by catalytic electrons (BBCE) mechanism [49,50,51].
In the small organic molecules treated herein, the dissociative processes usually lead to separation of radical site and the ion into separate species. The radical species that are produced in this way drive most of the ensuing chemistry that occurs in condensed phase. Section 3.2, Section 3.3 and Section 3.4 present an overview of the reactive species (ionic and/or radical) that were identified as being relevant to the electron-induced chemistry discussed in Section 4. Section 3.5 then summarizes effects of the condensed phase that modify the fundamental electron-molecule interactions.
3.2. Reactive Intermediates Formed by EI/DI
In the studies reviewed here, the energy of the impinging electrons typically ranges up to around 20 eV. This energy range spans the lower part of the typical energy distribution of LESEs and includes their maximum yield for diverse materials [1,52,53]. The reactive species discussed here thus do not include ions and radicals with an appearance energy above ~20 eV.
Evidence for EI/DI as initiating step of electron-induced reactions in mixed ices has been obtained for the prototypical reactants C2H4, CO, NH3, H2O, CH3OH and CH4. The reactive species relevant to our studies that can be formed by EI/DI are summarized in Table 2. We omit for simplicity propene and amines because the reactions identified in this case were analogous to those of the shorter analogues C2H4 and NH3 [29]. With the exception of CH3OH, where significant fragmentation in the gas phase sets in almost at the ionization threshold, we did not find evidence that dissociation following EI contributes to the electron-induced reactions described in Section 4. While EI fragments with appearance energies below 20 eV are known [54], apparently their formation cross sections are too low to play a significant role in the studies reviewed here. We thus refer to the conditions present in the experiments as “soft ionization”.
3.3. Reactive Intermediates from ND
In many of the studied systems, product formation was observed starting from energies well below the ionization threshold [24,25,26,31]. When the energy dependence reveals threshold-type processes and not resonances, the only primary interaction available in this energy range (usually between 6 and 10 eV) is ND. Contrary to EI thresholds, the exact thresholds for ND are often unknown, even in the gas phase [45]. This is in part due to the fact that ND as a source for reactive species under electron irradiation is difficult to assess due to the experimental difficulties in monitoring reactive neutral radical species [17]. The only recourse for the identification of ND processes in electron-driven chemistry is thus by exclusion of EI/DI processes on grounds of the lower observed threshold energy and exclusion of EA/DEA processes by their non-resonant energy dependence. If an observed process starts near the minimum energy for electronic excitation, it is attributed to ND. We have observed product formation triggered by ND in systems containing NH3, H2O, CH4, and especially CH3OH, which produces a number of very important radical species (see Table 3). Note that in the case of CO and C2H4, the formation of products could be rationalized without having to resort to ND. While we did observe the formation of butadiene in the latter case, this was at energies that also allow DI processes [28,29].
3.4. Reactive Intermediates from (D)EA
Resonant structures in the energy dependence pointing to the formation of products via EA/DEA processes was observed for ices containing C2H4, CO, H2O, CH3OH and CH3CN. The relevant reactive species are listed in Table 4. In contrast, no true evidence for product formation that starts with EA to NH3 and CH4 was found in our studies. While we have invoked DEA to NH3 as explanation for the resonant formation of formamide from NH3 and CO at around 10 eV [30], recent preliminary results from our lab have led us to question this assignment, as we now see it as much more likely that the process is instead triggered by (D)EA to CO, as observed in the formation of other compounds from CO at that energy [24,26,37]. For reference, an overview of gas phase EA/DEA processes in the investigated reactants is given in Figure 4.
3.5. Effect of Condensed Phase
Most studies on the fundamental electron-molecule interactions are conducted in the gas phase. These crossed beam experiments typically have an energy resolution in the meV range and very low detection limits of charged particles [1,44]. For the interpretation of condensed phase experiments, these data are very helpful, but need to be treated with a bit of caution [63]. In particular, the threshold energies needed to form a charged particle are often lowered by 1 to 2 eV in the condensed phase, because charged particles are stabilized by polarization of the matrix [10,64]. For the same reason, desorption of particles is often hindered and occurs at higher energies than expected from the gas phase, or is suppressed entirely [25,65].
On top of the changes in energy, additional effects come into play once an electron-molecule interaction occurs in a molecular matrix. The caging effect can suppress fragmentation, as particles quickly lose energy to the matrix and can then recombine. This was studied in detail for methanol, where it was shown that in clusters, DI fragmentation is almost entirely absent, compared to the gas phase [66,67]. This can also lead to DEA resonances disappearing in the condensed phase (see for instance Ref. [68]). EA resonances can also initiate fast reactions with the matrix [69]. The radical anions formed from EA to CO or C2H4, CO•− and C2H4•−, are very strong bases and can quickly react with acidic protons from matrix molecules like H2O, yielding HCO• and ethyl radicals C2H5• respectively [24,28].
In a similar vein, the radical cations produced from EI can undergo acid-base reactions with neutral molecules [70] leading to proton transfer reactions [71] between H2O•+ and H2O to yield H3O•+ and HO• radicals, or between NH3•+ and NH3 to yield NH4+ and NH2• radicals. Such acid-base chemistry has also been observed for larger molecules like acetaldehyde [36,72], primary and secondary amines [29] and alcohols [31]. It is also important for H− that is produced by DEA to numerous molecules (see, for instance, Figure 4). As a strong base, H− abstracts a proton from many reactants. This yields species such as HO− or CH3O− when reacting with H2O or CH3OH [73].
While radical recombination reactions are typically barrierless, the reaction between a radical and a closed-shell species usually comes with a (small) energy barrier [74]. In cryogenic ices, this energy might not be available, which would lead to the stabilization of radicals. Observation has, however, shown that in addition to anionic and cationic species, also neutral radicals can undergo further reactions in the condensed phase. For instance, the hydroxyl radical HO•, will react with CO from the matrix, yielding the radical species HOCO• [24]. This is mostly due to the fact that dissociative electron interactions usually impart some excess energy to fragments, which makes them hotter than their surroundings, enabling rapid diffusion as well as radical molecule interactions. When attempting to understand reaction mechanisms, not only HOCO• [24], but also other secondary radical species, such as HCO• [24,26,30], C2H5• [28] or HOC2H4• [28] need to be considered.
4. How Do Reactive Species Undergo Chemical Transformation in Condensed Phase?
Electron irradiation is not only destructive but can also lead to molecular synthesis, i.e., the formation of more complex molecules from smaller entities [16]. In many cases, the underlying mechanisms are remarkably similar to those known from organic chemistry including nucleophilic displacement reactions [75], acid-base reactions [28], addition reactions [27,28,29], and oligomerization [76,77]. This suggests that electron-induced synthesis is in fact not a random re-assembly of atomic and molecular fragments but that it follows certain reaction principles. This chapter discusses the reactions that the initially formed reactive species can undergo by outlining these principles. In Section 4.1 (Radical recombination) we summarize evidence that concurrent reactions may occur so that the products are not simply explained by stochastic encounters of radicals. In Section 4.2 (Transfer hydrogenation). we present syntheses of new molecules that are initiated by formation of molecular radical anions via low-lying EA processes and subsequent protonation of the latter. Section 4.3 (Reduction by hydrogen radicals) deals with reactions that are caused by hydrogen radicals released upon dissociative electron-molecule interactions. Section 4.4 (Hydrogenation by methoxy radicals) then presents reactions where the reducing H⦁ is instead supplied by transfer from a methoxy radical. In Section 4.5 (Addition reactions) we summarize reactions in which a radical or ion adds to multiple bonds of a reaction partner leading to atom-efficient synthesis of larger molecules. And finally, Section 4.6 (Oxidation of CO to CO2) discusses the mechanism by which CO is oxidized in electron-induced chemistry.
4.1. Radical Recombination
Current astrochemical models assume that radical recombination is the dominant mechanism by which molecular synthesis occurs in interstellar ices [34,78,79,80]. However, neutral radicals which are produced in ices by the interaction of molecules with electrons or UV photons are typically surrounded by closed-shell molecules only. Radical–molecule reactions thus require no diffusion through the ice but are limited by activation barriers [74] which might not be accessible at cryogenic temperatures. Diffusion, on the other hand, requires less energy allowing the radicals to travel until they encounter a second radical where a typically barrierless recombination reaction can occur. On this basis, recombination would be governed by the stochastic encounter of different radical species as might also be suggested, for instance, from the products observed upon electron irradiation of condensed CF2Cl2 [10,81]. As has been discussed in Section 3.5, this paradigm does not necessarily hold true because radicals released during dissociation of the parent molecule might have considerable excess energy which can be sufficiently high to overcome the activation barriers of radical-molecule reactions even at low temperatures. In fact, theoretical studies suggest that transfer of a hydrogen atom between two radicals, also referred to as disproportionation, might dominate over recombination [82,83,84,85]. An example of such a disproportionation reaction would be the reaction between two CH3O• radicals yielding formaldehyde (H2CO) and CH3OH [31]. A similar case has been observed in matrix isolation experiments for the reaction between two HCO• radicals which disproportionate to H2CO and CO rather than recombine to glyoxal (HC(O)CH(O)) [82].
As an example of a recombination reaction, the formation of N2 in ices containing NH3 (see also Section 4.1) relates to the encounter of NH2• radicals. This is supported by the detection of hydrazine (N2H4) as the immediate recombination product [29,86]. N2H4 was identified, together with diimide (N2H2), upon electron irradiation of pure NH3 layers [86]. The intermediate chemisorbed NH2• radicals were observed earlier upon electron-irradiation of a bilayer of NH3 at E0 = 50 eV as deduced from high-resolution electron energy loss spectroscopy (HREELS) and X-ray photoelectron spectroscopy (XPS) [87]. Loss of hydrogen from NH3 may result from DEA. However, the present results (Figure 5) reveal a lack of resonances in the energy dependent yield of both N2H4 and N2 from NH3 ice (Figure 5) which argues against a DEA-driven mechanism as dominant source of NH2• radicals.
Among the molecules reviewed in this work, radical recombination is particularly important in the case of CH3OH containing ices. In CH3OH, three different bonds can be cleaved yielding CH3O•, •CH2OH, CH3•, HO•, and H• radicals. Following electron irradiation of methanol ice, the recombination products methoxymethanol (CH3OCH2OH), ethanol (C2H5OH), ethylene glycol (HOC2H4OH), and dimethyl ether (CH3OCH3) were identified [31]. The similar energy dependencies for these products (Figure 6) suggest that the different radicals are produced by the same electron-molecule interactions. Assuming that radical recombination is in fact the dominant reaction, the relative product yields would reflect branching ratios with which the different radicals recombine. This approach was recently used in a photodissociation study of CH3OH [79]. The authors deduced branching ratios into CH3O•, •CH2OH, and CH3• radicals from the relative yields of ethanol, dimethyl ether, and ethylene glycol [79]. However, considering recent theoretical predictions that disproportionation dominates over radical recombination [82,83,84,85], it is obviously not possible to deduce the branching ratios by which CH3O•, •CH2OH, and CH3• radicals are formed because, unlike for recombination products, the products of such hydrogen transfer are not linked to specific primary radicals. For example, it is not possible to distinguish between H2CO that is formed by hydrogen transfer between two CH3O• radicals, two •CH2OH radicals, or between a CH3O• and a •CH2OH radical.
Considering the possible combinations of radical fragments of CH3OH listed above, it is obvious that more compounds would be expected upon recombination governed by stochastics such as dimethyl peroxide (CH3OOCH3) and ethane (C2H6). These, however, were not detected which calls for an explanation. Dimethyl peroxide, which is formed by recombination of two CH3O• radicals, could only be detected by the Kaiser group by use of a very sensitive photoionization reflectron time-of-flight mass spectrometer [88]. This suggests that dimethyl peroxide is produced in much smaller yields than other recombination products so that it is below the detection limit of our mass spectrometer. The competition of radical recombination with disproportionation provides a reasonable explanation why only certain recombination products are observed in high yields. Here, a higher efficiency of the disproportionation reaction between two CH3O• radicals yielding CH3OH and H2CO as compared to recombination can rationalize the low yield of dimethyl peroxide.
There are no reports on the formation of C2H6 under irradiation of CH3OH ice which is expected to form by coupling of two CH3• moieties, while its formation from the electron irradiation of CH4 (Ref. [89] and this work) and CH3CN [25] was indeed observed (Figure 7). In the case of CH4 ice (Figure 7, top), the threshold-type behavior of the yield of C2H6 points to formation through a reaction driven by ND or EI. In contrast, pronounced resonances are observed in the energy dependence of C2H6 yields upon irradiation of CH3CN [25]. They reflect known DEA processes that lead to release of CN− which is presumably accompanied by loss of CH3• radicals [62,90]. Öberg proposed that the lack of C2H6 formation under irradiation of CH3OH results from the higher abundances of CH3O• and •CH2OH radicals which favors reactions of CH3• with either CH3O• or •CH2OH over recombination of two CH3• radicals [79]. In addition, CH3⦁ radicals have a higher mobility at 35 K than CH3O⦁ and ⦁CH2OH [79,91] which might enable quick trapping of CH3⦁ radicals before the majority of CH3O⦁ and ⦁CH2OH radicals are consumed by other reactions. Disproportionation of two CH3• radicals would yield methylene (CH2) and CH4 which is endothermic by 0.21 eV as has been deduced from the standard enthalpies of formation [92] and should thus not be very efficient. However, CH3• radicals could react with CH3OH by abstracting a H• from the latter, which yields CH4 as also observed in the irradiation of methanol [31] and either CH3O• or •CH2OH. Thus, hydrogen radical abstraction, which competes with radical recombination, might be a crucial factor that prevents formation of C2H6, as will be discussed in the Section 4.2 in detail.
Recombination of radicals is also observed in CO/CH3OH [26] and C2H4/CH3OH mixed ices [32], although the yields may be lower compared to pure CH3OH ice samples with the same film thickness as is the case for the product ethylene glycol (HOC2H4OH) (Figure 8, shaded in grey. On one hand, this is because the amount of CH3OH is smaller when part of it has been replaced by a second reactant. On the other hand, radical recombination competes with concurrent reactions such as hydrogenation of CO and C2H4 which proceeds via different mechanisms (see Section 4.2, Section 4.3 and Section 4.4) and addition of radicals to CO or C2H4 (see Section 4.5). Because of these concurrent reactions, radical recombination is neither a purely stochastic process nor the only possible reaction by which radicals form stable products.
4.2. Transfer Hydrogenation
Transfer hydrogenation is an important reaction class in organic chemistry which subsumes all hydrogenation reactions where the reducing agent is not gaseous H2 [93]. Many of those reactions rely on the proton transfer between a radical anion and a proton donor, often an alcohol. A prominent example of such reactions is the Birch reduction [94,95] which can be used to reduce aromatic compounds. The reaction employs solvated electrons as reducing agents which are provided by dissolving either Na or Li in liquid NH3 [94,96]. Subsequently, these electrons add to an aromatic molecule yielding a radical anion. In a final reaction step, the radical anion is protonated by an alcohol, leading to hydrogenation. Depending on the reaction conditions, the molecule can undergo this reaction sequence multiple times leading to further hydrogenation. Several variations of the Birch reduction have been developed including electrochemical [97] and photochemical [98] all of which, however, rely on the initial formation of a radical anion.
Similar to the mechanism of the Birch reduction, EA to CO was observed at E0 = 4 eV in CO/H2O mixed ices. This was inferred from the formation of formaldehyde (H2CO) at this energy (Figure 9). EA yields an intact CO⦁− radical anion which can undergo proton transfer with H2O yielding HCO⦁ and HO−. Subsequently, HCO⦁ reacts with a nearby H2O molecule yielding H2CO and HO⦁ [24]. Recall that the resonance at 4 eV can only be due to EA to CO as DEA to H2O occurs at considerably higher energies (Figure 4). As noted in Section 3.5, an analogous reaction leading to the formation of ethanol and its side products ethane (C2H6) and butane (C4H10) was observed after EA to C2H4 in C2H4/H2O mixed ices at electron energies below 5–6 eV [28]. In this case, EA produces the C2H4⦁− radical anion which undergoes proton transfer with H2O to yield C2H5⦁ and HO− (Scheme 1). Subsequently, the C2H5⦁ radical can attack the double bond of another C2H4 moiety to form a dimer C4H9⦁ which can react further in a chain reaction. If either of the alkyl radical intermediates reacts with H2O, however, the chain propagation is terminated yielding C4H10 and a HO⦁ radical. The HO⦁ radical, in turn, can attack a nearby C2H4 moiety forming HOC2H4⦁. Finally, reaction with a nearby H2O molecule again yields ethanol and a further HO• (Scheme 1) potentially resulting in another chain reaction. Consideration of side products such as the different alkanes can help to identify the relevant intermediates and thus leads to a more comprehensive view of the reaction. We note, however, that such a reaction was not observed in C2H4/NH3 mixed ices [29] suggesting that a sufficiently high acidity of the proton donor is essential for the reaction to occur.
This type of reaction has also neither been observed in C2H4/CH3OH nor in CO/CH3OH mixed ices despite the fact that the gas phase acidity of CH3OH is in fact higher than that of H2O [99]. One reason for this lack of proton transfer from CH3OH could be the effect of the molecular environment. For example, the order of acidities of H2O and CH3OH is reversed in aqueous solution as HO− is more strongly stabilized than CH3O− [100,101]. Thus, depending on the stability of the CH3O− anion in a CH3OH matrix, the acidity of CH3OH might be too low to enable proton transfer. Another factor might be the effect of different molecular environments on the stability of the radical anion as has been reported for acetonylacetone [102]. A water matrix might be able to stabilize the CO•− and C2H4•− radical anions so that they have a sufficient lifetime to undergo subsequent chemistry whereas CH3OH is not. However, the finding that H2CO is formed via EA to CO in CO/H2O mixed ices [24] and the formation of ethanol in C2H4/H2O mixed ices via EA to C2H4 [28] demonstrate the relevance of transfer hydrogenation reactions, analogous to the Birch reduction, for electron-induced chemistry of molecular ices.
4.3. Reduction by Hydrogen Radicals
ESD and crossed beam experiments on DEA to H2O and CH3OH revealed the production of HO− [103] and CH3O− [61,104,105] (see Figure 4), respectively. This must be accompanied by the release of H•. In addition, there is evidence that hydrogen radicals are produced by ND of H2O [106]. Following these and possibly other initial electron-molecule interactions (see Section 3.3 and Section 3.4), the released H•, which are particularly mobile, can react with unsaturated compounds such as CO or C2H4 enabling reaction pathways that are neither accessible in pure H2O nor in CH3OH. Hydrogenation of CO has been reported to occur during electron irradiation of CO/H2O and CO/CH3OH mixed ices yielding H2CO, CH3OH, and intermediate radicals (Reaction 1) [78,80,107,108,109].
The hydrogenation of CO by H2O and CH3OH was also investigated by studying the energy dependence of H2CO formation [24,26]. In the case of CO/H2O mixed ices [24], the threshold around 7 eV (Figure 10) suggests that Reaction 1 is mainly triggered by ND of H2O into H• and HO•. In fact, the yield of H2CO does not exhibit resonances in the energy range from 6–13 eV suggesting that DEA of H2O into HO− and H• (Figure 4) does not significantly contribute to the hydrogenation of CO. Note that the resonant formation of H2CO at 4 eV results from EA to CO and subsequent protonation of the intermediate CO•− radical anion by H2O (Section 4.2).
In CO/CH3OH mixed ices [26], a more sophisticated approach is required to study Reaction 1 as H2CO is not only formed by hydrogenation of CO but also by fragmentation of CH3OH [26,31]. Other studies have differentiated between these two possibilities by using 13C and 18O labelled CH3OH and CO [78,80]. However, there is an alternative approach to isotopic labeling. Due to contributions of Reaction 1 to the overall yield, the energy dependence of H2CO formation in CO/CH3OH mixed ices differs from that in pure CH3OH, where it is dominated by a high-lying resonant process [31] (Figure 11a). The energy dependence of this additional formation channel shows an onset at ~7 eV, in good agreement with the electronic excitation energy of CH3OH which is about 6.4 eV in the gas phase [110] and 6.7 eV in the condensed phase [111]. The contribution of this channel to the H2CO yield increases steadily with electron energy (Figure 11a), indicative of a non-resonant process. This agrees well with the hypothesis that ND of CH3OH yields hydrogen radicals which then reduce CO according to Reaction 1. There is no further threshold near the ionization energy of CH3OH suggesting that EI does not produce significant amounts of H• which could contribute to hydrogenation of CO.
A similar result was observed at lower electron energies (Figure 11b). Here, the two DEA channels of CH3OH at 5–6 eV yielding CH3O– and H•, and CH3O• and H− (Figure 4) are responsible for the formation of H2CO [26]. In pure CH3OH ice, the H2CO yield increased linearly with electron energy. This linear increase is ascribed to an overlap of the DEA channel yielding CH3O• and H− (Figure 4) [105,112] with the onset of ND. [110,111] Subsequently, two CH3O• radicals can undergo disproportionation to yield H2CO and CH3OH. The DEA channel yielding CH3O− and H• (Figure 4) does not contribute to H2CO formation in pure CH3OH. In the presence of CO, the released H• can react with CO (Reaction 1), enabling formation of additional H2CO. This additional reaction channel is reflected in the energy dependence of H2CO where the linear increase of product yield is overlaid with a more intense DEA resonance at ~6 eV (Figure 11b).
In the CO/CH3OH mixed ice, the DEA channel at 5.5 eV yielding CH3O− and H• (Figure 4) does not only lead to the formation of H2CO but also produces methoxymethanol (CH3OCH2OH) which is not observed after electron irradiation of pure CH3OH at E0 = 5.5 eV (Figure 12). This is because this DEA channel only yields CH3O⦁ and H− but no ⦁CH2OH radicals [112]. The recombination reaction between CH3O⦁ and ⦁CH2OH radicals needed for formation of methoxymethanol in pure CH3OH thus does not occur at this low electron energy. In CO/CH3OH mixed ices, however, ⦁CH2OH radicals are formed by successive hydrogenation of CO (Reaction 1) which produces both types of radicals and thus enables the formation of methoxymethanol by recombination of CH3O⦁ and ⦁CH2OH radicals.
The DEA resonance of CH3OH at E0 = 5.5 eV is also observed in C2H4/CH3OH mixed ices where it causes hydrogenation of C2H4 yielding ethane (C2H6) (Figure 13a, shaded in grey) [32]. Note that no significant amounts of C2H6 are formed after irradiation of pure CH3OH (Section 4.1) or C2H4 at E0 = 5.5 eV (Figure 13a) suggesting that formation of C2H6 mainly occurs by a reaction between CH3OH and C2H4. Two reaction mechanisms contribute to the formation of C2H6 which are based on the two competitive DEA channels of CH3OH yielding CH3O− and H•, and CH3O• and H− (Figure 4) [104,112]. Following the first of these two DEA channels, the released hydrogen radicals can add to C2H4 producing an intermediate C2H5• radical. Subsequently, the C2H5• radical can abstract a H• from a nearby CH3OH molecule to yield C2H6 and CH3O•. The proposed mechanism is supported by the concomitant appearance of butane (C4H10) (Figure 13b) which is formed when the intermediate C2H5• radical reacts with a second C2H4 molecule to yield C4H9• before it abstracts a H• from a nearby CH3OH molecule. The reactions of the CH3O• radical which can also act as a reducing agent and the consequent outcome of the second DEA channel are discussed in Section 4.4. The relevance of reduction by H• has also been noted in the case of the electron-induced reactions of C2H4 with NH3 where electron-induced dissociation of NH3 releases H• [29] (Section 4.1 and Section 4.5). We note that related reactions of H• have also been observed in the cases of cisplatin, which is a complex carrying a NH3 ligand [113], and of η3-allyl ruthenium tricarbonyl chloride in the presence of NH3 [114] which underlines the wider perspective of the chemical concepts described herein.
4.4. Hydrogenation by Methoxy Radicals
Unsaturated molecules cannot only be reduced by free hydrogen radicals as discussed in Section 4.3, but also by CH3O• radicals. These are produced by DEA to CH3OH around E0 = 5.5 eV along with H– (Figure 4), by ND yielding CH3O• and H•, and by EI yielding a CH3OH•+ radical cation which can protonate a nearby CH3OH molecule to form CH3OH2+ and CH3O•. The CH3O• radical can transfer a hydrogen radical to C2H4 which produces H2CO and C2H5⦁ (Reaction 2). This is supported by the increase of the H2CO yield upon electron irradiation at E0 = 5.5 eV when CH3OH is mixed with C2H4 (see blue shaded signal in Figure 13a). Analogous to the reactions following addition of free hydrogen radicals to C2H4 (Section 4.3), the so formed C2H5• radical can react with a CH3OH molecule to yield C2H6 and regenerate the CH3O• radical (Reaction 3). In a concurrent process, the intermediate C2H5⦁ radical can add to a second C2H4 molecule before it reacts with CH3OH to finally yield butane (C4H10) (Reaction 4).
Note that the CH3O• radical is recovered at the end of Reactions 3 and 4 so that the net chemical equations simplify to:
The overall contribution of hydrogen transfer from CH3O• radicals to the hydrogenation of C2H4 can be estimated by comparing the relative yields of H2CO, C2H6, and butane (C4H10). Considering the stoichiometry of Reactions 5 and 6, the amount of H2CO should be equal to the total amount of C2H6 and butane (C4H10). A reanalysis of the data obtained after irradiation at 5.5 eV with 8000 µC/cm2 [32] revealed the yields of H2CO, C2H6, and butane (C4H10) to be 1.5%, 2.6%, and 1.3% with respect to the amount of CH3OH in a non-irradiated sample. Consequently, the total amount of C2H6 and butane was 2.6 times higher than the amount of H2CO. Thus, hydrogen transfer from CH3O• to C2H4 accounts for about 40% of the formed C2H6 and butane (C4H10), whereas the addition of free H• radicals to C2H4 (see Section 4.3) accounts for about 60% of these products. Reaction 2 has been studied in particular after DEA to CH3OH at 5.5 eV [32] but may also play a role for the reduction of C2H4 following CH3O⦁ formation after ND or EI of CH3OH. Recall that the amount of H2CO after irradiation of pure CH3OH at E0 = 5.5 eV was significantly lower than in C2H4/CH3OH mixed ices (Figure 13a) suggesting that unimolecular dissociation of CH3O⦁ into H2CO and H⦁ (Reaction 7) is not efficient under the experimental conditions of cryogenic ices.
Notably and in contrast to C2H4, CO is not reduced by CH3O⦁ radicals (Reaction 8) [26].
This has been inferred by comparing the energy dependence of H2CO with that of methyl formate (CH3OCHO). As the production of methyl formate relies on the addition of CH3O• radicals to CO (see Section 4.5), it can be used as a proxy for CH3O• formation. Notably, only the energy dependence of methyl formate but not that of H2CO shows a resonance at 10 eV (Figure 14). At this energy, CH3O• is produced through an ion-molecule reaction of CH3OH with O•− that is released by DEA to CO [115]. The lack of resonant enhancement in the yield of H2CO suggests that hydrogen transfer from CH3O• to CO is not efficient.
The different reactivity of CH3O⦁ with regard to hydrogen transfer to CO and C2H4 can be explained by considering the energy profiles of these reactions. To this end, quantum chemical calculations were performed (Figure 15). The calculated reaction energies and barriers for the decomposition of CH3O⦁ into H2CO and H⦁, and the hydrogen transfer from CH3O⦁ to CO to yield H2CO and HCO⦁ agree well with published and validated data (Figure 15a,b) [116,117]. The values of our present calculation for the hydrogen transfer from CH3O⦁ to C2H4, which have not been reported so far, should thus have a comparable accuracy as the results for the other two cases (Figure 15c).
The computations reveal that the unimolecular decomposition of CH3O⦁ into H2CO and H⦁ is endothermic by 0.89 eV and has a reaction barrier of 1.07 eV (Figure 15a). The presence of CO, which acts as a hydrogen acceptor, significantly lowers the reaction energy although the reaction is still endothermic by 0.28 eV. The reaction barrier, however, is not significantly lowered compared to dissociation of CH3O⦁ into H2CO and H⦁ (Figure 15b). In the case of CH3O⦁ and C2H4, the reaction is exothermic by −0.64 eV, and the reaction barrier is lowered to a value of 0.76 eV (Figure 15c). Thus, the trend seen in the calculated reaction barriers agrees with the experimental observation that only hydrogen transfer from CH3O• to C2H4 occurs. Note, however, that even in the case of a reaction with C2H4 the CH3O• radical must have sufficiently high excess energy to overcome the reaction barrier of 0.76 eV. The thermodynamic threshold for the formation of CH3O• and H− is 3.75 eV [112]. Under the assumption that the excess energy is only released as kinetic energy of the fragments, the kinetic energy of the CH3O• fragment can be calculated by [103] (1–31/32) × (E0 –3.75 eV) which yields only 0.055 eV at E0 = 5.5 eV, if vibrational excitation is neglected. This kinetic energy of CH3O• is not sufficient to overcome the reaction barrier suggesting that a considerable amount of the released excess energy must be stored in the rovibrational modes of CH3O•. Hydrogen transfer from CH3O• to C2H4 is thus relevant to the electron-induced chemistry in molecular ices under the condition that CH3O• is formed with sufficient excess energy to overcome the reaction barrier.
4.5. Addition Reactions Involving Radical Cations or Neutral Radicals
The formation of larger products from small ice components is most conceivable if the reactants do not dissociate upon interaction with the incident electron. This is the case when the electron energy is above the ionization threshold of a reactant but the excess energy is not sufficient to overcome any bond dissociation energy. The resulting intact radical cation of a reactant can then react with an adjacent molecule in the ice. If the two reactant molecules combine in an addition reaction to yield a larger product which contains all atoms of the reactants, the reaction is called atom-efficient.
An atom-efficient electron-induced reaction has first been identified in binary ice layers consisting of C2H4 and NH3. Formation of ethylamine (C2H5NH2) [27,29], i.e., an electron-induced hydroamination, was observed for electron energies above the ionization thresholds of C2H4 and NH3 [27]. However, the electron energies were still below the onset of DI at least for NH3 and not much above the threshold for C2H4 [54]. The proposed mechanism is shown in Scheme 2. Ionization of either C2H4 or NH3 leads to an attractive Coulomb force between the reactants which allows them to circumvent the reaction barrier that prevents a spontaneous reaction between the neutral molecules [27]. Reaction between C2H4 and NH3 after EI yields a cationic adduct (structure I in Scheme 2) as an intermediate. Intramolecular rearrangement of I into the ethylamine radical cation (structure II in Scheme 2) and subsequent neutralization by a thermalized electron eventually yields ethylamine. A theoretical study confirmed that bond formation between the reactants in the cation state is in fact barrierless in contrast to the neutral ground state where an activation energy of more than 2.2 eV must be overcome [120]. The mechanism in Scheme 2 has also been adopted to explain the formation of ethanol from C2H4/H2O mixed ices [28], the formation of formamide from CO/NH3 mixed ices [30], and the formation of ethyl methyl ether in C2H4/CH3OH mixed ices [32].
The hydroamination mechanism presented in Scheme 2 has been examined more comprehensively in a subsequent study [29] by investigating the dependence of product yield on electron energy and sample composition, as well as the formation of side products. Experiments were also performed using NH3 and propene (CH2=CHCH3), ethylamine and C2H4, and diethylamine ((C2H5)2NH) and C2H4 in order to probe possible steric effects. Hydroamination products were observed in all cases and the product yields exhibited a threshold-type behavior with onset around 8 eV (Figure 16). This is lower than the gas phase ionization energies of most of the investigated reactants (10.1 eV for NH3, 10.5 eV for C2H4, 9.7 eV for propene, values from 8.7 eV to 9.5 eV for ethylamine, and 7.9 eV to 8.6 eV for diethylamine [54]). The lower onset of product formation as compared to the gas phase ionization energies can be ascribed to the effect of the condensed phase (Section 3.5). This is illustrated by calculations performed on the B3LYP/6-311 + G(d) level of theory (Figure 17). In the first step, an isolated NH3 molecule was ionized resulting in a vertical ionization energy of 10.8 eV, which is in reasonable agreement with the experimental adiabatic gas phase value (Table 2). The NH3⦁+ was then placed near a C2H4 molecule, embedded in a cluster of further six NH3 molecules, and allowed to relax (MIN0 in Figure 17) resulting in a vertical ionization energy of 7.3 eV (MIN0⦁+ in Figure 17) which coincides roughly with the observed onset of ethylamine formation (Figure 16). As discussed in Section 3.5, further relaxation of this structure proceeds via proton transfer from the NH3⦁+ to an adjacent NH3 molecule producing NH4+ and a NH2⦁ radical and results in a major energy gain of 5.9 eV as compared to an isolated NH3⦁+ (MIN1⦁+ in Figure 17). From the relaxed state MIN1⦁+, a shallow barrier with a height of only 0.3 eV where the NH2⦁ radical approaches the second reactant C2H4 (TS1⦁+ in Figure 17) leads to another minimum (MIN2⦁+ in Figure 17). Here, a C-N bond has formed and another neutral NH3 interacts via one of its H atoms with the other C atom. Adding an electron to this cluster to simulate recombination with a thermalized electron in the condensed layer (MIN2 in Figure 17) then leads to bond flipping through the cluster which recovers the neutral NH3 molecules and forms a new C-H bond relaxing to the final product ethylamine (MIN3 in Figure 17). The previous theoretical study, that included only one NH3, placed that transition state on the cation potential energy surface only 1.38 eV below the isolated reactants NH3⦁+ and C2H4 [120]. The presence of more NH3 molecules at the reaction site stabilizes the transition state and thus helps to drive the reaction towards the product.
Considering the wealth of different electron-molecule interaction that yield reactive species (see Section 3), it is not surprising that other products were also observed. For example, ethane (C2H6) and butane (C4H10) were identified together with ethylamine in C2H4/NH3 mixed ices [29]. Although these products were also formed upon electron irradiation of pure C2H4, the product yields were considerably enhanced in the presence of NH3 suggesting that NH3 acts as a reducing agent. These reactions are governed by radical formation and recombination (Section 4.1), and are further driven by the action of free hydrogen radicals (Section 4.3).
It is also conceivable that the addition of a neutral radical to C2H4 is an alternative to the EI-driven hydroamination in the case of C2H4/NH3 mixed ices. Here, NH2⦁ radicals, that can for instance be formed by ND (Section 3.3), would add to the double bond of C2H4. This would yield an intermediate H2N–C2H4⦁ radical which may then abstract a H• from a nearby NH3 to convert to ethylamine (Scheme 3). This type of reaction is also closely related to the mechanism presented in Figure 17 where the NH2⦁ radical results from hydrogen transfer from a neutral NH3 to an ionized NH3⦁+ radical cation. Whether ND contributes to the yield of ethylamine can likely be unraveled by photon irradiation experiments on C2H4/NH3 mixed ices analogous to those reported previously for pure ND3 ice [122].
A similar reaction could occur whenever a radical intermediate is formed in a mixed ice that contains a species with multiple bonds, such as CO or C2H4. As discussed in Section 4.1, radical intermediates are particularly prominent when CH3OH is present as reactant in an ice layer but can also be formed from H2O (see Section 3). Therefore, a reaction mechanism involving radical addition is proposed for the formation of formic acid from CO and H2O, and methyl formate and ethyl methyl ether from C2H4 or CO and CH3OH, respectively. It is based on addition of a RO• radical (R = H, CH3) to CO or C2H4, respectively (Scheme 4). The energy dependences of product formation can then be traced back to the reaction channels that yield a RO• radical (R = H, CH3).
The energy dependences of methyl formate in CO/CH3OH mixed ices [26] and formic acid in CO/H2O mixed ices [24] reveal resonant product formation at E0 = 10 eV (Figure 18). Assignment of this resonance to a specific DEA process in a particular molecule is not directly possible because CO, H2O, C2H4 and CH3OH do all evince DEA resonances at around 10–11 eV in both gas phase (Figure 4) [56,112] and condensed phase [105,123,124,125,126,127] experiments. No resonances are observed at E0 = 10 eV for the formation of ethyl methyl ether in C2H4/CH3OH mixed ices (Figure 18b) and for the formation of ethanol in C2H4/H2O mixed ices (Figure 18a), suggesting that the 10 eV resonance is due to DEA to CO yielding C and O⦁−. The latter can react with a nearby H2O or CH3OH molecule to yield HO• or CH3O•, respectively (Reactions 9 and 10) [115].
DEA to CO thus clearly dominates over DEA to H2O and CH3OH at 10 eV, at least in the mixed ices investigated in the experiments summarized herein. This is in contrast to the gas phase where the cross sections for the formation of O•− by DEA at 10 eV are 2 × 10−19 cm2 for CO [45], 5.76 × 10−19 cm2 for H2O [60], and 4.5 × 10−20 cm2 for CH3OH respectively [128].This demonstrates that the relative DEA cross sections in the mixed ices differ significantly from those in the gas phase.
In the case of CO/CH3OH and C2H4/CH3OH mixed ices, a second resonant process was identified when exposures of 12,000 and 8000 µC/cm2, respectively, were applied (Figure 19). Here, DEA to CH3OH at E0 = 5.5 eV produces CH3O• and H− (Figure 4) which leads to the production of methyl formate and ethyl methyl ether, respectively. The complementary DEA channel that yields CH3O− and H• (Figure 4) also contributes to the production of methyl formate by hydrogenation of CO to CH3O• (Reaction 1, Section 4.3). DEA to C2H4 cannot be responsible for the observed resonance as the known DEA channels in the gas phase are located at considerably higher energies (Figure 4) [58,59,129], while the EA resonance of C2H4 which contributes to the formation of ethanol in C2H4/CH3OH mixed ices occurs at lower energy (Figure 18a) (see Section 4.2).
In addition to the 10 eV and 5.5 eV resonances, there are non-resonant contributions to the formation of formic acid, methyl formate, and ethyl methyl ether, which are due to ND and/or EI of H2O and CH3OH. In the case of CH3OH containing ices, EI clearly dominates over ND as can be seen by the onset for the formation of ethyl methyl ether at ~10 eV (Figure 18b). This value agrees nicely with the ionization threshold of CH3OH which is at 10.84 eV in the gas phase [54], and at 9.8 eV in the condensed phase [64]. In the case of methyl formate and formic acid, the non-resonant contributions to product formation cannot be unambiguously identified to be dominated by ND or EI as the onset of this second formation channel is masked by the DEA resonance at 10 eV (Figure 18). However, both ND and EI of H2O and CH3OH can produce RO• (R = H, CH3) radicals and thus contribute to some extent to the formation of formic acid and methyl formate.
After EI of H2O or CH3OH, the respective radical cation can transfer a proton to a nearby H2O or CH3OH molecule to yield a HO• or a CH3O• radical, respectively, along with a protonated H2O or CH3OH molecule (Reactions 11 and 12).
Furthermore, ND of H2O or CH3OH yields HO• or CH3O•, and a H• radical (Reactions 13 and 14) once the electron energy is higher than the electronic excitation energy.
In addition, DEA to CH3OH yields CH3O• and H− (Figure 4). Note that in CO/CH3OH mixed ices, additional CH3O• radicals are also produced by hydrogenation of CO (Reaction 1, Section 4.3) following DEA of CH3OH into CH3O− and H• (Figure 4) and by ND of CH3OH.
Regardless of how the HO• or CH3O• radicals are formed and at which electron energy (Reactions 9–14), they can subsequently react with CO to yield an intermediate HOCO• or CH3OCO• radical which can then abstract a H• from a nearby H2O or CH3OH molecule to yield formic acid or methyl formate, respectively (Scheme 4). Similarly, they can add to C2H4 to yield an intermediate CH3OC2H4• or HOC2H4• radical which can abstract a H• from a nearby CH3OH or H2O molecule to yield ethyl methyl ether or ethanol (Scheme 4). At the end of the reaction, the RO• radical is recovered, potentially inducing a chain reaction.
4.6. Oxidation of CO to CO2
In contrast to the reduction by transfer of hydrogen as discussed in Section 4.2, Section 4.3 and Section 4.4, electron irradiation can also initiate oxidation reactions. For instance, CO is readily oxidized to CO2 upon electron irradiation of CO/H2O [24] and CO/CH3OH [26] mixed ices. This CO2 formation has usually been attributed to either of two mechanisms: Formation and decay of an intermediate ROCO⦁ radical (R = H, CH3), or reaction of an oxygen atom with CO [107,108,130,131,132]. Considering the first mechanism, reaction of HO⦁ or CH3O⦁ with CO yields the intermediate HOCO⦁ or CH3OCO⦁ radicals, respectively. These intermediates can either be stabilized by the condensed phase which favors their subsequent conversion to formic acid (HCOOH) or methyl formate (CH3OCHO), respectively, or they can decay into CO2, and H⦁ or CH3⦁.
Results on electron-induced reactions in mixed CO/H2O mixed ices [24] suggest that HOCO⦁ is indeed sufficiently stabilized by the H2O matrix to enable its further reaction to formic acid (see Section 4.5). In contrast, decay of HOCO⦁ does not play a significant role for the formation of CO2. This has been inferred by comparing the energy dependences of CO2 with those of H2CO and formic acid (Figure 20). As discussed in Section 4.2, H2CO can only be formed via an intermediate HCO• radical. This process is enhanced around 4 eV (Figure 20), where formic acid is not observed. Therefore, the resonant formation of formic acid at 10 eV cannot be ascribed to HCO• but must proceed via the HOCO• radical. Conversely, it can be excluded that a significant amount of CO2 is formed by decay of HOCO• as the resonance at 10 eV is not reflected in the energy dependence of CO2 (Figure 20). It was thus proposed that CO2 is mainly formed by the competitive ND channel of H2O into H2 and O and subsequent reaction of the latter with CO [24].
In contrast to the result for HOCO•, the CH3OCO• radical which is formed upon irradiation of CO/CH3OH mixed ices [26] readily dissociates into CO2 and CH3•. This has been inferred from the formation of dimethyl ether (CH3OCH3) and methyl acetate (CH3C(O)OCH3) which are specific side products at E0 = 5.5 eV formed by recombination of the CH3• radical with CH3O• and CH3OCO•, respectively (Figure 21). At this energy, CH3O• is produced by DEA to CH3OH (Section 3.4) and CH3OCO• results from addition of the latter to CO (see also Section 4.5). Notably, dimethyl ether is not formed upon irradiation of pure CH3OH at E0 = 5.5 eV (Figure 21b) where CH3O• is resonantly produced together with H- (Figure 4) so that direct production of CH3• radicals by dissociation of CH3OH can be ruled out at this low energy [26,31]. Equally, methyl acetate cannot be formed in pure CH3OH (Figure 21a) because this requires not only CH3• radicals but also CO which is known to be produced by electron irradiation of CH3OH only at higher energies [48]. ND of CH3OH into CH4 and O may also contribute to CO2 formation to some extent as has been suggested previously but not directly observed [130,133].
Studies applying molecular mechanics suggest that in water ice, HOCO• loses all of its excess energy within a few picoseconds [134] which makes it impossible for HOCO• to overcome the barriers toward CO2 and H•. Thus, such a scenario provides a reasonable explanation why CO2 is not formed by dissociation of HOCO•. Instead, CO2 formation progresses by ND of H2O into H2 and O and subsequent addition of the latter to CO. In the case of CH3OCO•, stabilization in a CH3OH matrix is much less pronounced as the capacity to form hydrogen-bonds in CH3OH is lower compared to H2O, where molecules can act as donors of two as compared to one hydrogen bond in CH3OH. Furthermore, CH3OCO• can only act as an acceptor but not as a donor of hydrogen bonds. We propose that this reduced stabilization of the CH3OCO• radical enables it to readily dissociate and form CO2, which HOCO• cannot do.
5. Open Questions and Perspectives for Future Studies
As a conclusion to this review of our studies on electron-induced chemistry in ices, we discuss some cases where the reaction mechanisms remain to be resolved. This includes (a) the resonant formation of formamide from CO/NH3 mixed ices [30], (b) the formation of isocyanic acid (HNCO) from the same mixed ices, and (c) methylation reactions that start from the neutral dissociation of methane.
In analogy to the hydroamination reaction, formation of formamide in CO/NH3 mixed ices at electron energies above the ionization threshold was ascribed to an EI-driven reaction (see Section 4.5). However, the yield of formamide in CO/NH3 mixed ices is resonantly enhanced around 9–10 eV [30]. This reaction channel has been assigned to DEA to NH3 yielding H− and NH2⦁, or NH2− and H⦁ [135]. Following DEA, either NH2⦁ or H⦁ can attack a nearby CO molecule to yield H2NCO⦁ or HCO⦁. Finally, reaction with NH3 yields formamide and either a NH2⦁ or H⦁ radical, which potentially results in a chain reaction. However, a closer comparison of the formation of ethylamine in C2H4/NH3 mixed ices [29] and of formamide in CO/NH3 mixed ices [30] reveals that the proposed mechanisms may not be fully consistent, calling for a more comprehensive reevaluation. In fact, it could be expected that DEA to NH3 at 9–10 eV should also contribute to the formation of ethylamine and/or C2H6. However, in contrast to the energy dependence of formamide [30], the energy dependences of ethylamine and C2H6 do not evince a resonance at 9–10 eV [29]. There might be several reasons for this discrepancy. Firstly, the resonance may have been overseen in the data for ethylamine and C2H6 production as the electron exposure of 800 µC/cm2 used to study their formation was already above the regime where product formation depends linearly on the electron dose for E0 > 10 eV [29]. Secondly, ionization of C2H4 may already start to contribute to the formation of ethylamine at 10 eV if it is considered that the ionization energy of C2H4, which is about 10.5 eV in the gas phase [54], is very likely shifted to lower values in the condensed phase [44,136]. In contrast, the ionization energy of CO is 14.0 eV in the gas phase [54] and thus at considerably higher energies. Thus, resonant contributions to the overall product yields in the C2H4/NH3 mixed ices might be masked by higher contributions of the EI-driven mechanism.
As an alternative mechanism, the resonance observed in the formamide yield from CO/NH3 mixed ices may also stem from DEA to CO rather than NH3 [56,123,137]. Dissociation of CO into C and O⦁– could provide an alternative route to the formation of NH2⦁ via
Similar to Reaction 9, but with NH3 instead of H2O. From here, formation of formamide would again proceed by reaction of NH2⦁ with CO. However, all other products that form from NH2⦁ radicals should be resonantly enhanced when CO is present, but in CO/NH3 mixed ices, no such enhancement in the production of hydrazine (N2H4) is observed.
Non-dissociative EA to CO could lead to formamide via the reaction of the base CO⦁- with an adjacent NH3 to yield HCO⦁ and NH2−. HCO⦁ could then react with intact NH3, or possibly with the NH2− to eventually form formamide. However, so far, non-dissociative EA to CO at electron energies around 10 eV has not been reported, neither in gas phase nor in the condensed phase. A more conceivable scenario is that EI to NH3 creates a secondary electron with almost no kinetic energy, which could then attach to CO, triggering the low-lying non-dissociative resonance. At the same time, the NH3⦁+ radical cation could react with another intact NH3 to yield NH4+ and NH2⦁, as described in Section 3.5. This would provide both, a good acid to protonate CO⦁− and an NH2⦁ to complete the reaction to formamide. In conclusion, the interpretation of the experimental data with respect to the mechanism by which formamide is produced is still ambiguous which calls for further investigation.
The interpretation of the mechanism for formation of formamide from CO and NH3 is further confounded by the fact that formation of formamide is always accompanied by the formation of isocyanic acid (HNCO). Our preliminary experimental data show a very consistent ratio of about 2:1 moieties of HNCO to formamide (see Figure 1). Observation of extra-terrestrial occurrences of formamide have also always co-detected isocyanic acid [138], though ratios between the two vary. There is, to date, no good explanation for the formation of HNCO. We can only exclude that it is formed by radiolysis of formamide. Any substantial progress in the mechanistic study of the formation of formamide could thus be very helpful in understanding the origin of isocyanic acid as well.
The final suggestion for further study is a whole class of reactions, namely the methylation of compounds starting from ND of methane. Figure 7 shows the formation of ethane from methane, which could be considered the simplest methylation reaction, as it is the recombination of two methyl radicals. The energetic onset at around 10 eV hints at an ND process, rather than impact ionization, as the ionization threshold of methane in the gas phase lies at 12.6 eV and neutral dissociation of methane is known to have higher cross sections than EI for energies up to 30 eV, at least in the gas phase [139,140]. In the irradiation of CO/CH4 mixed ices, our preliminary results show that acetaldehyde (CH3CHO) is ormed. The dependence on electron energy of its formation is fairly similar to the formation of ethane from methane, which could be interpreted that acetaldehyde is formed by the methylation of CO with subsequent addition of a hydrogen radical. The methylation of small molecules is certainly a class of reactions that needs to be investigated in more detail.
These examples show that there is indeed plenty of room for future studies that aim at unravelling the mechanisms of electron-driven reactions in molecular ices. The dimension of this problem will certainly increase when more than two compounds are presents in the ice. However, the insight obtained so far demonstrates that the methodology presented in this review is a valuable approach to tackle these problems.
Author Contributions
Conceptualization J.H.B. and P.S.; investigation (theoretical calculations) T.B.; investigation (previously unpublished experiments NH3) M.P.M.; investigation (previously unpublished experiments CH4) S.B.; investigation (all other data) F.S.; data curation F.S.; writing—original draft preparation J.H.B. and F.S.; writing—review and editing all authors.; visualization F.S.; supervision J.H.B.; funding acquisition P.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft DFG grant number SW/26-15-2.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to them being in a proprietary data format.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ingólfsson, O. Low-Energy Electrons: Fundamentals and Applications; Pan Stanford Publishing: Singapore, 2019. [Google Scholar]
- Kumar, A.; Becker, D.; Adhikary, A.; Sevilla, M.D. Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci. 2019, 20, 3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.K.; Chaki, T.K.; Bhowmick, A.K. Electron Beam Processing of Polymers. In Advanced Polymer Processing Operations; Cheremisinoff, N.P., Ed.; William Andrew Publishing: Norwich, NY, USA, 1998; pp. 157–186. [Google Scholar]
- Dispenza, C.; Giacomazza, D.; Jonsson, M. Micro-to Nanoscale Bio-Hybrid Hydrogels Engineered by Ionizing Radiation. Biomolecules 2021, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Utke, I.; Hoffmann, P.; Melngailis, J. Gas-assisted focused electron beam and ion beam processing and fabrication. J. Vac. Sci. Technol. B 2008, 26, 1197–1276. [Google Scholar] [CrossRef] [Green Version]
- Huth, M.; Porrati, F.; Dobrovolskiy, O.V. Focused electron beam induced deposition meets materials science. Microelectron. Eng. 2018, 185–186, 9–28. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.; Harned, N. Lithography gets extreme. Nat. Photonics 2010, 4, 24–26. [Google Scholar] [CrossRef]
- Arumainayagam, C.R.; Garrod, R.T.; Boyer, M.C.; Hay, A.K.; Bao, S.T.; Campbell, J.S.; Wang, J.; Nowak, C.M.; Arumainayagam, M.R.; Hodge, P.J. Extraterrestrial prebiotic molecules: Photochemistry vs. radiation chemistry of interstellar ices. Chem. Soc. Rev. 2019, 48, 2293–2314. [Google Scholar] [CrossRef] [Green Version]
- Mason, N.J.; Nair, B.; Jheeta, S.; Szymańska, E. Electron induced chemistry: A new frontier in astrochemistry. Faraday Discuss. 2014, 168, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Arumainayagam, C.R.; Lee, H.-L.; Nelson, R.B.; Haines, D.R.; Gunawardane, R.P. Low-energy electron-induced reactions in condensed matter. Surf. Sci. Rep. 2010, 65, 1–44. [Google Scholar] [CrossRef]
- Boyer, M.C.; Rivas, N.; Tran, A.A.; Verish, C.A.; Arumainayagam, C.R. The role of low-energy (≤20 eV) electrons in astrochemistry. Surf. Sci. 2016, 652, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.H.; Naulleau, P.; Ahmed, M.; Kostko, O. Determination of effective attenuation length of slow electrons in polymer films. J. Appl. Phys. 2020, 127, 245301. [Google Scholar] [CrossRef]
- Thorman, R.M.; Kumar, T.P.R.; Fairbrother, D.H.; Ingólfsson, O. The role of low-energy electrons in focused electron beam induced deposition: Four case studies of representative precursors. Beilstein J. Nanotechnol. 2015, 6, 1904–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, J.A.; Rosenberg, S.G.; Barclay, M.; Wu, Y.-C.; McElwee-White, L.; Fairbrother, D.H. Understanding the electron-stimulated surface reactions of organometallic complexes to enable design of precursors for electron beam-induced deposition. Appl. Phys. A 2014, 117, 1631–1644. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Thakur, N.; Cartaya, R.; Castellanos, S.; Swiderek, P. Role of low-energy electrons in the solubility switch of Zn-based oxocluster photoresist for extreme ultraviolet lithography. Phys. Chem. Chem. Phys. 2021, 23, 16646–16657. [Google Scholar] [CrossRef] [PubMed]
- Böhler, E.; Warneke, J.; Swiderek, P. Control of chemical reactions and synthesis by low-energy electrons. Chem. Soc. Rev. 2013, 42, 9219–9231. [Google Scholar] [CrossRef]
- Moore, J.H.; Swiderek, P.; Matejcik, S.; Allan, M. Fundamentals of interactions of electrons with molecules. In Nanofabrication Using Focused Ion and Electron Beams: Principles and Applications; Russel, P., Moshkalev, S., Utke, I., Eds.; Oxford University Press: New York, NY, USA, 2012; pp. 184–225. [Google Scholar]
- Goesmann, F.; Raulin, F.; Bredehöft, J.H.; Cabane, M.; Ehrenfreund, P.; MacDermott, A.J.; McKenna-Lawlor, S.; Meierhenrich, U.J.; Muñoz Caro, G.M.; Szopa, C.; et al. COSAC prepares for sampling and in situ analysis of cometary matter from comet 67P/Churyumov–Gerasimenko. Planet. Space Sci. 2014, 103, 318–330. [Google Scholar] [CrossRef]
- Goesmann, F.; McKenna-Lawlor, S.; Roll, R.; Bredehöft, J.H.; Meierhenrich, U.; Raulin, F.; Thiemann, W.; Muñoz Caro, G.M.; Szopa, C. Interpretation of COSAC mass spectrometer data acquired during Rosetta’s Lutetia fly-by 10 July 2010. Planet. Space Sci. 2012, 66, 187–191. [Google Scholar] [CrossRef]
- Gardener, J.A.; Golovchenko, J.A. Ice-assisted electron beam lithography of graphene. Nanotechnology 2012, 23, 185302. [Google Scholar] [CrossRef]
- de Rooij, A. Corrosion in Space. In Encyclopedia of Aerospace Engineering; Blockley, R., Shyy, W., Eds.; John Wiley & Sons: Chichester, UK, 2010. [Google Scholar] [CrossRef]
- Lafosse, A.; Bertin, M.; Azria, R. Electron driven processes in ices: Surface functionalization and synthesis reactions. Prog. Surf. Sci. 2009, 84, 177–198. [Google Scholar] [CrossRef]
- Ipolyi, I.; Michaelis, W.; Swiderek, P. Electron-induced reactions in condensed films of acetonitrile and ethane. Phys. Chem. Chem. Phys. 2007, 9, 180–191. [Google Scholar] [CrossRef]
- Schmidt, F.; Swiderek, P.; Bredehöft, J.H. Formation of Formic Acid, Formaldehyde, and Carbon Dioxide by Electron-Induced Chemistry in Ices of Water and Carbon Monoxide. ACS Earth Space Chem. 2019, 3, 1974–1986. [Google Scholar] [CrossRef]
- Bass, A.D.; Bredehöft, J.H.; Böhler, E.; Sanche, L.; Swiderek, P. Reactions and anion desorption induced by low-energy electron exposure of condensed acetonitrile. Eur. Phys. J. D 2012, 66, 53. [Google Scholar] [CrossRef]
- Schmidt, F.; Swiderek, P.; Scheele, T.; Bredehöft, J.H. Mechanisms of methyl formate production during electron-induced processing of methanol–carbon monoxide ices. Phys. Chem. Chem. Phys. 2021, 23, 11649–11662. [Google Scholar] [CrossRef] [PubMed]
- Hamann, T.; Böhler, E.; Swiderek, P. Low-Energy-Electron-Induced Hydroamination of an Alkene. Angew. Chem. Int. Ed. 2009, 48, 4643–4645. [Google Scholar] [CrossRef] [PubMed]
- Warneke, J.; Wang, Z.; Swiderek, P.; Bredehöft, J.H. Electron-Induced Hydration of an Alkene: Alternative Reaction Pathways. Angew. Chem. Int. Ed. 2015, 54, 4397–4400. [Google Scholar] [CrossRef]
- Böhler, E.; Bredehöft, J.H.; Swiderek, P. Low-Energy Electron-Induced Hydroamination Reactions between Different Amines and Olefins. J. Phys. Chem. C 2014, 118, 6922–6933. [Google Scholar] [CrossRef]
- Bredehöft, J.H.; Böhler, E.; Schmidt, F.; Borrmann, T.; Swiderek, P. Electron-Induced Synthesis of Formamide in Condensed Mixtures of Carbon Monoxide and Ammonia. ACS Earth Space Chem. 2017, 1, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, F.; Swiderek, P.; Bredehöft, J.H. Electron-Induced Processing of Methanol Ice. ACS Earth Space Chem. 2021, 5, 391–408. [Google Scholar] [CrossRef]
- Schmidt, F.; Swiderek, P.; Bredehöft, J.H. Electron-Induced Formation of Ethyl Methyl Ether in Condensed Mixtures of Methanol and Ethylene. J. Phys. Chem. A 2019, 123, 37–47. [Google Scholar] [CrossRef]
- Öberg, K.I. Photochemistry and Astrochemistry: Photochemical Pathways to Interstellar Complex Organic Molecules. Chem. Rev. 2016, 116, 9631–9663. [Google Scholar] [CrossRef] [Green Version]
- Boamah, M.D.; Sullivan, K.K.; Shulenberger, K.E.; Soe, C.M.; Jacob, L.M.; Yhee, F.C.; Atkinson, K.E.; Boyer, M.C.; Haines, D.R.; Arumainayagam, C.R. Low-energy electron-induced chemistry of condensed methanol: Implications for the interstellar synthesis of prebiotic molecules. Faraday Discuss. 2014, 168, 249–266. [Google Scholar] [CrossRef]
- Burean, E.; Swiderek, P. Thermal desorption measurement of cross section for reactions in condensed acetaldehyde induced by low-energy electrons. Surf. Sci. 2008, 602, 3194–3198. [Google Scholar] [CrossRef]
- Burean, E.; Swiderek, P. Electron-Induced Reactions in Condensed Acetaldehyde: Identification of Products and Energy-Dependent Cross Sections. J. Phys. Chem. C 2008, 112, 19456–19464. [Google Scholar] [CrossRef]
- Schmidt, F.; Mues, M.P.; Bredehöft, J.H.; Swiderek, P. Molecular synthesis in ices triggered by dissociative electron attachment to carbon monoxide. Eur. Phys. J. D 2021, 75, 302. [Google Scholar] [CrossRef]
- Python (Version 3.8.1). Python Software Foundation. 2019. Available online: https://www.python.org/downloads/release/python-381/ (accessed on 19 December 2021).
- van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A. Savitzky-Golay Smoothing Filters. Comput. Phys. 1990, 4, 669–672. [Google Scholar] [CrossRef]
- Bald, I.; Langer, J.; Tegeder, P.; Ingólfsson, O. From isolated molecules through clusters and condensates to the building blocks of life. Int. J. Mass Spectrom. 2008, 277, 4–25. [Google Scholar] [CrossRef]
- McConkey, J.W.; Malone, C.P.; Johnson, P.V.; Winstead, C.; McKoy, V.; Kanik, I. Electron impact dissociation of oxygen-containing molecules-A critical review. Phys. Rep. 2008, 466, 1–103. [Google Scholar] [CrossRef]
- Christophorou, L.G.; Olthoff, J.K. Fundamental Electron Interactions with Plasma Processing Gases; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Szymańska, E.; Čadež, I.; Krishnakumar, E.; Mason, N.J. Electron impact induced anion production in acetylene. Phys. Chem. Chem. Phys. 2014, 16, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.; Michaud, M.; Sanche, L. Low Energy Electron Total Scattering Cross Section for the Production of CO Within Condensed Methanol. J. Chem. Phys. 1997, 107, 3478–3484. [Google Scholar] [CrossRef]
- Davis, D.; Vysotskiy, V.P.; Sajeev, Y.; Cederbaum, L.S. Electron Impact Catalytic Dissociation: Two-Bond Breaking by a Low-Energy Catalytic Electron. Angew. Chem. Int. Ed. 2011, 50, 4119–4122. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.; Vysotskiy, V.P.; Sajeev, Y.; Cederbaum, L.S. A One-Step Four-Bond-Breaking Reaction Catalyzed by an Electron. Angew. Chem. Int. Ed. 2012, 51, 8003–8007. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.; Kundu, S.; Prabhudesai, V.S.; Sajeev, Y.; Krishnakumar, E. Formation of CO2 from formic acid through catalytic electron channel. J. Chem. Phys. 2018, 149, 064308. [Google Scholar] [CrossRef] [PubMed]
- Pimblott, S.M.; LaVerne, J.A. Production of low-energy electrons by ionizing radiation. Radiat. Phys. Chem. 2007, 76, 1244–1247. [Google Scholar] [CrossRef]
- Völkel, B.; Gölzhäuser, A.; Müller, H.U.; David, C.; Grunze, M. Influence of secondary electrons in proximal probe lithography. J. Vac. Sci. Technol. B 1997, 15, 2877–2881. [Google Scholar] [CrossRef]
- Lias, S.G.; Rosenstock, H.M.; Draxl, K.; Steiner, B.W.; Herron, J.T.; Holmes, J.L.; Levin, R.D.; Liebman, J.F.; Kafafi, S.A. Ionization Energetics Data. In NIST Chemistry WebBook; Linstrom, P.J., Mallard, W.G., Eds.; NIST Standard Reference Database No. 69; National Institute for Standards and Technology: Gaithersburg, MD, USA.
- Rempt, R.D. Electron-Impact Excitation of Carbon Monoxide Near Threshold in the 1.5- to 3-eV Incident Energy Range. Phys. Rev. Lett. 1969, 22, 1034–1036. [Google Scholar] [CrossRef]
- Gope, K.; Tadsare, V.; Prabhudesai, V.S.; Mason, N.J.; Krishnakumar, E. Negative ion resonances in carbon monoxide. Eur. Phys. J. D 2016, 70, 134. [Google Scholar] [CrossRef]
- Walker, I.C.; Stamatovic, A.; Wong, S.F. Vibrational excitation of ethylene by electron impact: 1–11 eV. J. Chem. Phys. 1978, 69, 5532–5537. [Google Scholar] [CrossRef]
- Thynne, J.C.J.; MacNeil, K.A.G. Negative Ion Formation by Ethylene and 1,1-Difluoroethylene. J. Phys. Chem. 1971, 75, 2584–2591. [Google Scholar] [CrossRef]
- Trepka, L.V.; Neuert, H. Über die Entstehung von negativen Ionen aus einigen Kohlenwasserstoffen und Alkoholen durch Elektronenstoß. Z. Naturforsch. A Phys. Sci. 1963, 18, 1295–1303. [Google Scholar] [CrossRef]
- Itikawa, Y.; Mason, N. Cross Sections for Electron Collisions with Water Molecules. J. Phys. Chem. Ref. Data 2005, 34, 1–22. [Google Scholar] [CrossRef]
- Ibănescu, B.C.; May, O.; Monney, A.; Allan, M. Electron-induced chemistry of alcohols. Phys. Chem. Chem. Phys. 2007, 9, 3163–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heni, M.; Illenberger, E. Electron attachment by saturated nitriles, acrylonitrile (C2H3CN), and benzonitrile (C6H5CN). Int. J. Mass Spectrom. Ion Processes 1986, 73, 127–144. [Google Scholar] [CrossRef]
- Bredehöft, J.H. Electron-Induced Chemistry in the Condensed Phase. Atoms 2019, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.Y.; McMenamin, J.C.; Spicer, W.E. UPS measurements of molecular energy level of condensed gases. Surf. Sci. 1975, 50, 149–156. [Google Scholar] [CrossRef]
- Ellis-Gibbings, L.; Bass, A.D.; Cloutier, P.; García, G.; Sanche, L. Electron stimulated desorption from condensed pyrimidine and pyridazine. Phys. Chem. Chem. Phys. 2017, 19, 13038–13048. [Google Scholar] [CrossRef]
- Shukla, A.K.; Stace, A.J. Intermolecular Ion-Molecule Reactions in Clusters: The Reactions of Aliphatic Alcohols. J. Phys. Chem. 1988, 92, 2579–2583. [Google Scholar] [CrossRef]
- El-Shall, M.S.; Marks, C.; Sieck, L.W.; Meot-Ner, M. Reactions and Thermochemistry of Protonated Methanol Clusters Produced by Electron Impact Ionization. J. Phys. Chem. 1992, 96, 2045–2051. [Google Scholar] [CrossRef]
- Lengyel, J.; Papp, P.; Matejčík, Š.; Kočišek, J.; Fárník, M.; Fedor, J. Suppression of low-energy dissociative electron attachment in Fe(CO)5 upon clustering. Beilstein J. Nanotechnol. 2017, 8, 2200–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imhoff, M.; Parenteau, L.; Sanche, L.; Huels, M.A. Low energy electron and O− reactions in films of O2 coadsorbed with benzene or toluene. Phys. Chem. Chem. Phys. 2005, 7, 3359. [Google Scholar] [CrossRef] [PubMed]
- Schmittel, M.; Burghart, A. Understanding Reactivity Patterns of Radical Cations. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550–2589. [Google Scholar] [CrossRef]
- Matyushov, D.V.; Newton, M.D. Electron-Induced Proton Transfer. J. Phys. Chem. B 2021, 125, 12264–12273. [Google Scholar] [CrossRef] [PubMed]
- Borrmann, T.; Swiderek, P. Formation of 2-propanol in condensed molecular films of acetaldehyde following electron impact ionisation-induced proton transfer. Eur. Phys. J. D 2016, 70, 133. [Google Scholar] [CrossRef]
- Martinez, O.; Yang, Z.; Demarais, N.J.; Snow, T.P.; Bierbaum, V.M. Gas-phase reactions of hydride anion, H-. Astrophys. J. 2010, 720, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Garrett, B.C.; Dixon, D.A.; Camaioni, D.M.; Chipman, D.M.; Johnson, M.A.; Jonah, C.D.; Kimmel, G.A.; Miller, J.H.; Rescigno, T.N.; Rossky, P.J.; et al. Role of Water in Electron-Initiated Processes and Radical Chemistry: Issues and Scientific Advances. Chem. Rev. 2005, 105, 355–389. [Google Scholar] [CrossRef]
- Balog, R.; Langer, J.; Gohlke, S.; Stano, M.; Abdoul-Carime, H.; Illenberger, E. Low energy electron driven reactions in free and bound molecules: From unimolecular processes in the gas phase to complex reactions in a condensed environment. Int. J. Mass Spectrom. 2004, 233, 267–291. [Google Scholar] [CrossRef]
- Swiderek, P.; Deschamps, M.C.; Michaud, M.; Sanche, L. Bond Formation in Reactions of Solid Cyclopropane Induced by Low-Energy Electrons. J. Phys. Chem. B 2003, 107, 563–567. [Google Scholar] [CrossRef]
- Weeks, L.D.; Zhu, L.L.; Pellon, M.; Haines, D.R.; Arumainayagam, C.R. Low-Energy Electron-Induced Oligomerization of Condensed Carbon Tetrachloride. J. Phys. Chem. C 2007, 111, 4815–4822. [Google Scholar] [CrossRef]
- Maity, S.; Kaiser, R.I.; Jones, B.M. Formation of complex organic molecules in methanol and methanol-carbon monoxide ices exposed to ionizing radiation—A combined FTIR and reflectron time-of-flight mass spectrometry study. Phys. Chem. Chem. Phys. 2015, 17, 3081–3114. [Google Scholar] [CrossRef]
- Öberg, K.I.; Garrod, R.T.; van Dishoeck, E.F.; Linnartz, H. Formation rates of complex organics in UV irradiated CH3OH-rich ices. Astron. Astrophys. 2009, 504, 891–913. [Google Scholar] [CrossRef] [Green Version]
- Maity, S.; Kaiser, R.I.; Jones, B.M. Infrared and reflectron time-of-flight mass spectroscopic study on the synthesis of glycolaldehyde in methanol (CH3OH) and methanol-carbon monoxide (CH3OH-CO) ices exposed to ionization radiation. Faraday Discuss. 2014, 168, 485–516. [Google Scholar] [CrossRef] [PubMed]
- Rajappan, M.; Zhu, L.L.; Bass, A.D.; Sanche, L.; Arumainayagam, C.R. Chemical Synthesis Induced by Dissociative Electron Attachment. J. Chem. Phys. C 2008, 112, 17319–17323. [Google Scholar] [CrossRef]
- Butscher, T.; Duvernay, F.; Rimola, A.; Segado-Centellas, M.; Chiavassa, T. Radical recombination in interstellar ices, a not so simple mechanism. Phys. Chem. Chem. Phys. 2017, 19, 2857–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Ugliengo, P.; Balucani, N.; Skouteris, D. Reactivity of HCO with CH3 and NH2 on Water Ice Surfaces. A Comprehensive Accurate Quantum Chemistry Study. ACS Earth Space Chem. 2019, 3, 2158–2170. [Google Scholar] [CrossRef] [Green Version]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Balucani, N. The (impossible?) formation of acetaldehyde on the grain surfaces: Insights from quantum chemical calculations. Mon. Not. R. Astron. Soc. Lett. 2016, 459, L6–L10. [Google Scholar] [CrossRef] [Green Version]
- Rimola, A.; Skouteris, D.; Balucani, N.; Ceccarelli, C.; Enrique-Romero, J.; Taquet, V.; Ugliengo, P. Can Formamide Be Formed on Interstellar Ice? An Atomistic Perspective. ACS Earth Space Chem. 2018, 2, 720–734. [Google Scholar] [CrossRef]
- Shulenberger, K.E.; Zhu, J.L.; Tran, K.; Abdullahi, S.; Belvin, C.; Lukens, J.; Peeler, Z.; Mullikin, E.; Cumberbatch, H.M.; Huang, J.; et al. Electron-Induced Radiolysis of Astrochemically Relevant Ammonia Ices. ACS Earth Space Chem. 2019, 3, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.M.; Sloan, D.; Ihm, H.; White, J.M. Electron-induced surface chemistry: Production and characterization of NH2 and NH species on Pt(111). J. Vac. Sci. Technol. A 1996, 14, 1516–1521. [Google Scholar] [CrossRef]
- Zhu, C.; Frigge, R.; Bergantini, A.; Fortenberry, R.C.; Kaiser, R.I. Untangling the Formation of Methoxymethanol (CH3OCH2OH) and Dimethyl Peroxide (CH3OOCH3) in Star-forming Regions. Astrophys. J. 2019, 881, 156. [Google Scholar] [CrossRef]
- Esmaili, S.; Bass, A.D.; Cloutier, P.; Sanche, L.; Huels, M.A. Synthesis of complex organic molecules in simulated methane rich astrophysical ices. J. Chem. Phys. 2017, 147, 224704. [Google Scholar] [CrossRef] [PubMed]
- Sailer, W.; Pelc, A.; Limão-Vieira, P.; Mason, N.J.; Limtrakul, J.; Scheier, P.; Probst, M.; Märk, T.D. Low energy electron attachment to CH3CN. Chem. Phys. Lett. 2003, 381, 216–222. [Google Scholar] [CrossRef]
- Garrod, R.T.; Weaver, S.L.W.; Herbst, E. Complex Chemistry in Star-Forming Regions: An Expanded Gas-Grain Warum-up Chemical Model. Astrophys. J. 2008, 682, 283–302. [Google Scholar] [CrossRef] [Green Version]
- Afeefy, H.Y.; Liebman, J.F.; Stein, S.E.; Burgess, D.R., Jr. Neutral Thermochemical Data. In NIST Chemistry WebBook; Linstrom, P.J., Mallard, W.G., Eds.; NIST Standard Reference Database Number 69; National Institute of Standards and Technology: Gaithersburg, MD, USA.
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.J. Reduction by Dissolving Metals. Part I. J. Chem. Soc. 1944, 430–436. [Google Scholar] [CrossRef]
- Birch, A.J. The Birch reduction in organic synthesis. Pure Appl. Chem. 1996, 68, 553–556. [Google Scholar] [CrossRef]
- Birch, A.J.; Subba Rao, G.S. Reduction by dissolving metals. XV. Reactions of some cyclohexadienes with metal-ammonia solutions. Aust. J. Chem. 1970, 23, 1641–1649. [Google Scholar] [CrossRef]
- Peters, B.K.; Rodriguez, K.X.; Reisberg, S.H.; Beil, S.B.; Hickey, D.P.; Kawamata, Y.; Collins, M.; Starr, J.; Chen, L.; Udyavara, S.; et al. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science 2019, 363, 838–845. [Google Scholar] [CrossRef]
- Glaser, F.; Kerzig, C.; Wenger, O.S. Multi-Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods. Angew. Chem. Int. Ed. 2020, 59, 10266–10284. [Google Scholar] [CrossRef]
- Brauman, J.I.; Biairlb, L.K. Gas-Phase Acidities of Alcohols. J. Am. Chem. Soc. 1970, 92, 5986–5992. [Google Scholar] [CrossRef]
- Mackay, G.I.; Bohme, D.K. Bridging the Gap between the Gas Phase and Solution: Transition in the Relative Acidity of Water and Methanol at 296 ± 2 K. J. Am. Chem. Soc. 1978, 100, 327–329. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Ibrahim, M. Ab Initio Studies of RO−…HOR’ Complexes. Solvent Effects on the Relative Acidities of Water and Methanol. J. Comput. Chem. 1981, 2, 7–11. [Google Scholar] [CrossRef]
- Saenko, E.V.; Laikov, D.N.; Baranova, I.A.; Feldman, V.I. Communication: Stabilization of radical anions with weakly bound electron in condensed media: A case study of diacetonyl radical anion. J. Chem. Phys. 2011, 135, 101103. [Google Scholar] [CrossRef]
- Melton, C.E. Cross Sections and Interpretation of Dissociative Attachment Reactions Producing OH−, O−, and H− in H2O. J. Chem. Phys. 1972, 57, 4218–4225. [Google Scholar] [CrossRef]
- Kühn, A.; Fenzlaff, H.P.; Illenberger, E. Formation and dissociation of negative ion resonances in methanol and allylalcohol. J. Chem. Phys. 1988, 88, 7453–7458. [Google Scholar] [CrossRef]
- Boyer, M.C.; Boamah, M.D.; Sullivan, K.K.; Arumainayagam, C.R.; Bazin, M.; Bass, A.D.; Sanche, L. Dynamics of Dissociative Electron-Molecule Interactions in Condensed Methanol. J. Phys. Chem. C 2014, 118, 22592–22600. [Google Scholar] [CrossRef]
- Kimmel, G.A.; Orlando, T.M. Low-Energy (5–120 eV) Electron-Stimulated Dissociation of Amorphous D2O Ice: D(2S), O(3P2,1,0), and O(1D2) Yields and Velocity Distributions. Phys. Rev. Lett. 1995, 75, 2606–2609. [Google Scholar] [CrossRef]
- Petrik, N.G.; Monckton, R.J.; Koehler, S.P.K.; Kimmel, G.A. Distance-Dependent Radiation Chemistry: Oxidation versus Hydrogenation of CO in Electron-Irradiated H2O/CO/H2O Ices. J. Phys. Chem. C 2014, 118, 27483–27492. [Google Scholar] [CrossRef]
- Yamamoto, S.; Beniya, A.; Mukai, K.; Yamashita, Y.; Yoshinobu, J. Low-energy electron-stimulated chemical reactions of CO in water ice. Chem. Phys. Lett. 2004, 388, 384–388. [Google Scholar] [CrossRef]
- Petrik, N.G.; Monckton, R.J.; Koehler, S.P.K.; Kimmel, G.A. Electron-stimulated reactions in layered CO/H2O films: Hydrogen atom diffusion and the sequential hydrogenation of CO to methanol. J. Chem. Phys. 2014, 140, 204710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, K.; Hargreaves, L.R.; Ralphs, K.; Khakoo, M.A.; Winstead, C.; McKoy, V.; Rescigno, T.N.; Orel, A.E. Excitation of the 4 lowest electronic transitions in methanol by low-energy electrons. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 115208. [Google Scholar] [CrossRef]
- Michaud, M.; Fraser, M.-J.; Sanche, L. Low-energy electron-energy-loss spectroscopy of solid methanol: Vibrational and electronic excitations. J. Chim. Phys. 1994, 91, 1223–1227. [Google Scholar] [CrossRef]
- Curtis, M.G.; Walker, I.C. Dissociative Electron Attachment in Water and Methanol (5–14 eV). J. Chem. Soc. Faraday Trans. 1992, 88, 2805–2810. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Martinović, P.; Ahlenhoff, K.; Koch, S.; Emmrich, D.; Gölzhäuser, A.; Swiderek, P. Cisplatin as a Potential Platinum Focused Electron Beam Induced Deposition Precursor: NH3 Ligands Enhance the Electron-Induced Removal of Chlorine. J. Phys. Chem. C 2019, 123, 21774–21787. [Google Scholar] [CrossRef] [Green Version]
- Rohdenburg, M.; Boeckers, H.; Brewer, C.R.; McElwee-White, L.; Swiderek, P. Efficient NH3-based process to remove chlorine from electron beam deposited ruthenium produced from (η3-C3H5)Ru(CO)3Cl. Sci. Rep. 2020, 10, 10901. [Google Scholar] [CrossRef]
- Lee, J.; Grabowski, J.J. Reactions of the Atomic Oxygen Radical Anion and the Synthesis of Organic Reactive Intermediates. Chem. Rev. 1992, 92, 1611–1647. [Google Scholar] [CrossRef]
- Kamarchik, E.; Rodrigo, C.; Bowman, J.M.; Reisler, H.; Krylov, A.I. Overtone-induced dissociation and isomerization dynamics of the hydroxymethyl radical (CH2OH and CD2OH). I. A theoretical study. J. Chem. Phys. 2012, 136, 084304. [Google Scholar] [CrossRef]
- Wang, B.; Hou, H.; Gu, Y. Ab Initio/Density Functional Theory and Multichannel RRKM Calculations for the CH3O + CO Reaction. J. Phys. Chem. A 1999, 103, 8021–8029. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Sitha, S.; Jewell, L.L. Non-catalytic hydroamination of alkenes: A computational study. Tetrahedron 2010, 66, 3030–3036. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzweski, V.G.; Montgomery, J.A.; Stratman, R.E.; Burant, J.C.; et al. Gaussian 98, Revision A.7; Gaussian, Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Mullikin, E.; van Mulbregt, P.; Perea, J.; Kasule, M.; Huang, J.; Buffo, C.; Campbell, J.; Gates, L.; Cumberbatch, H.M.; Peeler, Z.; et al. Condensed-Phase Photochemistry in the Absence of Radiation Chemistry. ACS Earth Space Chem. 2018, 2, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Azria, R.; Parenteau, L.; Sanche, L. Mechanisms for O− electron stimulated desorption via dissociative attachment in condensed CO. J. Chem. Phys. 1988, 88, 5166–5170. [Google Scholar] [CrossRef]
- Kimmel, G.A.; Orlando, T.M. Observation of Negative Ion Resonances in Amorphous Ice via Low-Energy (5–40 eV) Electron-Stimulated Production of Molecular Hydrogen. Phys. Rev. Lett. 1996, 77, 3983–3986. [Google Scholar] [CrossRef]
- Klyachko, D.; Rowntree, P.; Sanche, L. Oxidation of hydrogen-passivated silicon surfaces induced by dissociative electron attachment to physisorbed H2O. Surf. Sci. 1996, 346, L49–L54. [Google Scholar] [CrossRef]
- Simpson, W.C.; Sieger, M.T.; Orlando, T.M.; Parenteau, L.; Nagesha, K.; Sanche, L. Dissociative electron attachment in nanoscale ice films: Temperature and morphology effects. J. Chem. Phys. 1997, 107, 8668–8677. [Google Scholar] [CrossRef] [Green Version]
- Lane, C.D.; Orlando, T.M. Inelastic electron scattering and energy-selective negative ion reactions in molecular films on silicon surfaces. Appl. Surf. Sci. 2007, 253, 6646–6656. [Google Scholar] [CrossRef]
- Prabhudesai, V.S.; Nandi, D.; Kelkar, A.H.; Krishnakumar, E. Functional group dependent dissociative electron attachment to simple organic molecules. J. Chem. Phys. 2008, 128, 154309. [Google Scholar] [CrossRef]
- Szymańska, E.; Mason, N.J.; Krishnakumar, E.; Matias, C.; Mauracher, A.; Scheier, P.; Denifl, S. Dissociative electron attachment and dipolar dissociation in ethylene. Int. J. Mass Spectrom. 2014, 365–366, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.J.; Chen, S.-H.; Sun, B.; Chang, A.H.H.; Kaiser, R.I. Mechanistical Studies on the Irradiation of Methanol in Extraterrestrial Ices. Astrophys. J. 2007, 660, 1588–1608. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.J.; Hama, T.; Kim, Y.S.; Kawasaki, M.; Kaiser, R.I. Laboratory studies on the formation of formic acid (HCOOH) in interstellar and cometary ices. Astrophys. J. 2011, 727, 27. [Google Scholar] [CrossRef] [Green Version]
- McCunn, L.R.; Lau, K.C.; Krisch, M.J.; Butler, L.J.; Tsung, J.W.; Lin, J.J. Unimolecular Dissociation of the CH3OCO Radical: An Intermediate in the CH3O + CO Reaction. J. Phys. Chem. A 2006, 110, 1625–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, C.J.; Kaiser, R.I. On the Formation of Glycolaldehyde (HCOCH2OH) and Methyl Formate (HCOOCH3) in Interstellar Ice Analogs. Astrophys. J. 2007, 661, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Arasa, C.; van Hemert, M.C.; van Dishoeck, E.F.; Kroes, G.J. Molecular Dynamics Simulations of CO2 Formation in Interstellar Ices. J. Phys. Chem. A 2013, 117, 7064–7074. [Google Scholar] [CrossRef] [Green Version]
- Tronc, M.; Azria, R.; Arfa, M.B. Differential cross section for H- and NH2- ions in NH3. J. Phys. B At. Mol. Opt. Phys. 1988, 21, 2497–2506. [Google Scholar] [CrossRef]
- Ingólfsson, O.; Weik, F.; Illenberger, E. The reactivity of slow electrons with molecules at different degrees of aggregation: Gas phase, clusters and condensed phase. Int. J. Mass Spectrom. Ion Processes 1996, 155, 1–68. [Google Scholar] [CrossRef]
- Nag, P.; Nandi, D. Fragmentation dynamics in dissociative electron attachment to CO probed by velocity slice imaging. Phys. Chem. Chem. Phys. 2015, 17, 7130–7137. [Google Scholar] [CrossRef] [Green Version]
- López-Sepulcre, A.; Jaber, A.A.; Mendoza, E.; Lefloch, B.; Ceccarelli, C.; Vastel, C.; Bachiller, R.; Cernicharo, J.; Codella, C.; Kahane, C.; et al. Shedding light on the formation of the pre-biotic molecule formamide with ASAI. Mon. Not. R. Astron. Soc. 2015, 449, 2438–2458. [Google Scholar] [CrossRef] [Green Version]
- Gadoum, A.; Benyoucef, D. Set of the Electron Collision Cross Sections for Methane Molecule. IEEE Trans. Plasma Sci. 2019, 47, 1505–1513. [Google Scholar] [CrossRef]
- Bouwman, D.; Martinez, A.; Braams, B.J.; Ebert, U. Neutral dissociation of methane by electron impact and a complete and consistent cross section set. Plasma Sources Sci. Technol. 2021, 30, 075012. [Google Scholar] [CrossRef]
Figure 1.
Procedure to derive product yields as function of electron energy from TDS experiments. The analysis is exemplified by data obtained from CO/NH3 mixed ices. (a) Raw data obtained as MS signals as function of substrate temperature. The m/z 45 signal is characteristic of formamide (H2NCHO) while m/z 43 contains contribution of both formamide and isocyanic acid (HNCO). A baseline correction is first applied to the desorption signals. (b) Contributions of formamide and isocyanic acid to the baseline corrected desorption signals at m/z 45 and m/z 43. The contribution of formamide to the m/z 43 signal was derived by scaling the m/z 45 curve according to the relative intensity of the two m/z signals in the MS of formamide. (c) Areas obtained from integration of the desorption peaks. The area of the signal at m/z 43 is divided into contributions of formamide (red) and isocyanic acid (blue). (d) Relative product yields obtained by correcting the peak areas by use of partial ionization cross sections for the formation of fragments with m/z 43 and m/z 45, respectively, from the products. The cross sections refer to the electron-induced formation of cation fragments at an electron energy of 70 eV as applied in the mass spectrometer. (e) Relative product yields in the mixed ice after different electron exposures. The curve reveals a roughly linear increase of the yields over an exposure range up to about 500 µC/cm2. (f) Relative product yields obtained for a range of electron energies (E0) after application of an electron exposure within the linear regime.
Figure 1.
Procedure to derive product yields as function of electron energy from TDS experiments. The analysis is exemplified by data obtained from CO/NH3 mixed ices. (a) Raw data obtained as MS signals as function of substrate temperature. The m/z 45 signal is characteristic of formamide (H2NCHO) while m/z 43 contains contribution of both formamide and isocyanic acid (HNCO). A baseline correction is first applied to the desorption signals. (b) Contributions of formamide and isocyanic acid to the baseline corrected desorption signals at m/z 45 and m/z 43. The contribution of formamide to the m/z 43 signal was derived by scaling the m/z 45 curve according to the relative intensity of the two m/z signals in the MS of formamide. (c) Areas obtained from integration of the desorption peaks. The area of the signal at m/z 43 is divided into contributions of formamide (red) and isocyanic acid (blue). (d) Relative product yields obtained by correcting the peak areas by use of partial ionization cross sections for the formation of fragments with m/z 43 and m/z 45, respectively, from the products. The cross sections refer to the electron-induced formation of cation fragments at an electron energy of 70 eV as applied in the mass spectrometer. (e) Relative product yields in the mixed ice after different electron exposures. The curve reveals a roughly linear increase of the yields over an exposure range up to about 500 µC/cm2. (f) Relative product yields obtained for a range of electron energies (E0) after application of an electron exposure within the linear regime.

Figure 2.
Schematic representation of the UHV chamber: (1) sample holder, (2) closed-cycle helium cryostat, (3) valve to gas handling manifold, (4) residual gas analyzer, (5) electron source, and (6) ion gauge.
Figure 2.
Schematic representation of the UHV chamber: (1) sample holder, (2) closed-cycle helium cryostat, (3) valve to gas handling manifold, (4) residual gas analyzer, (5) electron source, and (6) ion gauge.

Figure 3.
Schematic representation of the gas handling manifold with storage reservoirs (shaded in blue) and the mixing reservoir (shaded in red).
Figure 3.
Schematic representation of the gas handling manifold with storage reservoirs (shaded in blue) and the mixing reservoir (shaded in red).

Figure 4.
Gas phase EA/DEA channels of molecules for which the electron-induced chemistry in ices is reviewed herein. (D)EA to CO (2 eV to CO from Ref. [55] on charge trapping and 10 eV resonance from Ref. [56] on DEA ion yields), C2H4 (2 eV resonance from Ref. [57] derived from energy-dependent cross section for vibrational excitation and 10 eV resonance from Refs. [58,59] on DEA ion yields), H2O (from Ref. [60] on absolute DEA cross sections), CH3OH (from Ref. [61] on DEA ion yields) and CH3CN (from Ref. [62] on DEA ion yields). Each curve has been normalized to its maximum. The figure does not represent the relative yields of individual fragments from same molecule but merely serves to illustrate the resonance positions. Tick marks on the y-axis indicate the vertical offset. Adapted from Ref. [37] with kind permission of The European Physical Journal (EPJ).
Figure 4.
Gas phase EA/DEA channels of molecules for which the electron-induced chemistry in ices is reviewed herein. (D)EA to CO (2 eV to CO from Ref. [55] on charge trapping and 10 eV resonance from Ref. [56] on DEA ion yields), C2H4 (2 eV resonance from Ref. [57] derived from energy-dependent cross section for vibrational excitation and 10 eV resonance from Refs. [58,59] on DEA ion yields), H2O (from Ref. [60] on absolute DEA cross sections), CH3OH (from Ref. [61] on DEA ion yields) and CH3CN (from Ref. [62] on DEA ion yields). Each curve has been normalized to its maximum. The figure does not represent the relative yields of individual fragments from same molecule but merely serves to illustrate the resonance positions. Tick marks on the y-axis indicate the vertical offset. Adapted from Ref. [37] with kind permission of The European Physical Journal (EPJ).

Figure 5.
Energy dependence of the integrated desorption signals of N2 and N2H4 obtained from pure 10–14 monolayer thick NH3 ice for electron exposures of 500 and 50 µC/cm2, respectively (this work). The values for N2H4 were obtained by TDS while N2 was monitored both in ESD and TDS. The solid lines serve as a guide to the eye.
Figure 5.
Energy dependence of the integrated desorption signals of N2 and N2H4 obtained from pure 10–14 monolayer thick NH3 ice for electron exposures of 500 and 50 µC/cm2, respectively (this work). The values for N2H4 were obtained by TDS while N2 was monitored both in ESD and TDS. The solid lines serve as a guide to the eye.

Figure 6.
Dependence of the integrated desorption signals of methoxymethanol, ethanol, ethylene glycol, and dimethyl ether on electron energy obtained by TDS after irradiating pure CH3OH with 32 000 µC/cm² in the range from 3.5–9 eV. The solid lines serve as a guide to the eye. Adapted with permission from Ref. [31]. Copyright 2021 American Chemical Society.
Figure 6.
Dependence of the integrated desorption signals of methoxymethanol, ethanol, ethylene glycol, and dimethyl ether on electron energy obtained by TDS after irradiating pure CH3OH with 32 000 µC/cm² in the range from 3.5–9 eV. The solid lines serve as a guide to the eye. Adapted with permission from Ref. [31]. Copyright 2021 American Chemical Society.

Figure 7.
Energy dependence of the integrated desorption signals of ethane C2H6 from the recombination of methyl radicals formed during electron irradiation of CH4 (this work) or CH3CN [25]. Error bars in the upper series were estimated to be 10% of most intense peak. The solid lines serve as a guide to the eye. Adapted from Ref. [25] with kind permission of The European Physical Journal (EPJ).
Figure 7.
Energy dependence of the integrated desorption signals of ethane C2H6 from the recombination of methyl radicals formed during electron irradiation of CH4 (this work) or CH3CN [25]. Error bars in the upper series were estimated to be 10% of most intense peak. The solid lines serve as a guide to the eye. Adapted from Ref. [25] with kind permission of The European Physical Journal (EPJ).

Figure 8.
Thermal desorption spectra of pure CH3OH (black lines) and CO/CH3OH (1:1) mixed ices (red lines) after irradiation with 250 µC/cm2 and without electron exposure (denoted as 0 µC/cm2). The signals at 160 K and 195 K in the m/z 33 curves are assigned to methoxymethanol and ethylene glycol, respectively. The desorption signal at ~130 K is due to the 13CH3OH isotopologue. The TDS data show that signal intensities of the products decrease upon mixing CH3OH with CO. Adapted from Ref. [26] with permission from the PCCP Owner Societies.
Figure 8.
Thermal desorption spectra of pure CH3OH (black lines) and CO/CH3OH (1:1) mixed ices (red lines) after irradiation with 250 µC/cm2 and without electron exposure (denoted as 0 µC/cm2). The signals at 160 K and 195 K in the m/z 33 curves are assigned to methoxymethanol and ethylene glycol, respectively. The desorption signal at ~130 K is due to the 13CH3OH isotopologue. The TDS data show that signal intensities of the products decrease upon mixing CH3OH with CO. Adapted from Ref. [26] with permission from the PCCP Owner Societies.

Figure 9.
Energy dependence of the integrated desorption signal of H2CO obtained by TDS after electron irradiation of CO/H2O (1:1) mixed ices with 250 µC/cm². The resonance at 4 eV was assigned to EA to CO yielding CO⦁−. The solid line serves as a guide to the eye. Adapted with permission from Ref. [24]. Copyright 2019 American Chemical Society.
Figure 9.
Energy dependence of the integrated desorption signal of H2CO obtained by TDS after electron irradiation of CO/H2O (1:1) mixed ices with 250 µC/cm². The resonance at 4 eV was assigned to EA to CO yielding CO⦁−. The solid line serves as a guide to the eye. Adapted with permission from Ref. [24]. Copyright 2019 American Chemical Society.

Scheme 1.
Mechanism for the electron-induced formation of ethanol in C2H4/H2O mixed ices by EA to C2H4 at E0 < 6 eV. Side products of the reaction are C2H6 and butane [28].
Scheme 1.
Mechanism for the electron-induced formation of ethanol in C2H4/H2O mixed ices by EA to C2H4 at E0 < 6 eV. Side products of the reaction are C2H6 and butane [28].

Figure 10.
(Left panel) Dependence of the integrated desorption signal of H2CO on electron energy obtained by TDS after irradiation of CO/H2O (1:1) mixed ices with 250 µC/cm2. Note that the data points below 10 eV are identical to those in Figure 9. (Right panel) Reactions leading to the formation of H2CO by ND of H2O. The solid line serves as a guide to the eye. Adapted with permission from Ref. [24]. Copyright 2019 American Chemical Society.
Figure 10.
(Left panel) Dependence of the integrated desorption signal of H2CO on electron energy obtained by TDS after irradiation of CO/H2O (1:1) mixed ices with 250 µC/cm2. Note that the data points below 10 eV are identical to those in Figure 9. (Right panel) Reactions leading to the formation of H2CO by ND of H2O. The solid line serves as a guide to the eye. Adapted with permission from Ref. [24]. Copyright 2019 American Chemical Society.

Figure 11.
Dependence of the integrated desorption signal of H2CO on electron energy obtained by TDS after irradiation of pure CH3OH (▲), and CO/CH3OH (1:1) mixed ices (●) with (a) 250 µC/cm² and (b) 12,000 µC/cm². At lower electron energies, higher exposures can be used as product degradation is negligible. This enhances the detection of products with smaller yields. The difference curve (✕) in panel (a) is reflective of an additional reaction channel that is not accessible in pure CH3OH. The reaction schemes (bottom) denote the processes that lead to the enhanced production rate of H2CO in CO/CH3OH mixed ices. The solid and broken lines serve as a guide to the eye. Adapted from Ref. [31] with permission from the PCCP Owner Societies.
Figure 11.
Dependence of the integrated desorption signal of H2CO on electron energy obtained by TDS after irradiation of pure CH3OH (▲), and CO/CH3OH (1:1) mixed ices (●) with (a) 250 µC/cm² and (b) 12,000 µC/cm². At lower electron energies, higher exposures can be used as product degradation is negligible. This enhances the detection of products with smaller yields. The difference curve (✕) in panel (a) is reflective of an additional reaction channel that is not accessible in pure CH3OH. The reaction schemes (bottom) denote the processes that lead to the enhanced production rate of H2CO in CO/CH3OH mixed ices. The solid and broken lines serve as a guide to the eye. Adapted from Ref. [31] with permission from the PCCP Owner Societies.

Figure 12.
Identification of methoxymethanol by its desorption signal at 161 K (shaded in grey) in the m/z 33, 45, and 61 curves of the TDS after irradiation of CO/CH3OH (1:1) mixed ices with 12,000 µC/cm² at 5.5 eV. Notably, methoxymethanol is not observed in pure CH3OH. Adapted from Ref. [26] with permission from the PCCP Owner Societies.
Figure 12.
Identification of methoxymethanol by its desorption signal at 161 K (shaded in grey) in the m/z 33, 45, and 61 curves of the TDS after irradiation of CO/CH3OH (1:1) mixed ices with 12,000 µC/cm² at 5.5 eV. Notably, methoxymethanol is not observed in pure CH3OH. Adapted from Ref. [26] with permission from the PCCP Owner Societies.

Figure 13.
Thermal desorption spectra obtained after irradiation of C2H4/CH3OH (1:1) mixed ices and of pure C2H4 and pure CH3OH at 5.5 eV with 8000 µC/cm2. The desorption signal at 60 K in the m/z 30 curve (shaded in grey, panel (a)) was assigned to ethane (C2H6) whereas the signal at 100 K in the m/z 43 curve (shaded in grey, panel (b)) was assigned to butane (C4H10). Notably, no significant amounts of ethane and butane are formed in pure C2H4 and CH3OH. Furthermore, the intensity of the desorption signal of H2CO (shaded in blue, panel a) in irradiated C2H4/CH3OH mixed ices is significantly higher than in pure CH3OH. The reaction scheme (bottom) denotes the formation of ethane and butane following DEA to CH3OH yielding CH3O− and H•. Adapted with permission from Ref. [32]. Copyright 2019 American Chemical Society.
Figure 13.
Thermal desorption spectra obtained after irradiation of C2H4/CH3OH (1:1) mixed ices and of pure C2H4 and pure CH3OH at 5.5 eV with 8000 µC/cm2. The desorption signal at 60 K in the m/z 30 curve (shaded in grey, panel (a)) was assigned to ethane (C2H6) whereas the signal at 100 K in the m/z 43 curve (shaded in grey, panel (b)) was assigned to butane (C4H10). Notably, no significant amounts of ethane and butane are formed in pure C2H4 and CH3OH. Furthermore, the intensity of the desorption signal of H2CO (shaded in blue, panel a) in irradiated C2H4/CH3OH mixed ices is significantly higher than in pure CH3OH. The reaction scheme (bottom) denotes the formation of ethane and butane following DEA to CH3OH yielding CH3O− and H•. Adapted with permission from Ref. [32]. Copyright 2019 American Chemical Society.

Figure 14.
Comparison of the dependencies of the integrated desorption signals of methyl formate (●) and H2CO (△) on electron energy obtained by TDS after irradiation of CO/CH3OH (1:1) mixed ices with 250 µC/cm2. The solid lines serve as a guide to the eye. Adapted from Ref. [26] with permission from the PCCP Owner Societies.
Figure 14.
Comparison of the dependencies of the integrated desorption signals of methyl formate (●) and H2CO (△) on electron energy obtained by TDS after irradiation of CO/CH3OH (1:1) mixed ices with 250 µC/cm2. The solid lines serve as a guide to the eye. Adapted from Ref. [26] with permission from the PCCP Owner Societies.

Figure 15.
Zero-point energy corrected energy profiles for the (a) hydrogen loss from CH3O⦁ yielding H2CO and H⦁, (b) hydrogen transfer reaction from CH3O⦁ to CO yielding H2CO and HCO⦁, and (c) hydrogen transfer reaction from CH3O⦁ to C2H4 yielding H2CO and C2H5⦁. All geometries were optimized at the DFT B3LYP/aug-cc-pVTZ level of theory using RB3LYP and UB3LYP methods for closed shell and open shell species, respectively. The optimized geometries were characterized by their harmonic frequencies as minima (reactants or products) and saddle points (transition states). Improved single point energies were calculated for all minima and saddle points at the CCSD(T)/aug-cc-pVTZ level of theory. These were corrected by the zero-point vibrational energies (ZPEs) which were obtained during the DFT calculation. Calculations were performed with the ORCA 5 software package [118,119]. Previous values are given in parentheses and were reported for (a) by Kamarchik et al. (calculated at the RCCSD(T)/CBS level of theory) [116], and (b) by Wang et al. (calculated with the G2(B3LYP/MP2/CC) method) [117].
Figure 15.
Zero-point energy corrected energy profiles for the (a) hydrogen loss from CH3O⦁ yielding H2CO and H⦁, (b) hydrogen transfer reaction from CH3O⦁ to CO yielding H2CO and HCO⦁, and (c) hydrogen transfer reaction from CH3O⦁ to C2H4 yielding H2CO and C2H5⦁. All geometries were optimized at the DFT B3LYP/aug-cc-pVTZ level of theory using RB3LYP and UB3LYP methods for closed shell and open shell species, respectively. The optimized geometries were characterized by their harmonic frequencies as minima (reactants or products) and saddle points (transition states). Improved single point energies were calculated for all minima and saddle points at the CCSD(T)/aug-cc-pVTZ level of theory. These were corrected by the zero-point vibrational energies (ZPEs) which were obtained during the DFT calculation. Calculations were performed with the ORCA 5 software package [118,119]. Previous values are given in parentheses and were reported for (a) by Kamarchik et al. (calculated at the RCCSD(T)/CBS level of theory) [116], and (b) by Wang et al. (calculated with the G2(B3LYP/MP2/CC) method) [117].

Scheme 2.
Mechanism for the ionization-driven formation of ethylamine in C2H4/NH3 mixed ices upon electron irradiation [27,29].

Figure 16.
Dependence of the relative product yields of ethane (C2H6) and ethylamine (C2H5NH2) on electron energy obtained by TDS after irradiation of a C2H4/NH3 (1:1) mixed ices with 800 µC/cm2. The solid lines serve as a guide to the eye. Adapted with permission from Ref. [29]. Copyright 2014 American Chemical Society.
Figure 16.
Dependence of the relative product yields of ethane (C2H6) and ethylamine (C2H5NH2) on electron energy obtained by TDS after irradiation of a C2H4/NH3 (1:1) mixed ices with 800 µC/cm2. The solid lines serve as a guide to the eye. Adapted with permission from Ref. [29]. Copyright 2014 American Chemical Society.

Figure 17.
Reaction pathway of the ionization-driven hydroamination reaction of ethylene (C2H4) and NH3 leading to ethylamine (C2H5NH2) calculated on the B3LYP/6-311 + G(d) level of theory. The condensed phase environment was modelled by embedding the reactants in a cluster of six additional NH3 molecules. For comparison, the energies of the isolated molecules relative to their ground states is included on the left side of the energy diagram. The calculations were performed with the Gaussian 98 package [121]. All energies are given relative to the sum of the isolated reactant molecules. The geometries are snapshots on the potential energy surface along the proposed reaction path. Note, that the geometries are no true minima or transition states. However, the remaining imaginary modes are very small and can all be associated with rotational modes of the NH3 moieties. The true minima and transition states thus should not deviate much from the geometries presented herein.
Figure 17.
Reaction pathway of the ionization-driven hydroamination reaction of ethylene (C2H4) and NH3 leading to ethylamine (C2H5NH2) calculated on the B3LYP/6-311 + G(d) level of theory. The condensed phase environment was modelled by embedding the reactants in a cluster of six additional NH3 molecules. For comparison, the energies of the isolated molecules relative to their ground states is included on the left side of the energy diagram. The calculations were performed with the Gaussian 98 package [121]. All energies are given relative to the sum of the isolated reactant molecules. The geometries are snapshots on the potential energy surface along the proposed reaction path. Note, that the geometries are no true minima or transition states. However, the remaining imaginary modes are very small and can all be associated with rotational modes of the NH3 moieties. The true minima and transition states thus should not deviate much from the geometries presented herein.

Scheme 3.
Reaction of a neutral NH2⦁ radical with ethylene to yield ethylamine.

Scheme 4.
Reaction of H2O or CH3OH with CO yielding formic acid and methyl formate, respectively, and reaction of CH3OH with C2H4 yielding ethyl methyl ether. The reactions can be initiated by DEA to CO at E0 = 10 eV yielding C and O⦁−, or by ND, EI, or DEA to CH3OH. Note that DEA to CO is not accessible in C2H4/CH3OH mixed ices.
Scheme 4.
Reaction of H2O or CH3OH with CO yielding formic acid and methyl formate, respectively, and reaction of CH3OH with C2H4 yielding ethyl methyl ether. The reactions can be initiated by DEA to CO at E0 = 10 eV yielding C and O⦁−, or by ND, EI, or DEA to CH3OH. Note that DEA to CO is not accessible in C2H4/CH3OH mixed ices.

Figure 18.
Dependence of the integrated desorption signals of products formed upon electron irradiation of different mixed ices as obtained by TDS after the short exposures stated in the graphs: (a) HCOOH in CO/H2O mixed ices (bottom, from Ref. [24]) and C2H5OH in C2H4/H2O mixed ices (top, from Ref. [28]), (b) CH3OCHO in CO/CH3OH mixed ices (top, from Ref. [26]) and C2H5OCH3 in C2H4/CH3OH mixed ices (bottom, from Ref. [32]). The solid lines serve as a guide to the eye. A set of experiments with longer exposures at lower electron energies is presented in Figure 19. For details of the experiments see Refs. [24,26,28,32]. Adapted from Ref. [37] with kind permission of The European Physical Journal (EPJ).
Figure 18.
Dependence of the integrated desorption signals of products formed upon electron irradiation of different mixed ices as obtained by TDS after the short exposures stated in the graphs: (a) HCOOH in CO/H2O mixed ices (bottom, from Ref. [24]) and C2H5OH in C2H4/H2O mixed ices (top, from Ref. [28]), (b) CH3OCHO in CO/CH3OH mixed ices (top, from Ref. [26]) and C2H5OCH3 in C2H4/CH3OH mixed ices (bottom, from Ref. [32]). The solid lines serve as a guide to the eye. A set of experiments with longer exposures at lower electron energies is presented in Figure 19. For details of the experiments see Refs. [24,26,28,32]. Adapted from Ref. [37] with kind permission of The European Physical Journal (EPJ).

Figure 19.
Energy dependences for the formation of methyl formate in CO/CH3OH (1:1) mixed ices, and of ethyl methyl ether in C2H4/CH3OH (1:1) mixed ices after electron exposures of 12,000 and 8000 µC/cm2, respectively. The solid lines serve as a guide to the eye. Upper data are adapted from Ref. [26] with permission from the PCCP Owner Societies. Lower data are adapted with permission from Ref. [32]. Copyright 2019 American Chemical Society. Data for shorter exposures over a wider energy range are presented in Figure 18b.
Figure 19.
Energy dependences for the formation of methyl formate in CO/CH3OH (1:1) mixed ices, and of ethyl methyl ether in C2H4/CH3OH (1:1) mixed ices after electron exposures of 12,000 and 8000 µC/cm2, respectively. The solid lines serve as a guide to the eye. Upper data are adapted from Ref. [26] with permission from the PCCP Owner Societies. Lower data are adapted with permission from Ref. [32]. Copyright 2019 American Chemical Society. Data for shorter exposures over a wider energy range are presented in Figure 18b.

Figure 20.
Dependence of the integrated desorption intensities of the m/z 45 signal of formic acid (HCOOH, ☐), the m/z 30 signal of H2CO (△), and of the m/z 44 signal of CO2 (●) on electron energy obtained by TDS after irradiation of CO/H2O (1:1) mixed ices with 250 µC/cm2. Individual curves are scaled by an arbitrary factor to facilitate comparison. The solid and broken lines serve as a guide to the eye. Adapted with permission from Ref. [24]. Copyright 2019 American Chemical Society.
Figure 20.
Dependence of the integrated desorption intensities of the m/z 45 signal of formic acid (HCOOH, ☐), the m/z 30 signal of H2CO (△), and of the m/z 44 signal of CO2 (●) on electron energy obtained by TDS after irradiation of CO/H2O (1:1) mixed ices with 250 µC/cm2. Individual curves are scaled by an arbitrary factor to facilitate comparison. The solid and broken lines serve as a guide to the eye. Adapted with permission from Ref. [24]. Copyright 2019 American Chemical Society.

Figure 21.
Thermal desorption spectra obtained after irradiation of CO/CH3OH (1:1) mixed ices and of pure CH3OH with 12,000 µC/cm2 at 5.5 eV. Panel (a) shows the formation of methylacetate in the mixed films but not in pure CH3OH. Panel (b) shows the formation of dimethylether (shaded in blue in the mixed ices but not in pure CH3OH. The second desorption signal in the m/z 45 trace at around 170 K is a fragment of methoxymethanol. Adapted from Ref. [26] with permission from the PCCP Owner Societies.
Figure 21.
Thermal desorption spectra obtained after irradiation of CO/CH3OH (1:1) mixed ices and of pure CH3OH with 12,000 µC/cm2 at 5.5 eV. Panel (a) shows the formation of methylacetate in the mixed films but not in pure CH3OH. Panel (b) shows the formation of dimethylether (shaded in blue in the mixed ices but not in pure CH3OH. The second desorption signal in the m/z 45 trace at around 170 K is a fragment of methoxymethanol. Adapted from Ref. [26] with permission from the PCCP Owner Societies.


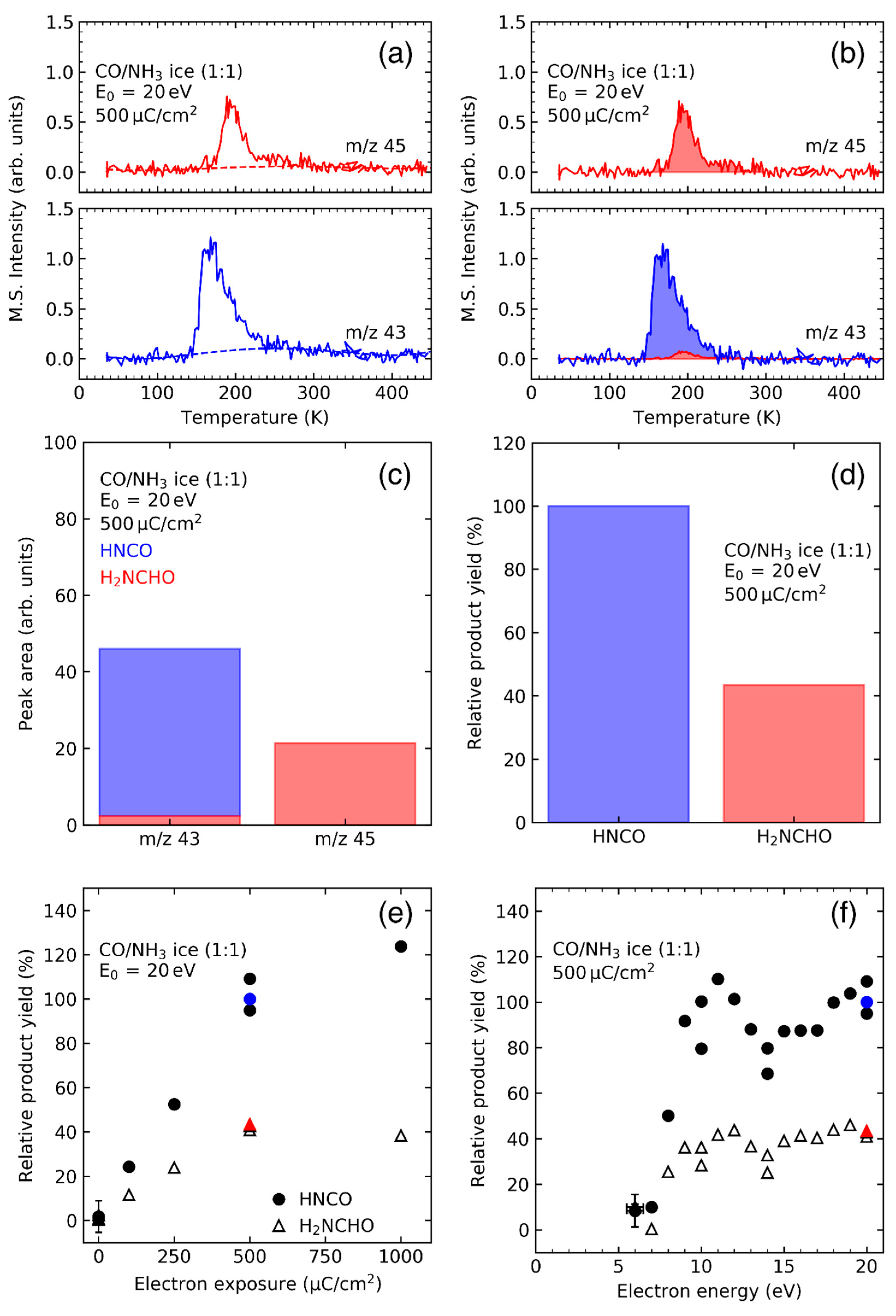{kind=link}
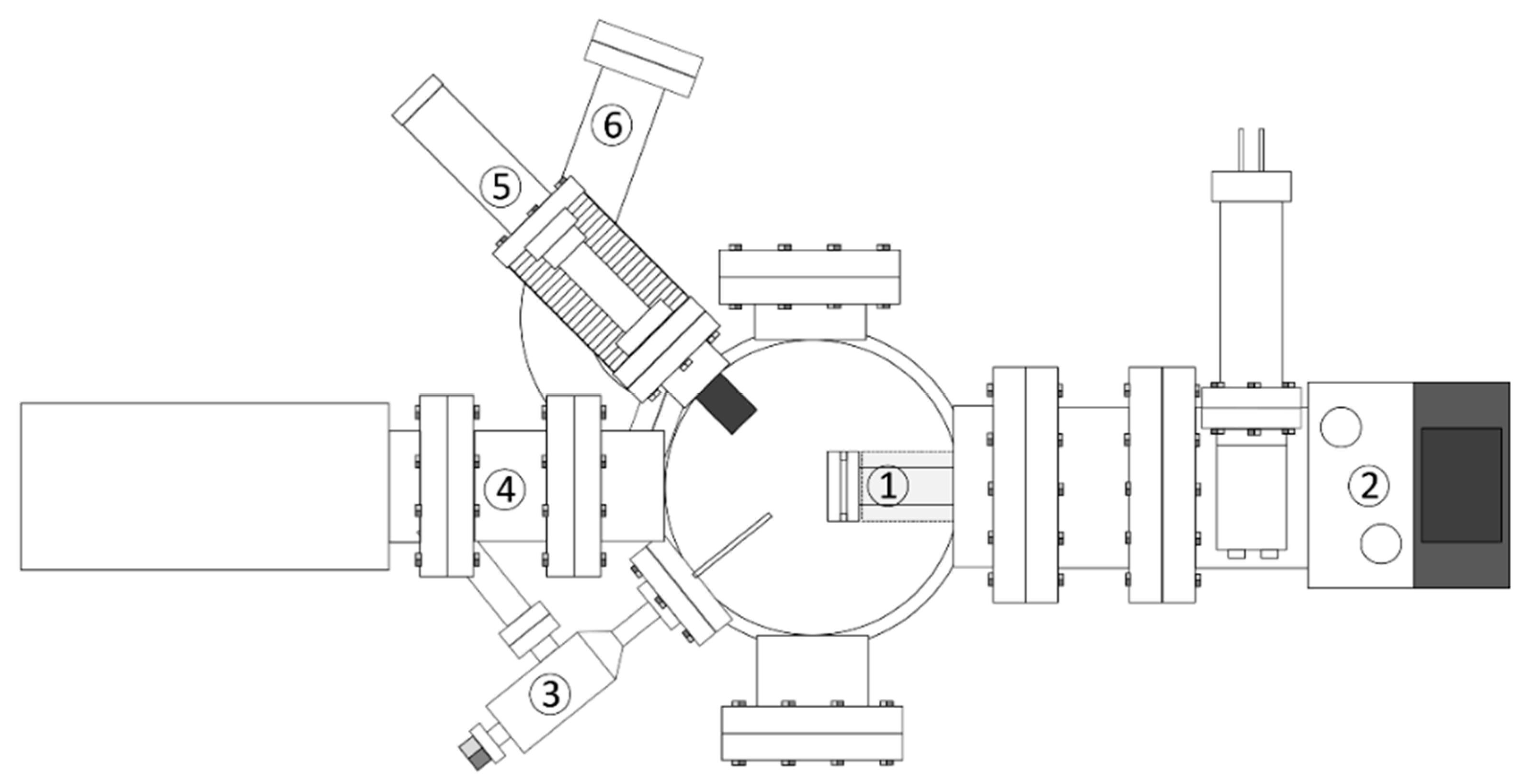{kind=link}
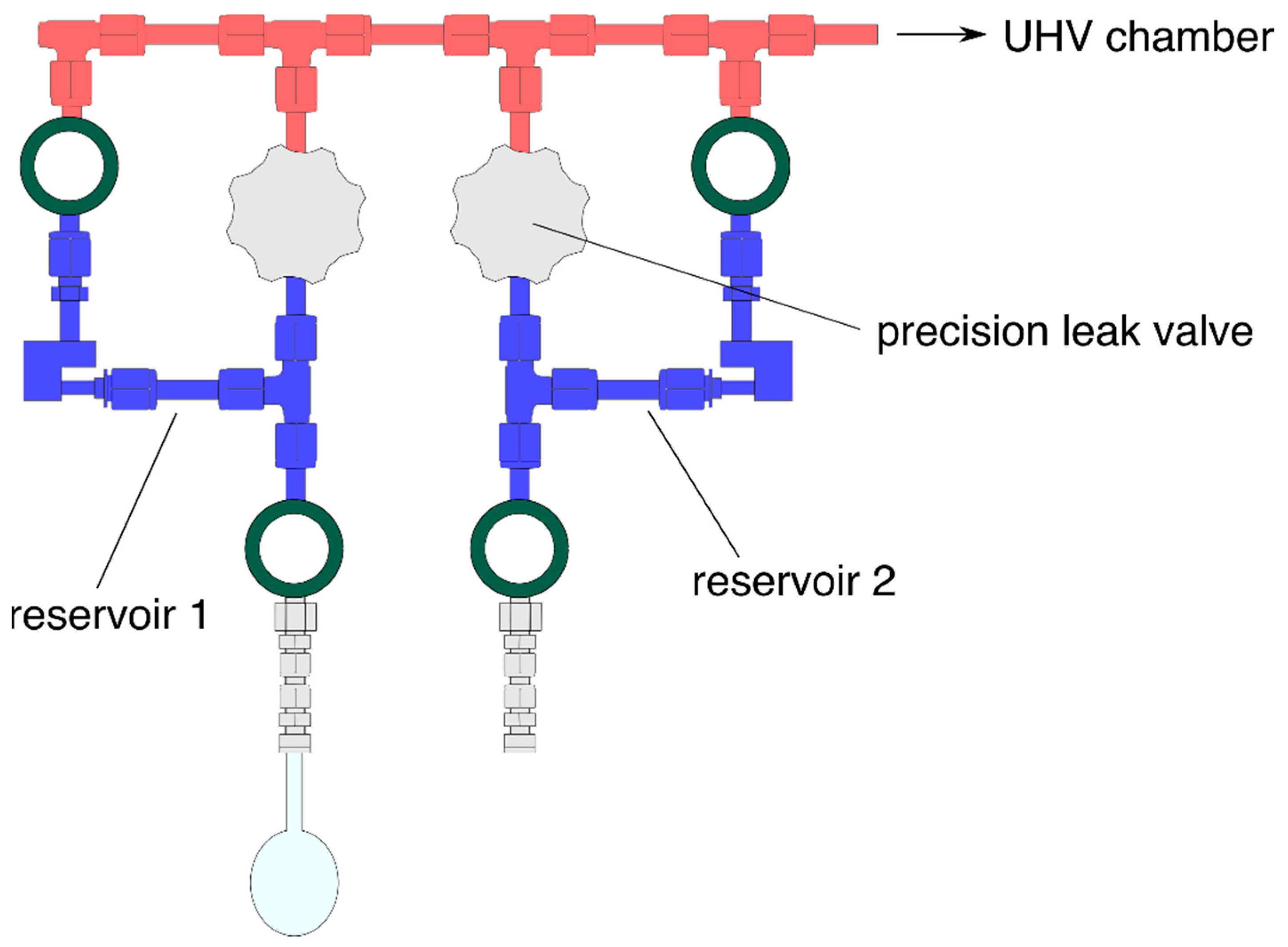{kind=link}
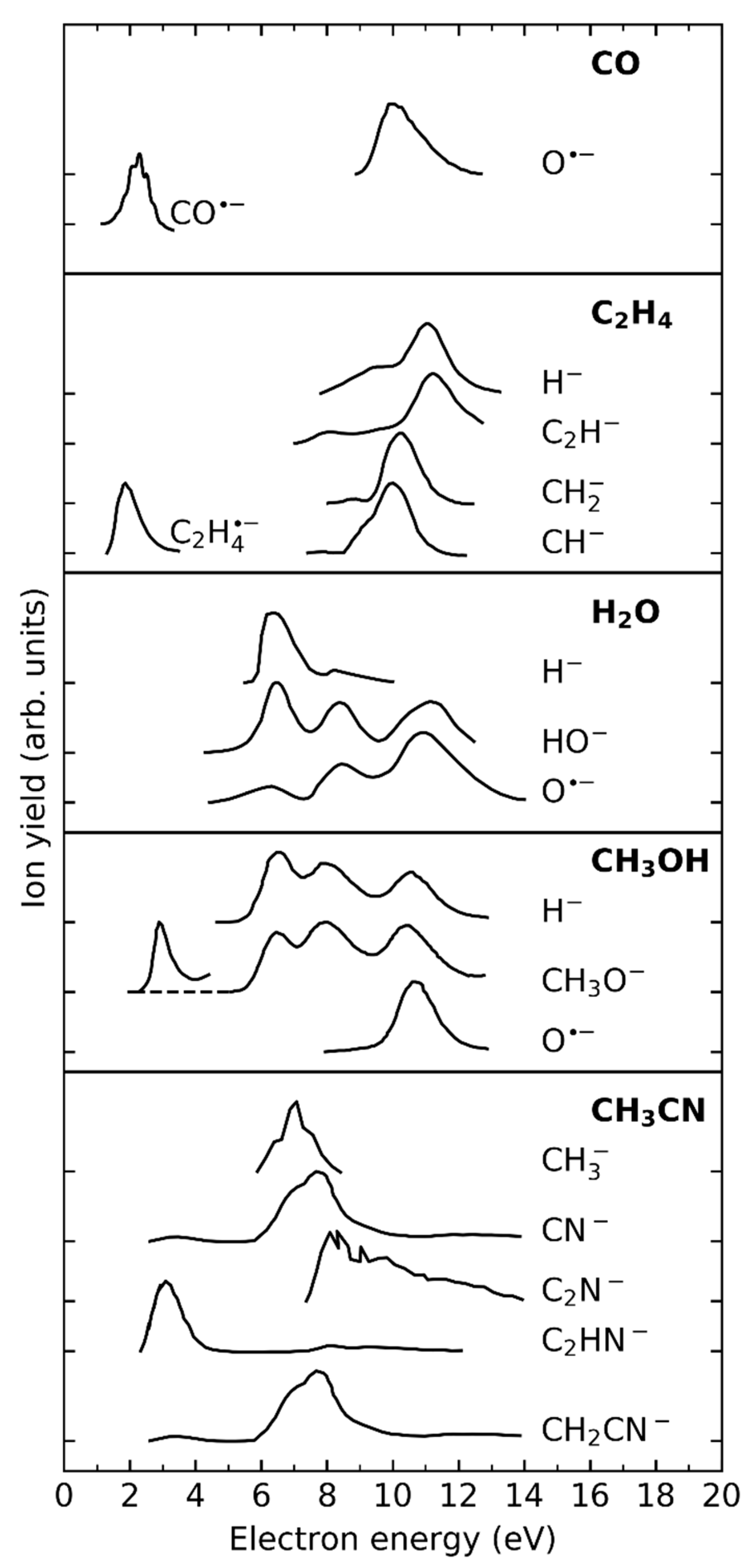{kind=link}
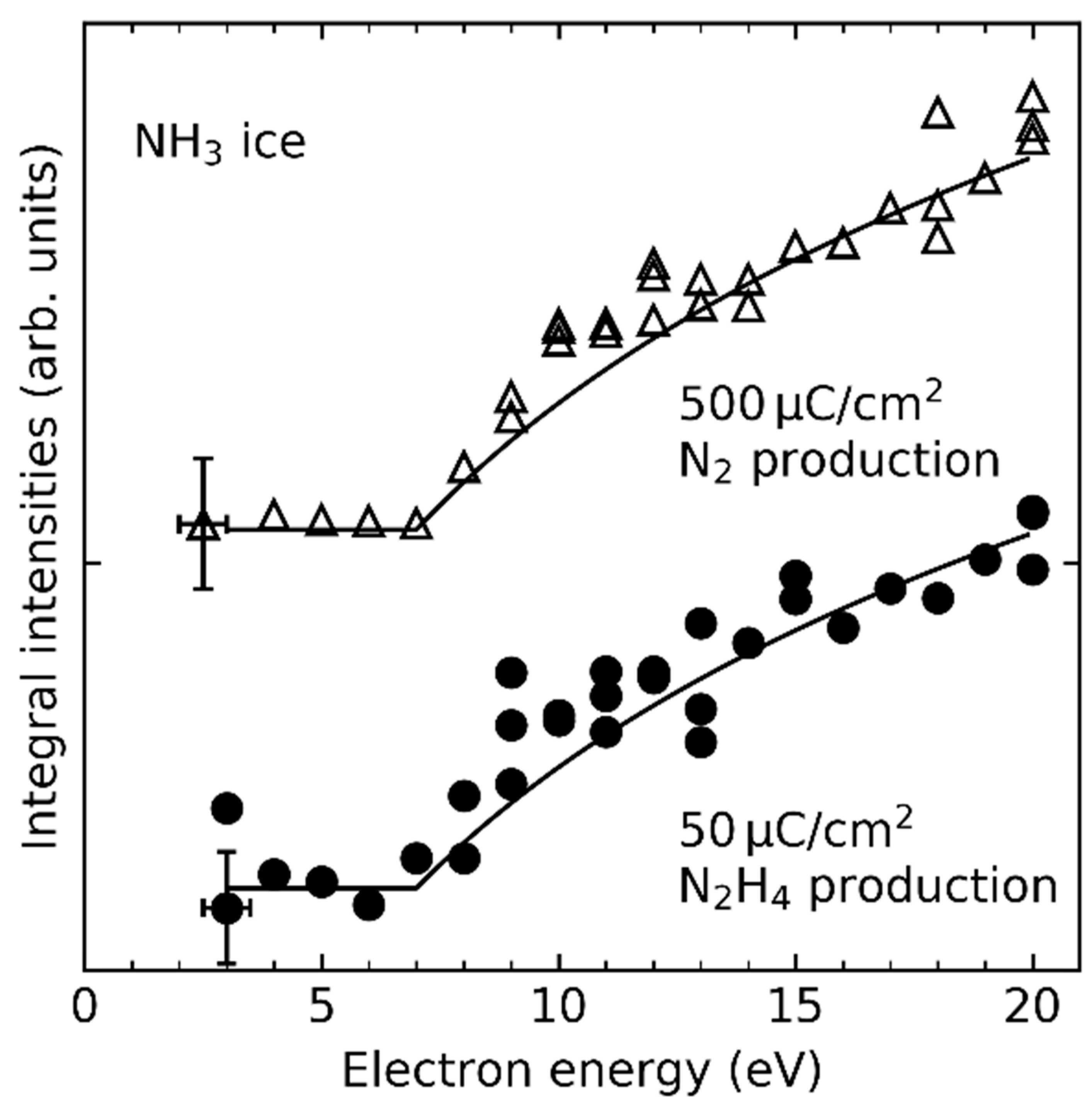{kind=link}
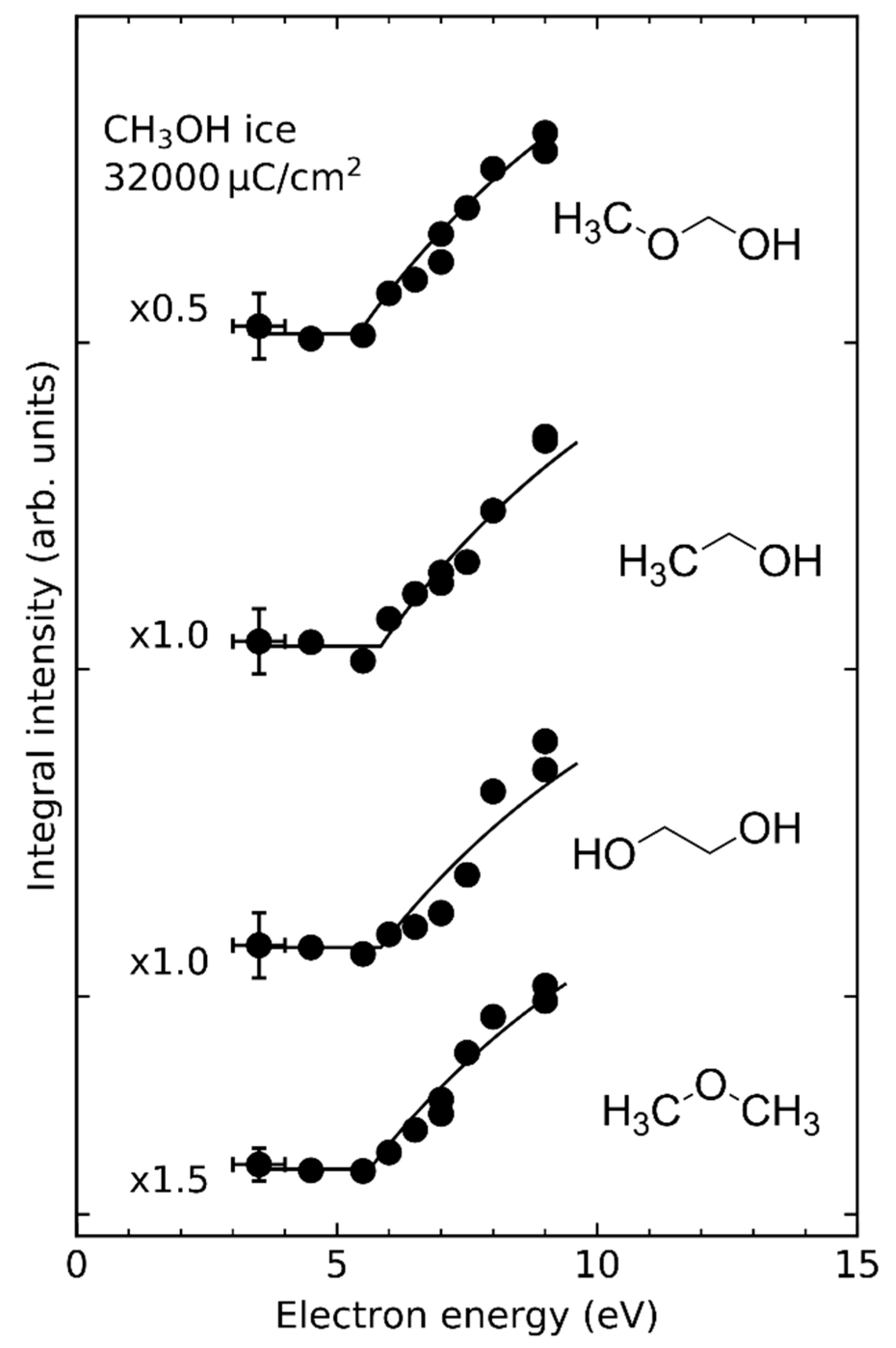{kind=link}
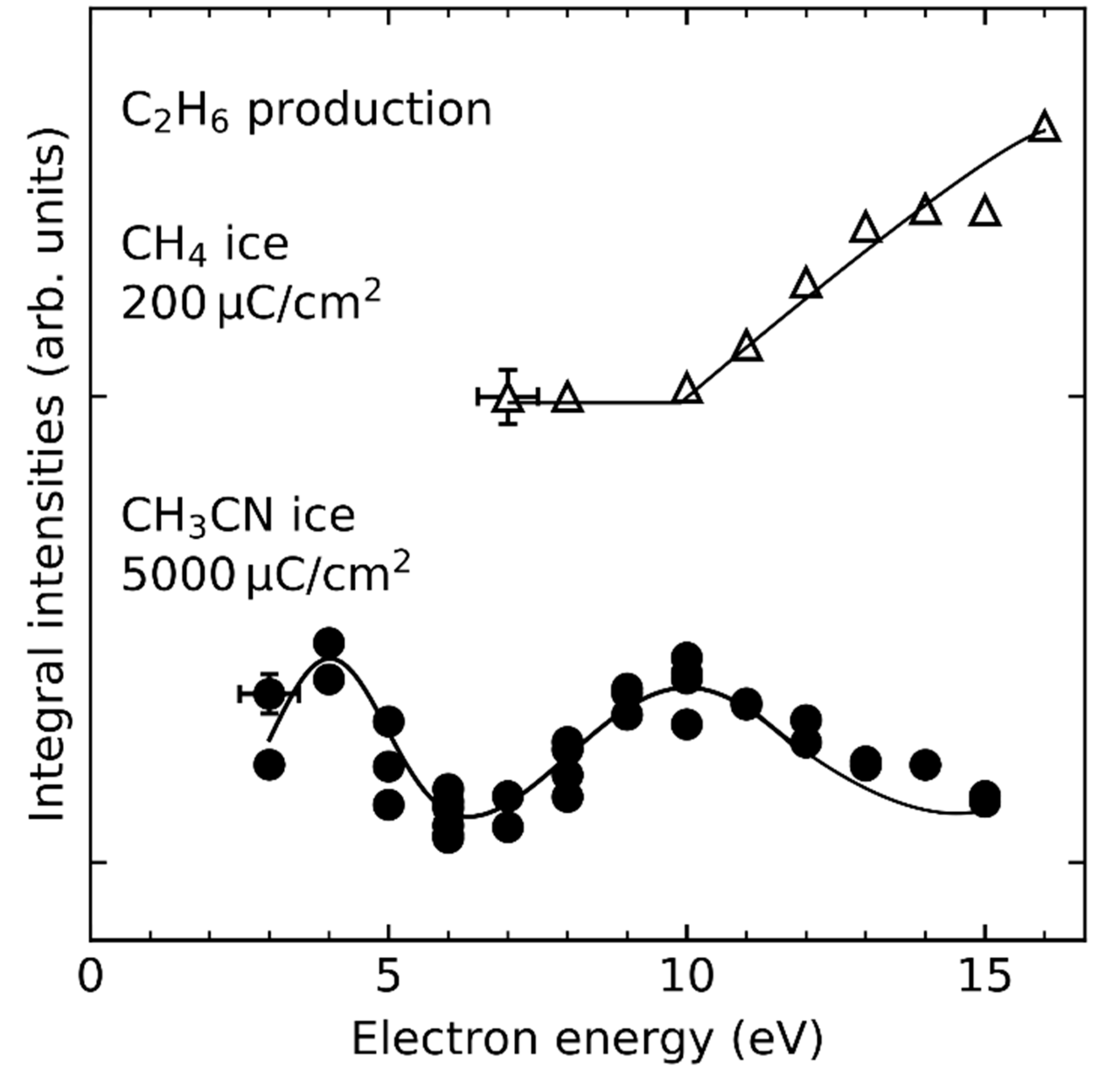{kind=link}
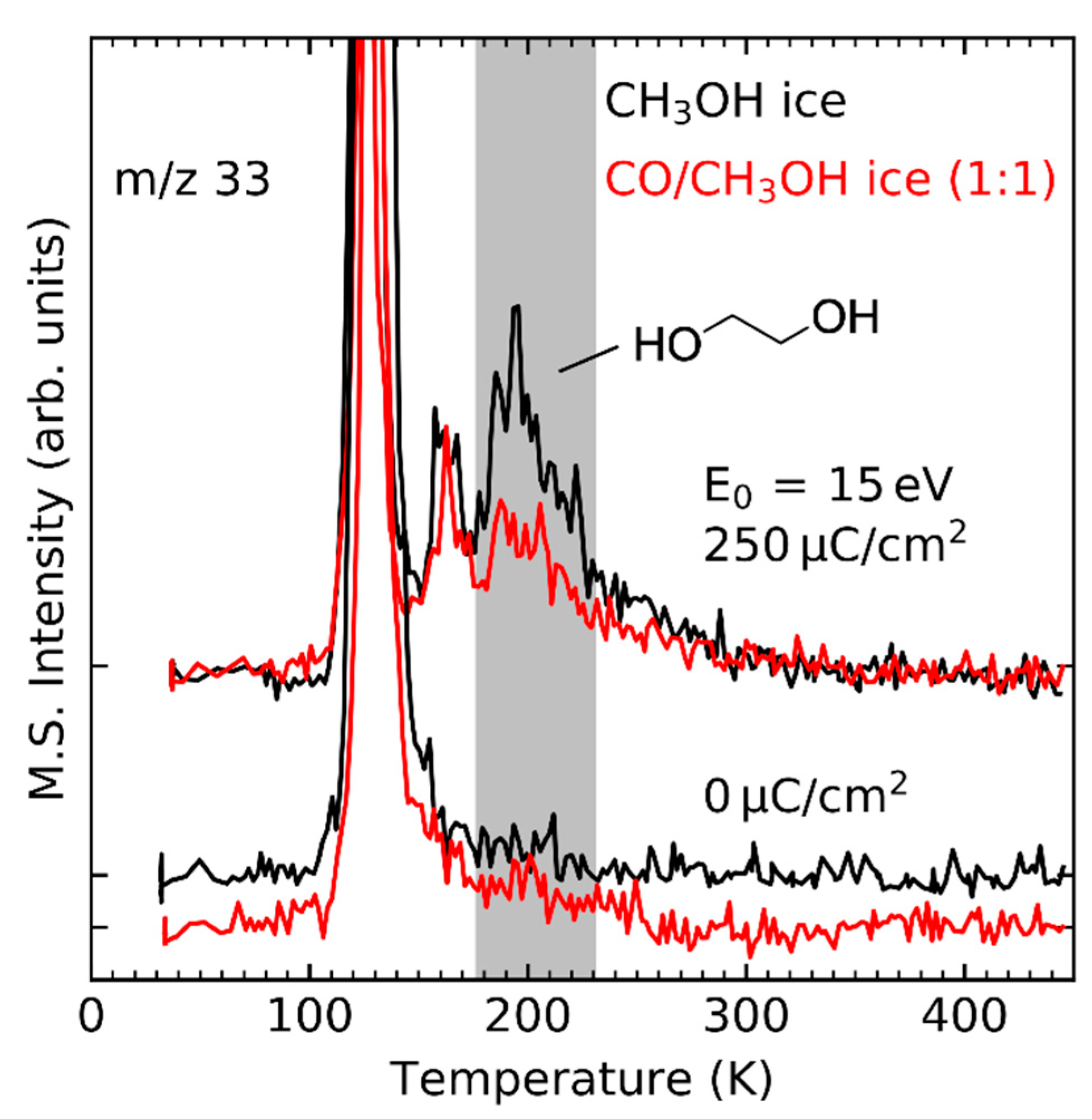{kind=link}
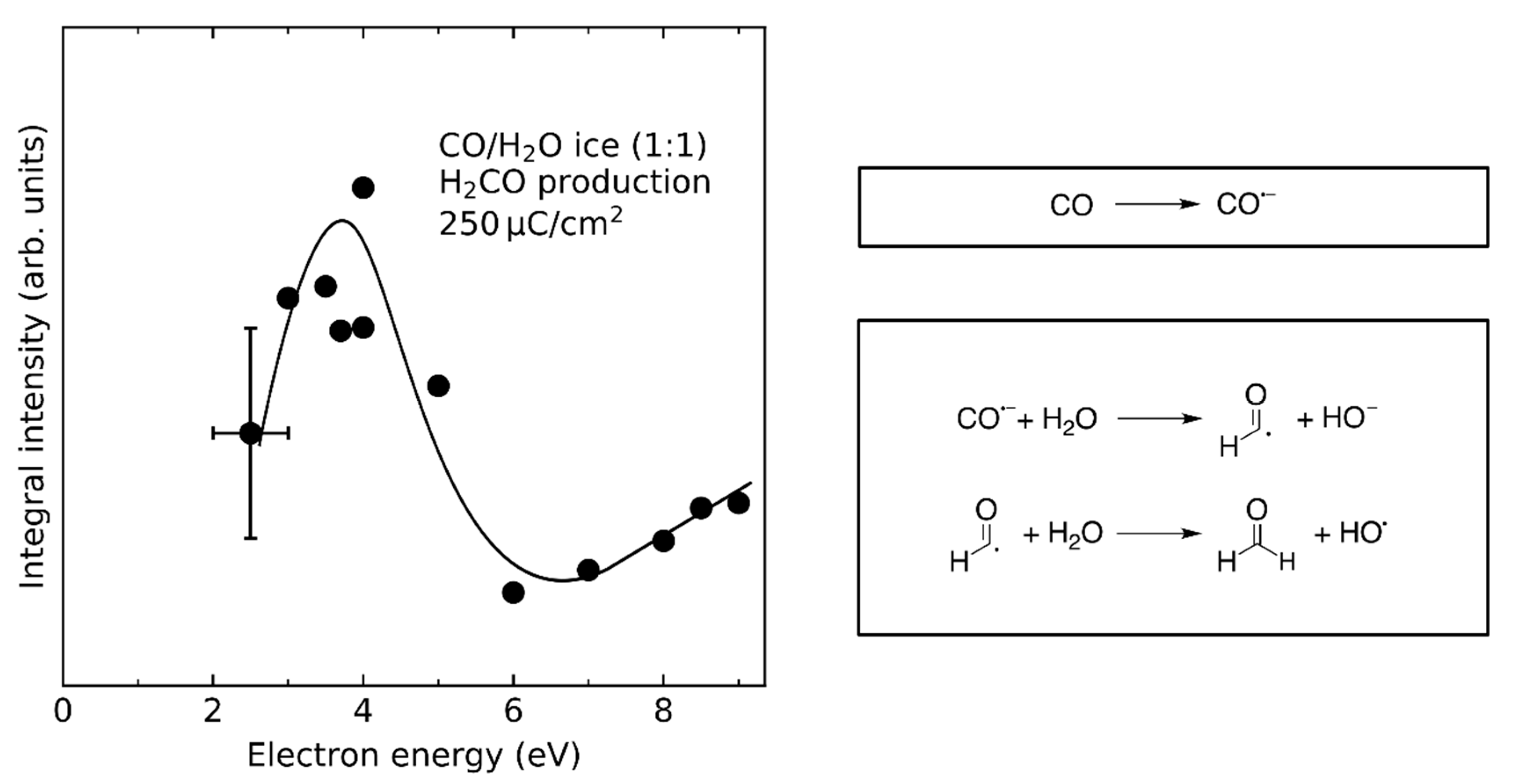{kind=link}
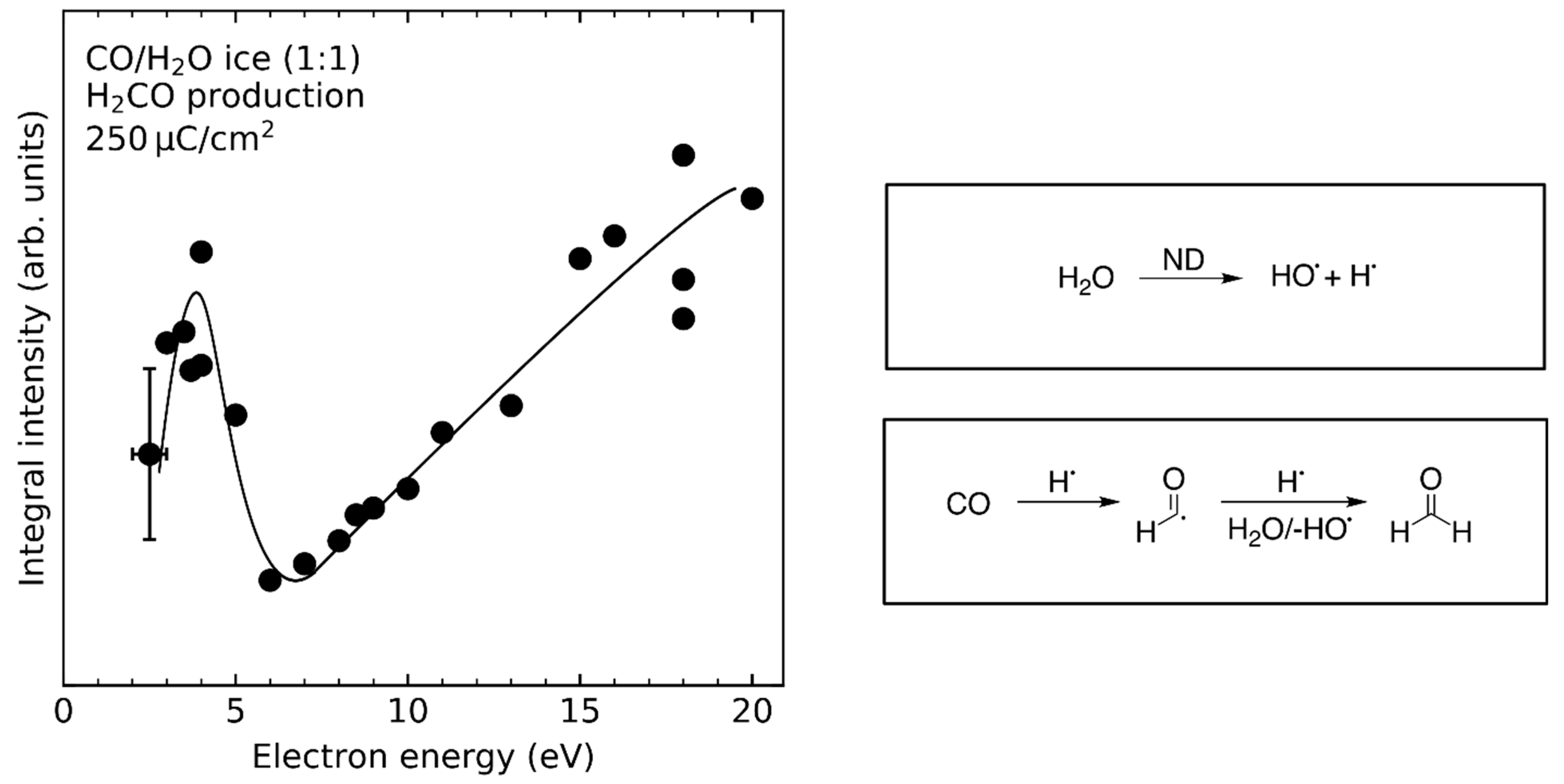{kind=link}
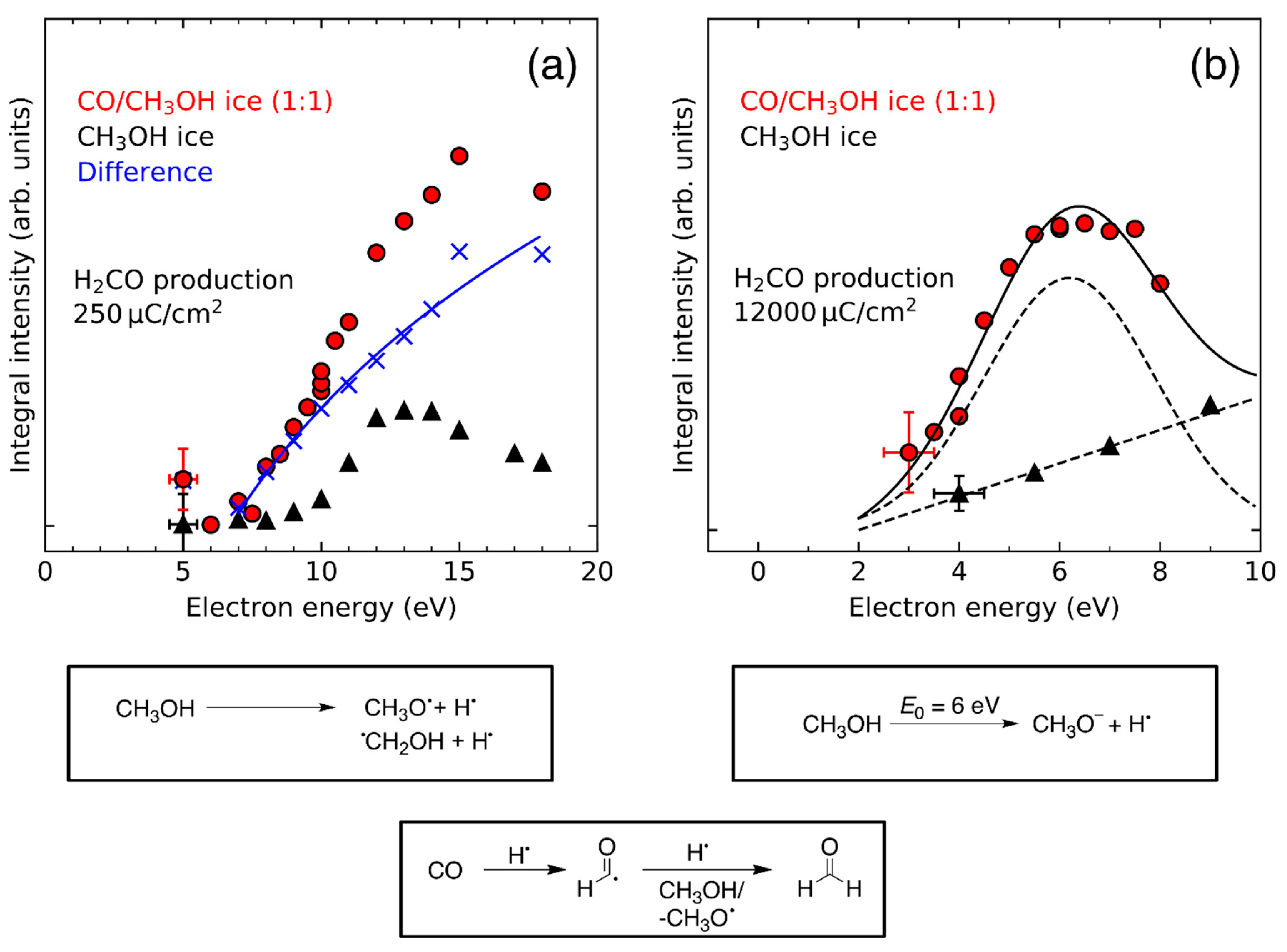{kind=link}
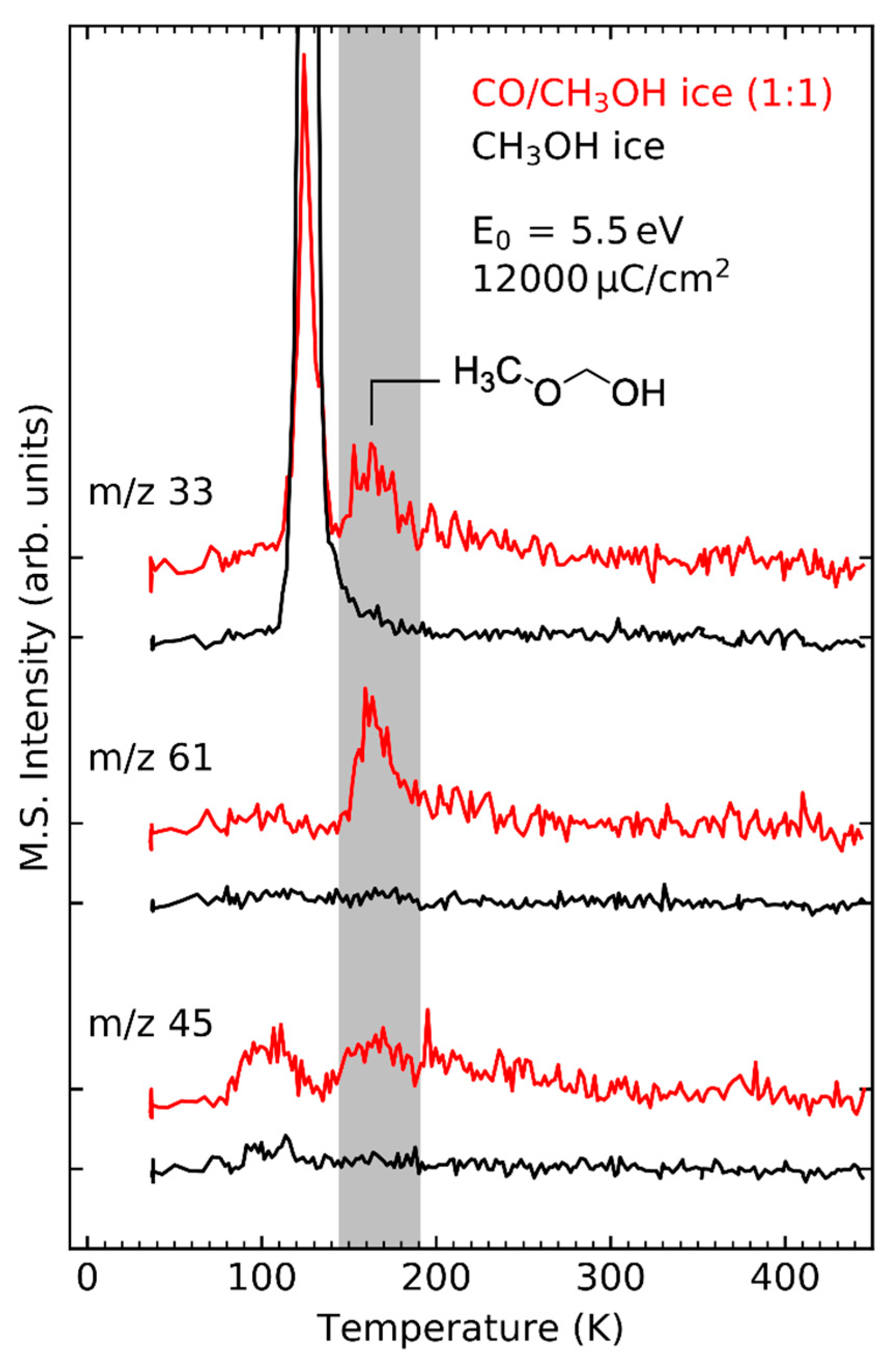{kind=link}
{kind=link}
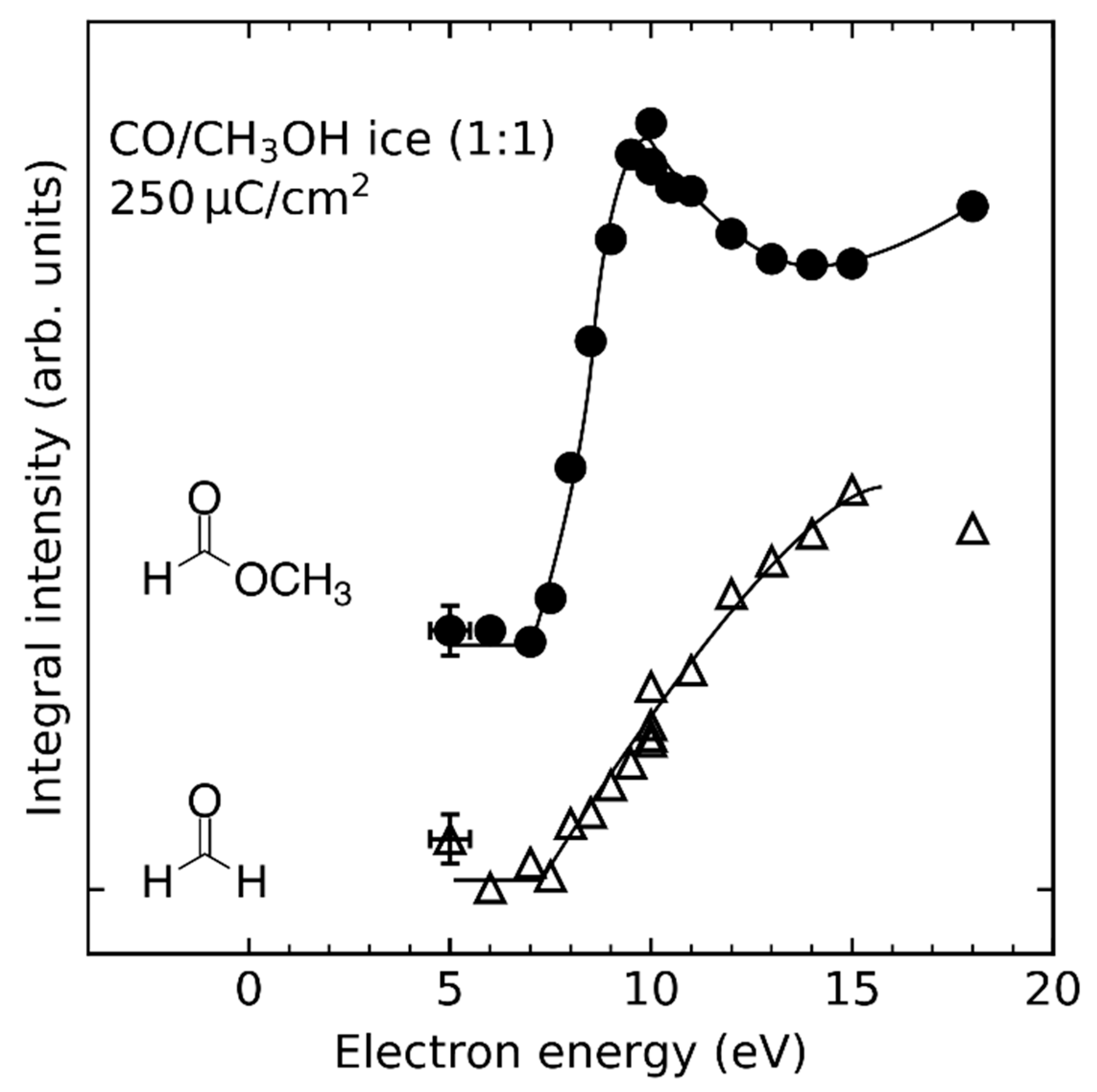{kind=link}
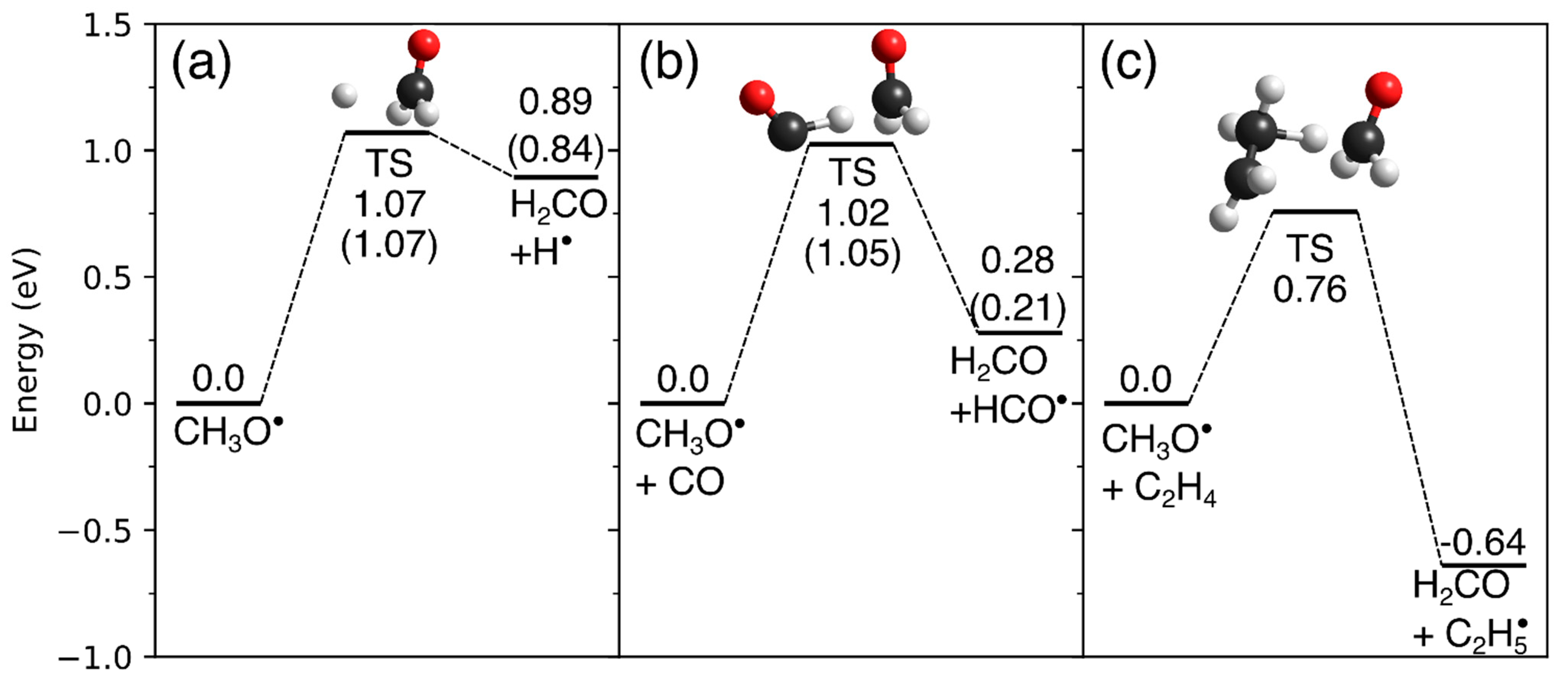{kind=link}
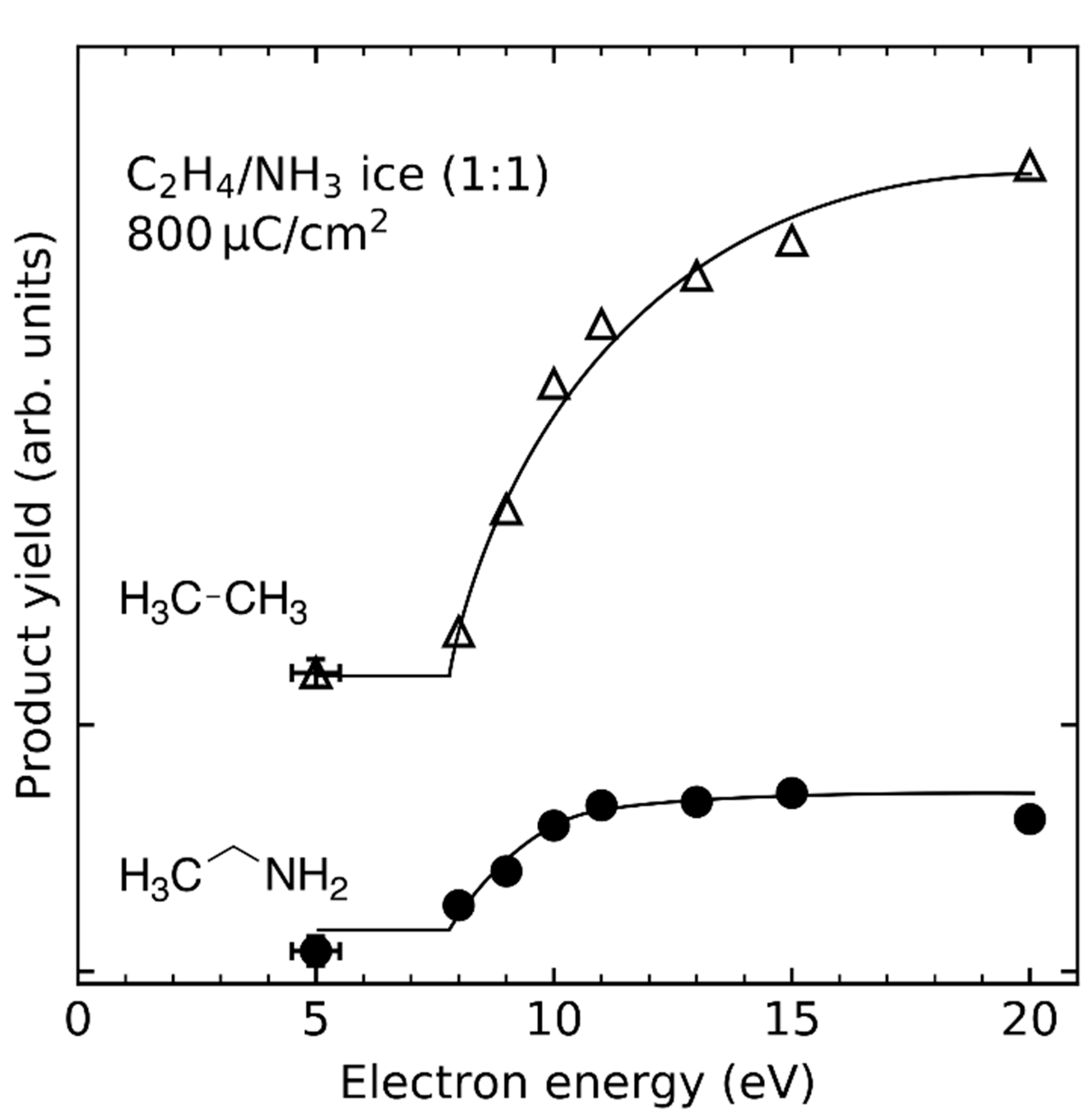{kind=link}
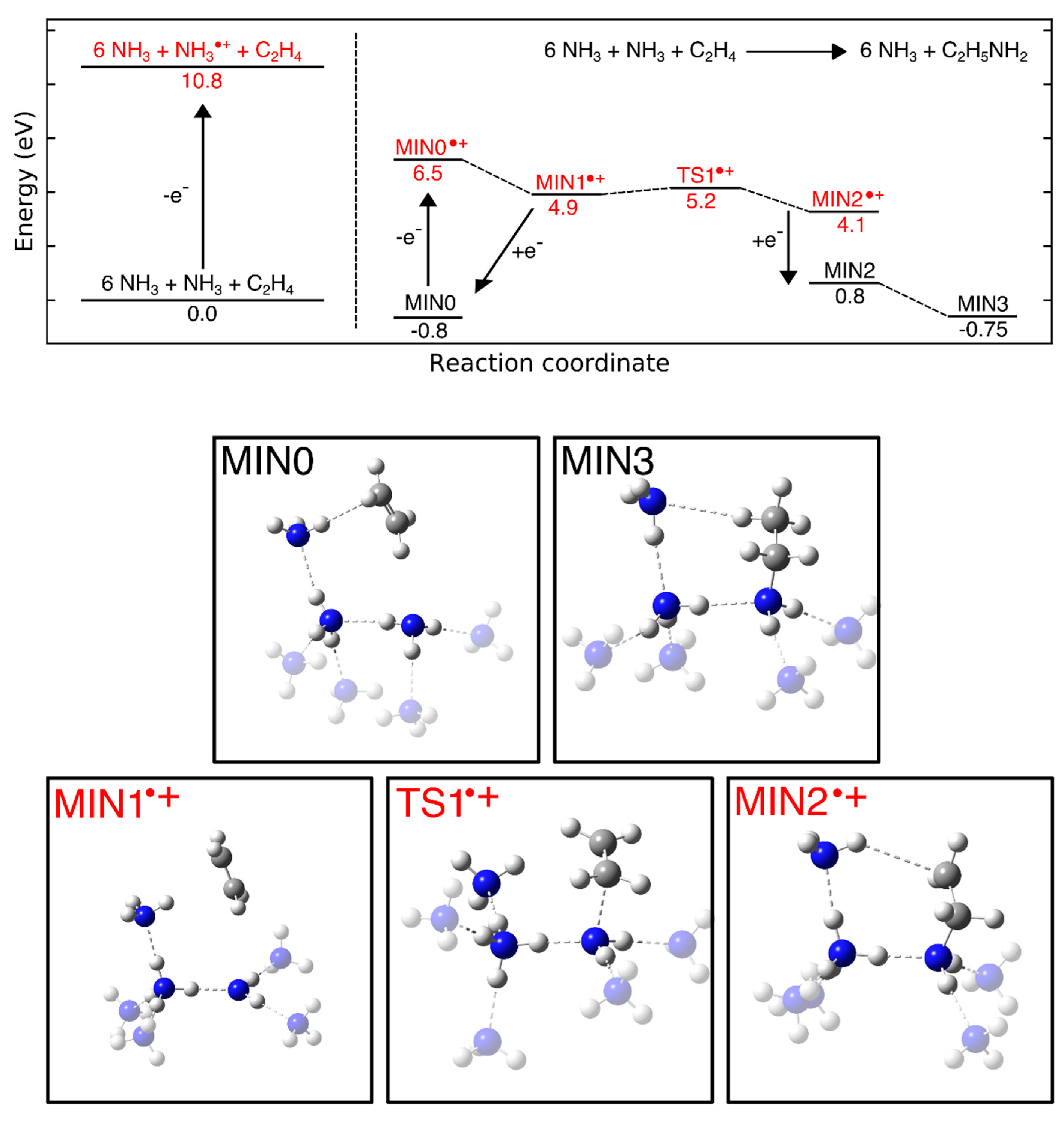{kind=link}
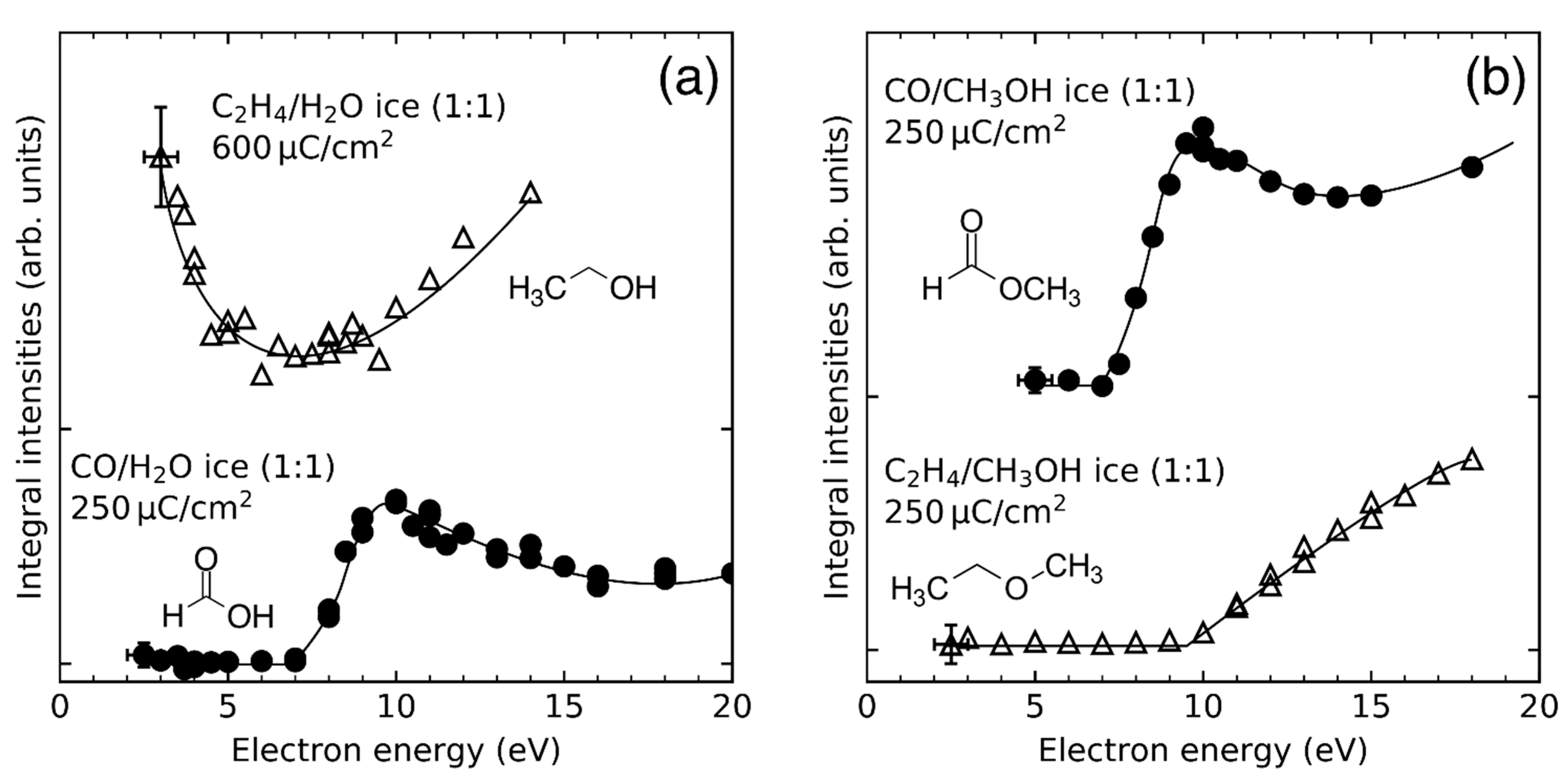{kind=link}
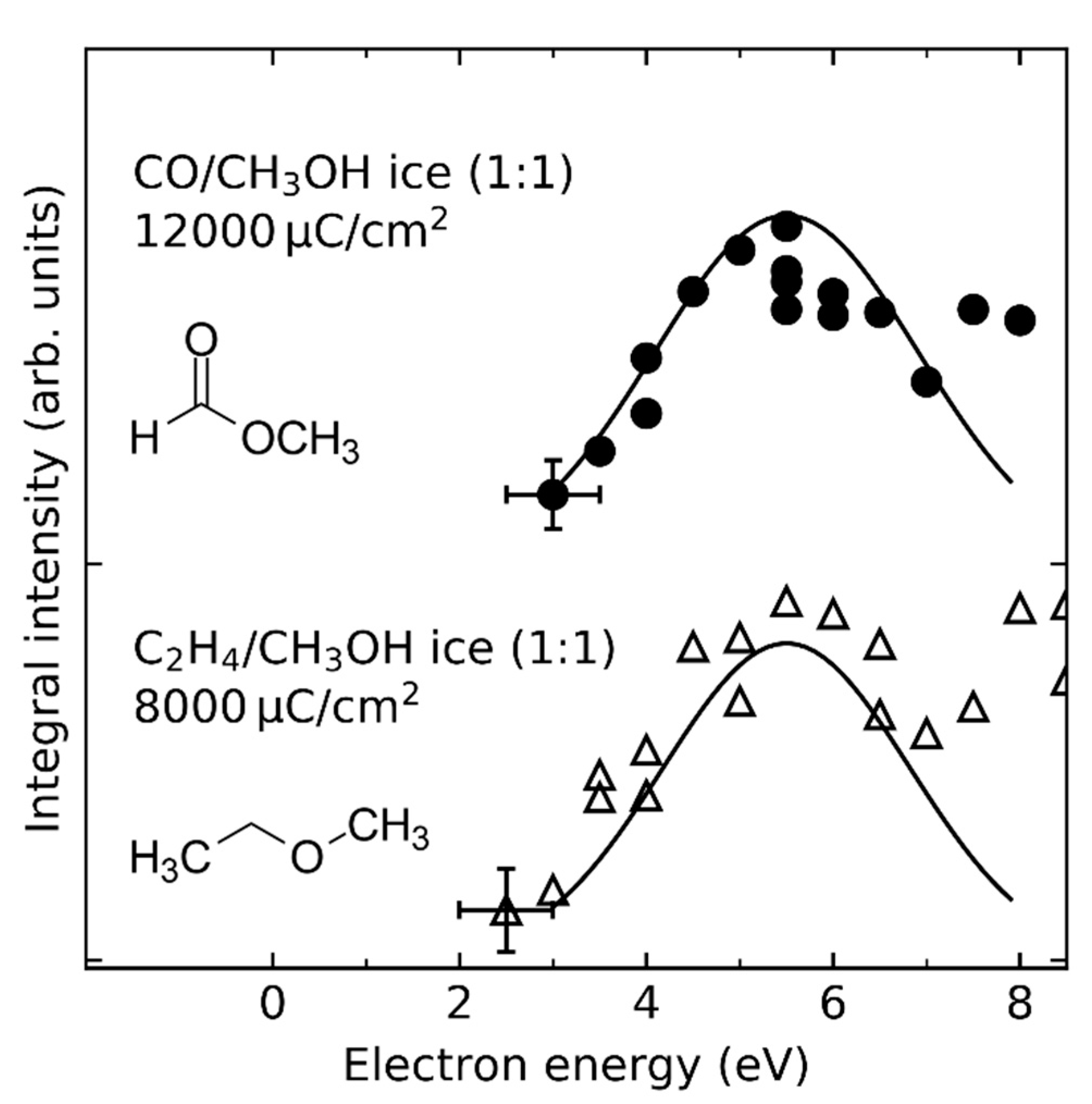{kind=link}
{kind=link}
{kind=link}
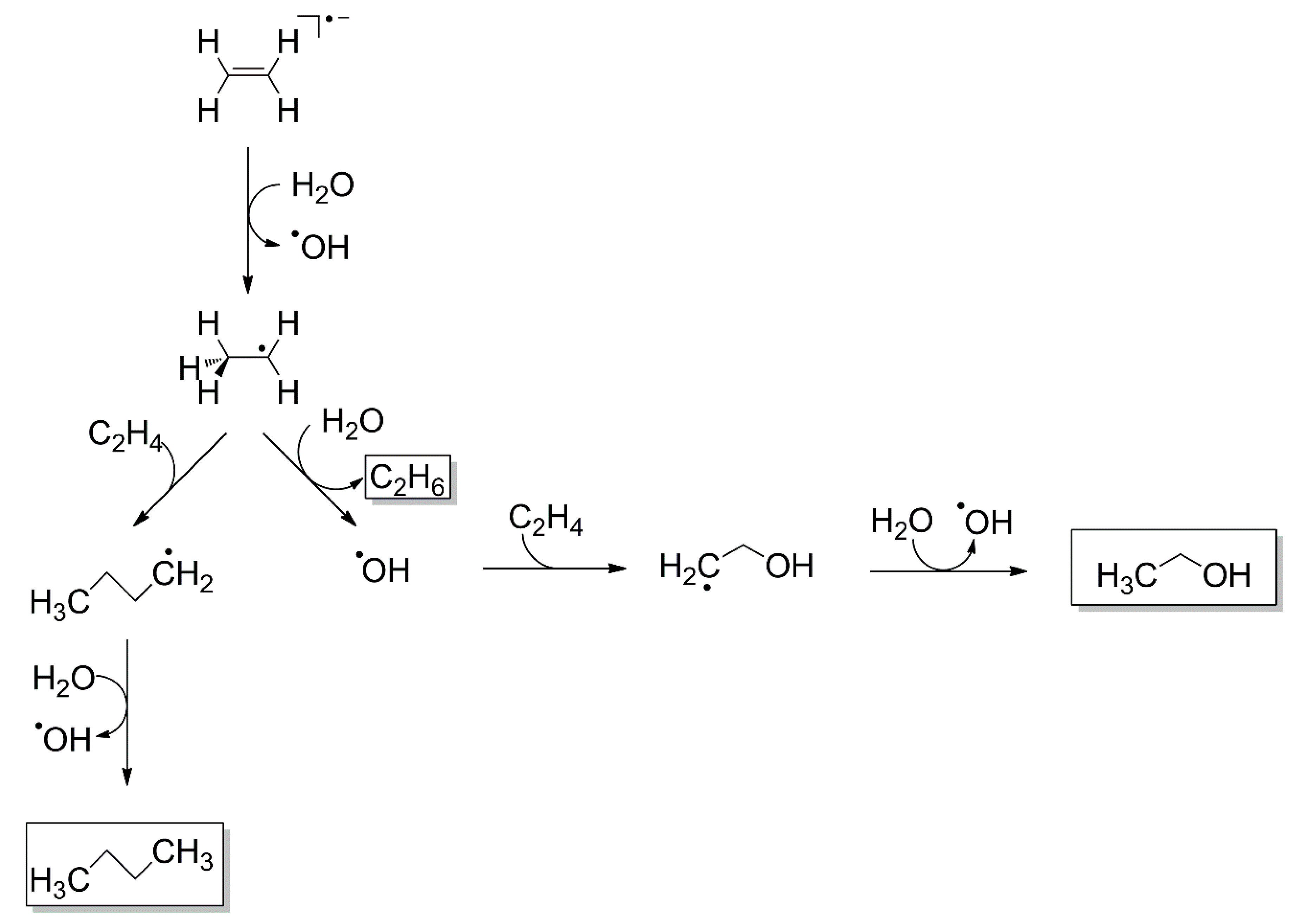{kind=link}
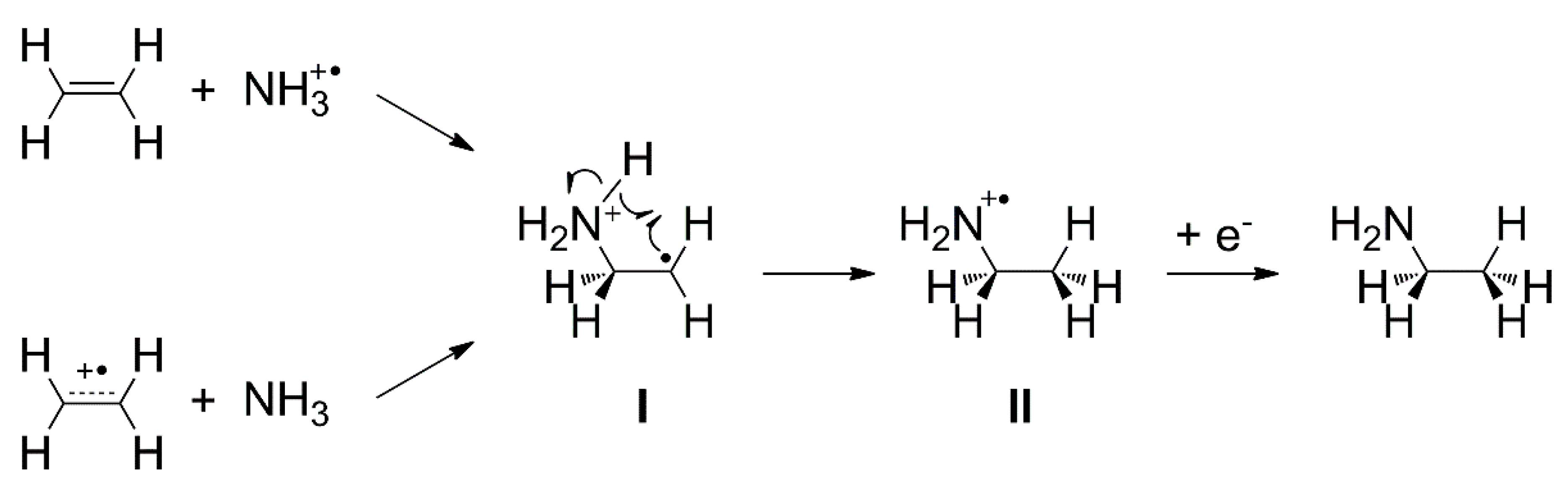{kind=link}
{kind=link}
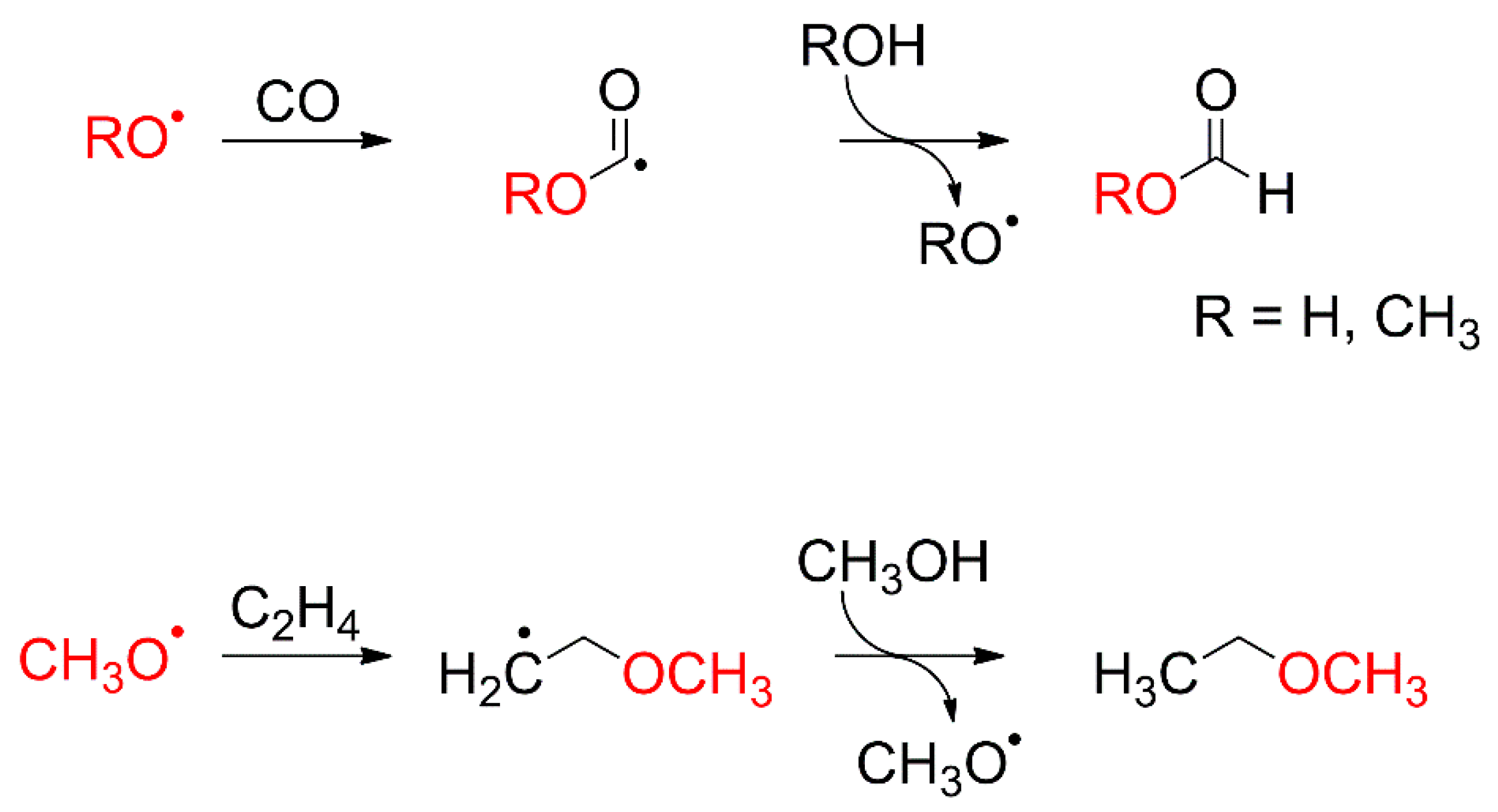{kind=link}
Table 1.
Products that can be formed in reactions of C2H4 and CO with NH3, H2O, and CH3OH when electron interaction leads to addition of one reactant to the other so that all atoms of the initial reactants become incorporated in the product. Such reactions are also referred to as atom-efficient. The products shown here are those that are formed with the smallest possible number of bonds being broken and reformed as compared to the reactants.
Table 1.
Products that can be formed in reactions of C2H4 and CO with NH3, H2O, and CH3OH when electron interaction leads to addition of one reactant to the other so that all atoms of the initial reactants become incorporated in the product. Such reactions are also referred to as atom-efficient. The products shown here are those that are formed with the smallest possible number of bonds being broken and reformed as compared to the reactants.
| NH3 | H2O | CH3OH | |
| C2H4 |  Ethylamine [29] |  Ethanol [28] |  Ethyl methyl ether [32] |
| CO |  Formamide [30] |  Formic acid [24] |  Methyl formate [26] |
Table 2.
Reactive species formed by EI and DI as relevant to the electron-induced reactions in molecular ices discussed in Section 4. Note that the listed species were not directly detected but their contribution to the reactions is deduced indirectly from the identified final reaction products. Other electron-molecule interactions may contribute as well as summarized in Section 3.3 and Section 3.4.
Table 2.
Reactive species formed by EI and DI as relevant to the electron-induced reactions in molecular ices discussed in Section 4. Note that the listed species were not directly detected but their contribution to the reactions is deduced indirectly from the identified final reaction products. Other electron-molecule interactions may contribute as well as summarized in Section 3.3 and Section 3.4.
| Source Molecule | Reactive Species | Ionization Threshold (Gas Phase) 1 | Threshold for Product Formation (Ice) | References (Ice) |
|---|---|---|---|---|
| C2H4 | C2H4•+ | 10.5 eV | 8–10 eV | [28,29,32] |
| CO | CO•+ | 14.014 eV | 13–14 eV | [30] |
| NH3 | NH3•+ | 10.070 eV | ≤12 eV/10 eV | [27,30] |
| H2O | H2O•+ | 12.621 eV | 11 eV | [28] |
| CH4 | CH4•+ | 12.61 eV | 12 eV | this work |
| CH3OH | CH3OH•+ CH3O•+ | 10.84 eV 11.67 eV | 9.8 eV ≤11 eV | [31,32] [32] |
1 Values for gas phase ionization energy are taken from NIST Webbook database [54].
Table 3.
Reactive species formed by ND as relevant to the electron-induced reactions in molecular ices discussed in Section 4. Note that the listed species were not directly detected but their contribution to the reactions is deduced indirectly from the identified final reaction products. Other electron-molecule interactions may contribute as well as summarized in Section 3.2 and Section 3.4.
Table 3.
Reactive species formed by ND as relevant to the electron-induced reactions in molecular ices discussed in Section 4. Note that the listed species were not directly detected but their contribution to the reactions is deduced indirectly from the identified final reaction products. Other electron-molecule interactions may contribute as well as summarized in Section 3.2 and Section 3.4.
| Source Molecule | Reactive Species | Threshold for Product Formation (Ice) | References (Ice) |
|---|---|---|---|
| NH3 | H• + NH2• | ~7 eV | this work |
| H2O | H2 + O H• + HO• | 6–7 eV 6–7 eV | [24] [24,28] |
| CH4 | H• + CH3• | ~10 eV | this work |
| CH3OH | CH3O• + H• H• + •CH2OH CH3• + HO• | ~6 eV ~6 eV ~6 eV | [31] [31] [31] |
Table 4.
Reactive species formed by (D)EA as relevant to the electron-induced reactions in molecular ices discussed in Section 4. Note that the listed species were not directly detected but their contribution to the reactions is deduced indirectly from the identified final reaction products. Other electron-molecule interactions may contribute as well as summarized in Section 3.2 and Section 3.3.
Table 4.
Reactive species formed by (D)EA as relevant to the electron-induced reactions in molecular ices discussed in Section 4. Note that the listed species were not directly detected but their contribution to the reactions is deduced indirectly from the identified final reaction products. Other electron-molecule interactions may contribute as well as summarized in Section 3.2 and Section 3.3.
| Source Molecule | Reactive Species | Resonance in Product Formation (Ice) | References (Ice) |
|---|---|---|---|
| C2H4 | C2H4•− | <5 eV | [28] |
| CO | CO•− C + O•− | ~4 eV 11 eV/16 eV | [24] [37] |
| H2O | H• + HO− O•− + H2 | 6.5 eV 10 eV | [24] [24] |
| CH3OH | CH3O• + H− CH3O− + H• | 5.5 eV 5.5 eV | [32] [26,31] |
| CH3CN | CH3• + CN− | 4 eV/10 eV | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schmidt, F.; Borrmann, T.; Mues, M.P.; Benter, S.; Swiderek, P.; Bredehöft, J.H. Mechanisms of Electron-Induced Chemistry in Molecular Ices. Atoms 2022, 10, 25. https://doi.org/10.3390/atoms10010025
AMA Style
Schmidt F, Borrmann T, Mues MP, Benter S, Swiderek P, Bredehöft JH. Mechanisms of Electron-Induced Chemistry in Molecular Ices. Atoms. 2022; 10(1):25. https://doi.org/10.3390/atoms10010025
Chicago/Turabian StyleSchmidt, Fabian, Tobias Borrmann, Martin Philipp Mues, Sanna Benter, Petra Swiderek, and Jan Hendrik Bredehöft. 2022. "Mechanisms of Electron-Induced Chemistry in Molecular Ices" Atoms 10, no. 1: 25. https://doi.org/10.3390/atoms10010025
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.