
Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies

1
Department of Medical and Surgical Sciences, University of Bologna, 40138 Bologna, Italy
2
Medical Genetics Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy
*
Author to whom correspondence should be addressed.
Antioxidants 2023, 12(2), 353; https://doi.org/10.3390/antiox12020353
Submission received: 28 December 2022
/
Revised: 27 January 2023
/
Accepted: 31 January 2023
/
Published: 2 February 2023
(This article belongs to the Special Issue The Versatility of Mitochondrial Calcium: Insights in the Regulation of Redox Signaling)
Abstract
:Mitochondria are key players in energy production, critical activity for the smooth functioning of energy-demanding organs such as the muscles, brain, and heart. Therefore, dysregulation or alterations in mitochondrial bioenergetics primarily perturb these organs. Within the cell, mitochondria are the major site of reactive oxygen species (ROS) production through the activity of different enzymes since it is one of the organelles with the major availability of oxygen. ROS can act as signaling molecules in a number of different pathways by modulating calcium (Ca2+) signaling. Interactions among ROS and calcium signaling can be considered bidirectional, with ROS regulating cellular Ca2+ signaling, whereas Ca2+ signaling is essential for ROS production. In particular, we will discuss how alterations in the crosstalk between ROS and Ca2+ can lead to mitochondrial bioenergetics dysfunctions and the consequent damage to tissues at high energy demand, such as the heart. Changes in Ca2+ can induce mitochondrial alterations associated with reduced ATP production and increased production of ROS. These changes in Ca2+ levels and ROS generation completely paralyze cardiac contractility. Thus, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells. These interplays in the cardiovascular system are the focus of this review.
1. Introduction
Mitochondria are crucial to maintaining the regulation and performance of different important cellular activities, in particular, with their critical role in energy production for the smooth functioning of energy-demanding organs such as muscles, the brain, and the heart [1]. As a consequence, any dysregulation or alteration in mitochondrial bioenergetics primarily perturbs these organs. Within several cell types and particularly in cardiac myocytes, mitochondria are the major site of reactive oxygen species (ROS) production through the activity of different enzymes (including NADPH oxidase and uncoupled nitric oxide synthase) [2] since they are organelles with great oxygen availability [3].
ROS can act as signaling molecules in a number of different pathways by modulating calcium (Ca2+) signaling. Intracellular calcium (Ca2+) is the most common second messengers of living cells. This versatility allows it to control diverse processes mediated by rapid Ca2+ fluxes, such as contractility, secretion, proliferation, apoptosis, protein folding, and energy metabolism [1,4]. Calcium and ROS mutually influence each other, ROS regulate cellular calcium signaling, whereas calcium signaling is crucial for ROS production [5]. These interplays have been studied in depth in the cardiovascular system and are the focus of this review. In particular, we will discuss how alterations in the crosstalk between ROS and Ca2+ can lead to mitochondrial bioenergetics dysfunctions, and the consequent damages for tissues at high energy demand such as heart.
In the heart, maintenance of cellular homeostasis is ensured by a low basal concentration of ROS, as they regulate multiple signaling pathways and physiological processes such as differentiation, proliferation, and excitation–contraction (E-C) coupling [6].
In cardiac cells, mitochondria and the sarcoplasmic reticulum (SR) are closely interconnected, and Ca2+ is crucial for optimal function of these two organelles [7]. The controlled release of Ca2+ from the SR is necessary for excitation–contraction (E–C) coupling [8]. Mitochondrial dysfunctions are associated with alterations in mitochondrial Ca2+ levels [9] and a dysregulated calcium handling is a hallmark in cardiac dysfunction. Ca2+ and, ADP, together with the redox state of pyridine nucleotides, actively regulate ROS production in cardiac mitochondria [10]. In fact, a reduction in Ca2+ uptake in these organelles and an increased energy demand at the cardiac level induce the oxidation of NADH and NADPH. Thus, the drastically impaired redox potential of the matrix results in increased H2O2 release [11]. In this context, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells [12,13]. Finally, in this review we will discuss genes that, when altered, cause cardiomyopathies with mitochondrial dysfunctions.
2. ROS
Redox (reduction–oxidation) homeostasis is the dynamic equilibrium of electron transfer reactions, and is related to the concept of free radicals, fundamental to redox signaling and biological function [14]. Free radicals can be oxidants and they are unstable molecular entities with an unpaired electron on the outer layer [14]. As a result of this unpaired electron the free radical undergoes electron transfer reactions, being a reductase when it donates the electron, or an oxidase when it takes an electron from another molecule. The most abundant form of free radicals in the cell originates from diatomic oxygen (O2) [15], and they are known as ‘reactive oxygen species’ (ROS).
The term “ROS” does not refer to a specific species, however it is a wide-range term and comprehends different oxygen species with different reactivities and half-lives. The term ROS might refer to both free radicals such as superoxide anion (O2•−) and hydroxyl radical (•OH), and non-radical oxidants, such as hydrogen peroxide (H2O2) [16]. Since these molecules are highly reactive, they can react with lipids, proteins, DNA and even other ROS [17].
It is worth noting that the sites of ROS production and Ca2+ storage coincide in the cells, at the interface between the plasma membrane and the endoplasmic reticulum (ER) and between this last one and mitochondria.
The major redox signaling agents are the superoxide anion radical (O2•−) and the hydrogen peroxide (H2O2). Superoxide is produced mainly through the electron transfer chains (ETC) in mitochondria, but also in other organelles such as the ER and the plasma membrane, by NADPH oxidases (NOX), that catalyze the transfer of an electron from NADPH to O2 [18]. In mitochondria, the superoxide radicals (O2•−) are generated by electrons that escape from complex I and III and that reduce O2 [19]. When O2•− reacts with another superoxide radical, generates the hydrogen peroxide (H2O2), which can be reduced to water or partially reduced to hydroxyl radical (•OH), reactions catalyzed by the enzyme superoxide dismutase [20].
Together with O2•−, H2O2 is generated in healthy cells at a controlled steady-state level [21] and is physiologically produced by NOXs in the plasma membranes, mitochondria, endoplasmic reticulum, peroxisomes in response to external stimuli [14]. H2O2 is not a hazardous molecule itself, however it can undergo Fenton reactions through which there is the generation of hydroxyl radical (HO2), consequent to the reaction of H2O2 with the reduced metal ions (Fe2+ or Cu+). HO2 is the most aggressive form of ROS and is the initiator of lipid peroxidation, in fact, can diffuse in lipids, and can produce a carbon-centered radical of polyunsaturated lipids [22].
Like calcium (Ca2+) [23], H2O2 is also a crucial redox signaling agent [14] and the pivotal molecule in homeostatic metabolism, according to the third principle of the Redox Code [24]. In physiological conditions, H2O2 can oxidize target proteins through reversible reactions—as occur in the reversible oxidation of specific cysteine residues of proteins [25,26,27]—regulating protein activity, localization, and interactions, contributing to organizing cellular processes such as cell proliferation, differentiation, and autophagy [20,28,29].
The Alpha and Omega of Mitochondrial ROS
Within the cell, the mitochondria are the major site of ROS production, through the activity of different enzymes (complex I and III; oxoglutarate dehydrogenase (OGDH); pyruvate dehydrogenase (PDH); complex II (site IIF) [30]), however, the ETC is the site with the highest production of ROS [31], in particular complexes I and III of the respiratory chain [20,32,33]. In particular, complex I and to a lesser extent complex II release O2•−/H2O2 toward the mitochondrial matrix, whereas release from complex III is toward the cristae lumen and the intermembrane space [34,35]. This single electron might reduce oxygen and generate superoxide anion (O2•−), which is then converted to H2O2−. H2O2 is generated in the mitochondrial matrix by the action of SOD2 (manganese superoxide dismutase) matrix and in the intermembrane space by SOD1 (Cu, Zn-superoxide dismutase) [3,35,36]. The generated H2O2 is highly permeable and can be reduced by peroxidases such as glutathione peroxidases (GPx), peroxiredoxins 3 and 5 [37], and catalase (CAT) [38].
Independently of ETC, other mitochondrial enzymes are responsible for ROS production. ROS can be produced in the outer mitochondrial membrane by enzymes such as monoamine oxidase (MAO) and cytochrome b5 reductase (Cb5R) [39], and in the mitochondrial matrix by enzymes of the Krebs cycle such as pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (αKGDH), which produce both superoxide and hydrogen peroxide [40,41], and in the inner mitochondrial membrane by glycerol-3-phosphate dehydrogenase and various cytochrome P450 monooxygenases [39]. Interestingly, the activity of the latter enzymes in ROS production is dependent on mitochondrial membrane potential (ΔΨ) [42,43]. Mitochondria produce more ROS at high membrane potential [44]. The closure of the mitochondrial permeability transition pore, the inhibition of complex I and III with rotenone and antimycin A, respectively, and the inhibition of ATP synthase, can all lead to an increase ΔΨ and to an increased ROS production [45,46,47]. However, in pathological conditions, an opposite situation might be also observed, for example, in a neuronal cell model carrying a loss of function SPART variant (c.892dupA), cause of Troyer syndrome where a reduced ΔΨ and decreased respiratory activity with a concurrent increase in ROS production have been reported [48].
3. Calcium
The crucial role of mitochondria for normal cell physiology is evident in the natural history of various disorders. Indeed, the presence of calcium in mitochondria acts as a double-edged sword: whereas cellular homeostasis is maintained by optimal Ca2+ levels, an excess of this ion is reportedly found in many diseases, including neurodegenerative and muscular diseases, such as Huntington’s disease (HD) [49] and Alzheimer’s disease (AD) [50].
Mitochondrial calcium influx is driven by differences in electric changes across the inner mitochondrial membrane and resulting from the proton pumping of the respiratory chain.
The diffusion of Ca2+ within the cell is tightly controlled by the elaborate mechanism of cytosolic Ca2+ chelation. Under basal conditions, cytosolic calcium concentrations are maintained low and controlled (100 nM) by continuous extrusion to the extracellular environment or uptake by intracellular stores, thus creating a gradient of rapidly increasing Ca2+ upon opening of ion pumps and specialized channels [51]. This finely regulated balance allow the genesis of localized Ca2+ signals that coordinate the function of target proteins/organs with great spatio-temporal precision [4].
3.1. Influx and Efflux of Ca2+ in Mitochondria
A variety of targets and Ca2+ transport systems are present on the mitochondrial membrane that modulate several mitochondrial functions. The endoplasmic reticulum (ER) represents the primary intracellular Ca2+ store and the release of Ca2+ occurs through the inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs), located in its membranes.
The close proximity and juxtaposition of the ER to mitochondria grants a direct and selective transmission of physiological and pathological Ca2+ signals [52]). The membrane contact sites between the endoplasmic reticulum and mitochondria are called mitochondria-associated membranes (MAMs) [53].
The Ca2+ transfer between ER and mitochondria through the MAMs depends on a tripartite protein complex that includes IP3R, localized on the ER membrane, voltage-dependent anion channel 1 (VDAC1) residing on the outer mitochondrial membrane (OMM), and the cytosolic glucose-regulated chaperone protein 75 (GRP75) that forms a tether between the two organelles [54].
When the cytosolic Ca2+ level is high, the cation is passively transported through the OMM. The presence and function of VDAC1, which enables the transport of all energy metabolites (pyruvate, malate, succinate, NADH, ATP, ADP, and phosphate) from the cytosol to the mitochondria, provides high membrane permeability.
In contrast, the transit of Ca2+ across the inner mitochondrial membrane (IMM) is driven by the negative membrane potential and the MCU channel protein [55].
The key components of the MCU channel protein complex include MCU, EMRE, MICU1, and MICU2 (MEMMS) [56]. MCU is the main protein of the holo-complex responsible for the transfer of Ca2+ into the mitochondrial matrix. The transmembrane domain of each of the four subunits of MCU (TMD) forms a tetrameric conformation and shapes a pore in the inner mitochondrial membrane effective for Ca2+ transfer [57,58,59]. In fact, the TMD contains a ‘DIME’ motif, with conserved amino acids required for MCU-mediated Ca2+ uptake: the two residues of aspartic acid and glutamic acid form two parallel side-chain carboxylate rings that act as Ca2+ selectivity filter [55,60].
MICU1 forms a large interaction surface area with MCU to seal the intermembrane pore space entrance, while MICU2 binds to MICU1 from the side without contacting MCU [61]. According to literature data, it has been found that MICU1 and MICU2 form a plug to occlude the MCU channel under conditions of low Ca2+ concentrations. In the presence of high Ca2+ concentrations, these two regulators undergo conformational changes through their EF-handed motif, which result in pore opening and Ca2+ permeation into the mitochondria [60]. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex [60]. EMRE interacts with MICU1 in the intermembrane space and MCU oligomers in the inner membrane. Thus, EMRE appears to operate as a bridge between the channel properties of MCU and Ca2+ sensing activity of MICU1/MICU2 [59].
In addition to the Ca2+ uptake mechanism, mitochondria present Ca2+ release systems, mediated by Na+/Ca2+ (NCLX, Na+/Ca2+/Li+ exchanger) and H+/Ca2+ (mHCX, mitochondrial H+/Ca2+ exchanger) exchangers, which export Ca2+ outside the mitochondria. In this way, the organelle limits the accumulation of Ca2+ within the matrix and regulates its homeostasis [62].
3.2. Key Ca2+ Targets and Roles in the Regulation of Mitochondrial Bioenergetics
The controlled uptake of Ca2+ in mitochondria regulates the rate of energy production and metabolism, shapes the amplitude and spatio-temporal patterns of intracellular Ca2+ signals, and is crucial for programmed cell death [5]. Calcium in mitochondria is critical for the regulation of four dehydrogenases (glycerol phosphate, pyruvate, α-ketoglutarate, and isocitrate dehydrogenase), F0-F1 ATP synthase and two isoforms of the mitochondrial aspartate/glutamate transporter, aralar1 and citrin [56,63]. Of these protein complexes, the two transporters and the glycerol phosphate dehydrogenase have Ca2+-binding domains facing the intermembrane space and are affected by changes in the cytoplasmic concentration of calcium ions [56].
In addition, when Ca2+ activates the complex F1-F0-ATP synthase, by replacing its natural cofactor (Mg2+), the increased steric bulk within the catalytic sites of F1 triggers conformational changes that reverse the function of the complex, and thus ATP synthase hydrolyzes ATP [64].
The other three dehydrogenases are rate-limiting enzymes in feeding electrons at complex I of the ETC [65]. In vertebrates, the mechanisms of activation of these enzymes are all dependent on the accumulation of Ca2+ in the mitochondrial matrix [66]. Pyruvate dehydrogenase (PDH) depends on de-phosphorylation of the catalytic subunit by a Ca2+-dependent phosphatase [67], while α-ketoglutarate and isocitrate dehydrogenases are activated directly by Ca2+ binding [68]. As a result of high mitochondrial Ca2+ levels, PDH, α-ketoglutarate, and isocitrate dehydrogenases are activated and stimulate the synthesis of ATP by the mitochondria.
These enzymes are very responsive to changes in Ca2+ in the matrix, but the increase in this ion is not the only mechanism that induces their activation; in particular, PDH is also regulated by other allosteric modulators such as pyruvate, ATP, NADH, and matrix pH [66].
Recently, Foskett and collaborators proposed a new regulatory mechanism for cellular bioenergetics, showing that a constitutive reduced Ca2+ release through IP3R is crucial for the maintenance of optimal cellular bioenergetics under normal basal conditions because it provides sufficient reducing equivalents to support oxidative phosphorylation [69,70]. In fact, inhibition of IP3R-dependent Ca2+ release and, consequently, of mitochondrial Ca2+ uptake, causes an overall impairment of cellular bioenergetics. If Ca2+ transfer to the mitochondria is absent, an increase in pyruvate dehydrogenase phosphorylation is observed, resulting in its inactivation and decreased TCA (tricarboxylic acid) cycle activity. The slowdown of the TCA cycle reduces NADH and FADH2 production, affecting the activity of the ETC and causing less ATP production. This reduction is detected by the cellular energy sensor AMPK, which, in the presence of a higher AMP/ATP ratio, determines the activation of autophagic processes [70].
Similarly, Filadi et al. described for the first time a new role for TOM70 in modulating ER–mitochondria communication and cellular bioenergetics in mammalian cells [71]. TOM70 is a subunit of the translocase of the outer membrane (TOM) complex and, with the translocase of the inner membrane (TIM), is responsible for the post-translational import of mitochondrial proteins encoded by the nucleus. TOM70 forms clusters along the OMM frequently associated with ER–mitochondria contact sites. Here, it interacts with IP3R isoform 3 and GRP75 (chaperone 75 kDa glucose-regulated protein), stabilizing the functional IP3R-3/GRP75/VDAC1 complex and promoting Ca2+ shuttling. This, in turn, promotes and sustains the Krebs cycle and mitochondrial respiration. In fact, the downregulation of TOM70 reduces Ca2+ uptake and alters mitochondrial function by reducing ETC activity and ATP synthesis, thereby activating autophagy [71,72].
Regardless of its link to the juxtaposition of ER and mitochondria, the mitochondrial Ca2+ uptake process itself plays a crucial role. In 2012, Mallilankaraman and coauthors identified a regulator of the MCU complex (MCUR1), an integral membrane protein required for mitochondrial Ca2+ uptake, which was found to be important for maintaining a normal cell metabolism. Knockdown of MCUR1 did not alter MCU localization, but prevented Ca2+ uptake by mitochondria. Ablation of MCUR1 also disrupted oxidative phosphorylation, reduced cellular ATP production and oxygen consumption, and finally activated AMP kinase [73].
4. The Interplay between Ca2+ and ROS
The capacity of mitochondria to accumulate Ca2+ is critical for maintaining proper tissue homeostasis. However, Ca2+ accumulation in mitochondria leads to decreased ATP production and prolonged opening of the mPTP (permeability transition pore), a high-conductance channel, whose opening allows the release of proapoptotic mitochondrial components [74].
mPTP opening depends not only on Ca2+ concentration, but also on other factors including high phosphate concentrations, low adenine nucleotide concentrations and oxidative stress. In fact, Madesh and Hajnoczky showed that O2•− is able to induce mPTP opening in a Ca2+-dependent manner [75]. The opening of the channel triggers the mitochondrial permeability transition (mPT), which is characterized by a drastic increase in mitochondrial membrane permeability, causing the entry of any molecule with a weight less than 1.5 kDa. This event in turn causes the immediate collapse of the mitochondrial membrane potential (ΔΨ m), membrane depolarization, and ATP depletion. The initial uncoupling effect is followed by the reduction in respiratory activity caused by the loss of pyridine nucleotides and cytochrome c [76]. Swelling of the mitochondrial matrix causes disruption of the outer membrane. Subsequent inhibition of electron flow could explain the increase in ROS formation generated by PTP opening; since the last event is promoted by ROS, a vicious cycle of damage amplification is triggered (Figure 1) [77].
In addition, the continuous release of ROS from mitochondria allows mitochondrial ROS peaks to be maintained during apoptosis. This mechanism may be necessary for signaling to adjacent mitochondria, resulting in global activation of cell death by apoptosis [78].
Proteins, lipids, and nucleic acids can be altered by the accumulation of ROS in mitochondria, which result in covalent modifications and profoundly alter their structure and function [77].
One of the most susceptible targets is cardiolipin, a highly abundant phospholipid in the inner mitochondrial membrane. It has been proposed that oxidation of cardiolipin contributes to complex I impairment [79] and cytochrome c release [80]. Oxidative alterations of mitochondrial lipids and proteins can result in true dysfunction due to alterations in mitochondrial DNA (mtDNA) in the long term. Among the DNA products generated by ROS attack, 8-oxo-deoxyguanosine is the most prevalent [81].
Ben-Kasus Nissim and coworkers have shown that NCLX knockdown increases the mitochondrial Ca2+ levels and leads to an stimulates l ROS production in mitochondria [82]. Furthermore, the consequences of specific redox alterations of different isoforms of VDAC are only beginning to be investigated, and it is unclear whether these play a role in VDAC function or might have a role in the pathophysiology of disorders [83].
5. Cardiac Muscle, ROS, and Ca2+ Signaling
The cardiomyocyte is a cell type that requires high amounts of energy to function efficiently. For these cells, ATP production by oxidative phosphorylation is essential, especially during contraction, after the increase in beating frequency/speed, and for all energy-intensive processes. About one-third of the volume of cardiomyocytes is occupied by mitochondria, which play a crucial role in the efficient coupling between energy production and cell requests [72].
During cardiomyocyte development, the sarcoplasmic reticulum (SR) and mitochondria undergo profound changes in coupling, shape, and distribution. In myoblasts (poorly differentiated muscle cells) mitochondria are few and elongated, much like a reticulum, while in myotubes (differentiated muscle fibers) they appear more spotted and globular. In addition, in myoblasts, mitochondria are coupled to Ca2+ release from the ER; in contrast, in myotubes, Ca2+ release is driven by SR. in this context, SR is crucial in the regulation of cytoplasmic calcium dynamics and cellular activity.
During differentiation, SR progressively change its shape as a beehive with tubular structure [84]. Ca2+ transfer between SR and mitochondria is particularly important in cardiac muscle, where ATP demand is high due to the energy required for excitation–contraction coupling (ECC). Ca2+ is uploaded into the mitochondria to invigorate metabolism, generate the ATP needed for contraction, and mediate Ca2+ elimination from the cytosol in the relaxation phase [85].
During ECC, the increase in intracytoplasmic calcium required for contraction induction is generated by Ca2+-dependent activation of ryanodine receptors (RyRs). These are Ca2+ release channels located on the SR, which is the main intracellular storage of this ion. In cardiomyocytes both RyRs and inositol 1,4,5-trisphosphate receptors (IP3Rs) are present. In myoblast mitochondria, Ca2+ release from the ER is mediated by IP3R. During cell differentiation, the expression of RyRs (particularly isoform 2) increases dramatically in cardiac muscle, and this is critical for excitation–contraction coupling. Simultaneously, IP3Rs play different roles, such as regulating gene transcription and hypertrophy [84,86], although the physiological contribution of IP3R-mediated Ca2+ release is not completely elucidated [85].
As it is the case in other cell types, also in cardiomyocytes the transfer of Ca2+ from SR to mitochondria involving IP3Rs in MAMs is mediated by the VDAC1-GRP75 (a mammalian heat shock stress protein 70). In this context, IP3R1 is called up, binds VDAC1 through GRP75 that tethers the two proteins by binding to their regions exposed in the cytosol and therefore forming a channel complex through which there is Ca2+ transfer between SR and mitochondria, [87].
5.1. Calcium and ROS in Heart Failure
Mitochondria and SR are interrelated and connected, and Ca2+ is crucial for the functioning of both organelles. Mitochondrial dysfunction is associated with alterations in Ca2+ levels [87,88,89,90,91,92,93,94,95].
Dysregulated calcium handling is a hallmark in cardiac dysfunction. In particular, changes in Ca2+ can induce mitochondrial alterations with reduced ATP production and increased production of ROS. Thus, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells [96,97,98,99,100,101]. Different studies reported a correlation between an increased level of ROS (in plasma and heart) and the gravity of left ventricular dysfunction [102,103,104].
Heart failure induces the activation of a number of mechanisms that result in a reduction in mitochondrial calcium transfer. Specifically, there is an increase Ca2+ influx in through the NCLX due to increased Na+ in the cytosol, reduction in MCU opening, SR Ca2+-ATPase activity, and ryanodine receptor expression. The final combined effect of these events is the negative alteration of the Krebs cycle [105,106,107,108,109,110]. As a result, the accumulation of oxidized pyridine nucleotides prevents the production of ATP from NADH and the detoxification of ROS by NADPH. The antioxidative enzyme responsible for the regeneration of NADPH from NADH is the nicotinamide nucleotide transhydrogenase (NNT) [111]: under pathological conditions, when the metabolic demand increases, the direction of the NNT reaction is reversed, oxidizing NADPH to regenerate NADH and produce ATP, and interfering with the NADPH-dependent antioxidative capacity [10]. Because mitochondria represent the main ROS scavenging system of the cell [103], the oxidation of NADPH by NNT could cause excessive mitochondrial ROS release, that leads to necrosis, left ventricular dysfunction, and death [112].
5.2. Mitochondrial ROS vs. ER ROS
Mitochondria have been considered the primary source of ROS [7,113,114,115,116,117,118], but several lines of evidence propose an important role also for ROS generated in the ER in cardiovascular diseases.
MAMs are the contact sites between the endoplasmic reticulum or sarcoplasmic reticulum. Our knowledge of calcium signaling in cardiac pathologies, where ER and oxidative stresses are predominant [5,119,120,121], suggests that calcium may, in fact, be the cause, rather than the effect, of altered mitochondrial ROS. This implies that calcium overload signals mitochondria to produce lethal levels of ROS.
The ER is a primary site of protein synthesis and post-translational processing, and the protein folding process is influenced by the redox state of the ER. A well-known condition of the ER, called ER stress, due to the accumulation of misfolded polypeptides, is caused by the accumulation of oxidant equivalents in the ER. ER stress activates the unfolded protein response (UPR), which raises the folding protein capacity, resulting in an increased production of oxidative equivalents and an additional deterioration of the redox state [122]. During ER stress calcium channels, both ryanodine and inositol-3-phosphate receptors open [123] with the release of calcium, which is crucial for the contraction of the muscles. To sustain Ca2+ homeostasis, Ca2+ returns to the SR/ER through SERCA (sarco-endoplasmic reticulum calcium ATPase). In pathological conditions, Ca2+ homeostasis in the ER is dysregulated, with an enhanced Ca2+ release from mitochondria [124,125]. Ca2+ within the mitochondria generates superoxide, a marker for oxidative stress [126]. Cardiac contractility is entirely paralyzed by Ca2+ leakage, Ca2+ overload, and ROS generation.
5.3. In Vivo Mouse Model of Postmyocardial Infarction
Type 2 ryanodine receptor (RyR2) and type 2 inositol 1,4,5-trisphosphate receptor (IP3R2) [127,128,129] are the two intracellular Ca2+ channels in SR of cardiac cells, and the RyR2 receptor is crucial for cardiac excitation–contraction (E–C) coupling in cardiomyocytes.
In several murine models of postmyocardial infarction that have been generated so far, it has been shown that, based on the physical and functional association between the SR and mitochondria, when the SR calcium was released in cardiomyocytes by the two main channels, it accumulated in mitochondria, bringing to mitochondrial dysfunction, oxidative stress and decreased ATP production [88,130,131].
To investigate whether Ca2+ accumulation in mitochondria of failing hearts was caused by Ca2+ leak through the RyR2 receptor, Santulli et al. generated a murine model carrying a homozygous RyR2 mutation that renders the channel leaky (RyR2S2808D/S2808D) and a mouse model with a homozygous RyR2 mutation that renders the channel protected against leak (RyR2S2808A/S2808A) [129]. In cardiomyocytes derived from the RyR2S2808D/S2808D mice, there was an increased mitochondrial Ca2+ and ROS production, the presence of dysmorphic and malfunctioning mitochondria, a reduction in mitochondrial size, and low fusion-to-fission ratio compared with wild-type (WT) and RyR2S2808A/S2808A cardiomyocytes [129].
In opposition, cardiac-specific deletion of the IP3R2 receptor did not produce consequences on mitochondrial Ca2+ accumulation, as observed in the murine model (IP3R2CVKO), in which IP3R2 expression was suppressed in ventricular cardiomyocytes. IP3R2CVKO mice survived to adulthood without showing alteration in mitochondrial function. Ca2+ sparks, SR Ca2+ load, mitochondrial Ca2+ level, and ROS production were not affected in IP3R2CVKO ventricular cardiomyocytes. Moreover, IP3R2CVKO mice exhibited normal myocardial mitochondria and no significant effect after postmyocardial infarction [129].
5.4. Drug Targeting of Mitochondrial Ca2+ and Homeostasis
Based on the observed experimental data, the mitochondrial redox state might be a plausible drug target in cardiac alterations. Two distinct approaches have been used to modulate ROS and Ca2+ in mitochondria: (i) direct targeting of mitochondrial ROS by agents that accumulate in mitochondria; (ii) normalization of mitochondrial Ca2+ signaling to guarantee a balance between mitochondrial ROS emission and detoxification.
For the direct targeting of mitochondrial ROS, drugs that have been developed to accumulate in mitochondria or scavenge ROS (MitoQ) or directly act at the ETC level to reduce ROS production (SS-31). As an example, CGP-37157 is a benzodiazepine compound selectively inhibiting NCLX, the mitochondrial Na+/Ca2+ exchanger by which mitochondrial Ca2+ is extruded. CGP-37157 suppresses mitochondrial Ca2+ efflux, abolishes NADH oxidation, and reduces ROS production in failing cardiomyocytes [110]. Overall, experimental studies performed with CGP-37157 showed the positive effects of mitochondrial NCLX inhibition in animal models of heart failure and in isolated heart cells. However, currently, there are no clinical trials to test the effect of these drugs.
A different strategy to normalize mitochondrial Ca2+ signaling is to reduce the concentration of intracellular Na+. In heart failure, [Na+]i is increased [132], and during excitation–contraction coupling, it hinders mitochondrial Ca2+ accumulation since it increases mitochondrial Ca2+ efflux via the NCLX [102]. It has been shown that an elevated late Na+ current (late INa) is required to increase cytosolic Na+. An enhanced late INa has been related to elevated mitochondrial ROS emission. This, in turn, worsens late INa through Ca2+-/calmodulin-dependent kinase II (CaMKII) oxidation. CaMKII [133,134] can enhance late Na+ current and cause arrhythmias during HF [135].
Ranolazine is a piperazine derivative inhibitor of late Na+ current [136] that was approved by the FDA in 2006 for chronic angina and cardiac ventricular dysrhythmias [137]. MERLIN-TIMI 36 trials provided the hypothesis that ranolazine may be particularly beneficial in patients with HF [138]. In this trial, patients with acute coronary syndrome profited from ranolazine, particularly when they had elevated levels of brain natriuretic peptide as a marker of HF [138]. Evidence suggested that the mechanisms of action could be the blocking of the late Na+ current that occurs during ischemia, the blocking of mitochondrial complex I activity, or by modulating mitochondrial metabolism [139]. In mitochondria isolated from the hearts of patients treated with ranolazine, there was decreased cytochrome c release and mild resistance to the opening of mPTP when compared with control hearts. Through its late Na+ current blocking action, ranolazine protected the heart during IR injury by decreasing the load of cytosolic (c)Ca2+ and mitochondrial (m)Ca2+. The final effect was a reduction in necrosis and apoptosis [139].
Recently, empagliflozin has demonstrated its positive effects in regulating cytosolic Na+ and mitochondrial Ca2+. It selectively inhibits the sodium glucose cotransporter 2 channel (SGLT-2) [140], which reabsorbs the filtered glucose in the renal early proximal tubule. Its main role is to reabsorb the majority of filtered glucose, decreasing renal glucose reabsorption and, thus, hyperglycemia. A clinical trial in 7020 patients, the EMPA-REG OUTCOME study, showed that patients with type 2 diabetes at high cardiovascular risk and treated with empagliflozin showed ameliorated cardiovascular endpoints and reduced death from HF when the study drug was added to standard care [141,142]. Empagliflozin might reduce [Na+]i in cardiac myocytes independently of SGLT inhibition, possibly by interacting with the Na+/H+ exchanger [143]. Thereby, this drug empowered mitochondrial Ca2+ in cardiac myocytes.
Antioxidants such as vitamins E and C have been used in animal experiments, but promising results were observed in preclinical studies only [144]. In subsequent clinical trials, none of these compounds led to any significant benefit. Despite evidence that mitochondria are crucial for oxidative damage, compartmentalization and the interaction between compartments probably explain why “generic” antioxidants, such as vitamins, failed to target dysfunctional mitochondria and altered Ca2+ levels.
A more targeted and specific approach has been focused on NOXs, which have the biological role of producing ROS for signaling purposes and transferring electrons across biological membranes to produce O2− [145]. Along with the already well-known lipid-lowering effect of statins in preventing cardiovascular disease, their antioxidant effect through NOX2 inhibition has been demonstrated. Statin treatment provided positive antioxidant effects in vitro in isolated cardiomyocytes and in animal models of HF [141,142]. However, in the CORONA study, comprising 5011 HF patients, statin therapy did not show to reduce cardiovascular death [146].
In January 2021, the FDA approved vericiguat, a new soluble oral guanylate cyclase stimulator, which enhances the production of cyclic guanosine monophosphate, for the treatment of chronic HF. It is well known that the cardioprotective pathway of soluble nitric oxide guanylate cyclase-cyclic guanosine monophosphate is compromised in patients with HF [147].
Based on data from the SOCRATES-REDUCED and VICTORIA trials in adult patients with chronic HF, this drug was approved for medical treatment. In the VICTORIA trial, the dose of vericiguat was initially 2.5 mg/day and increased after 2 weeks to 5 mg/day and then to 10 mg/day. Placebo doses were administered in the same manner. After about 1 year, 90% of patients were treated with the 10 mg target dose. The median follow-up for the primary endpoint was 11 months. The annualized absolute risk reduction with the drug treatment was ~4.2% over the course of the study [148]. The recommended initial oral dose of vericiguat was 2.5 mg/day, with food intake, and should be doubled approximately every 2 weeks to reach the target maintenance dose of 10 mg/day. Based on the VICTORIA trial, vericiguat significantly reduced the rate of hospitalization and cardiovascular death attributed to HF. In fact, regardless of patients’ atrial fibrillation status, vericiguat was better in preventing all causes of death, cardiovascular death, hospitalization, and HF.
The main advantage of this drug is that it avoids the risk of electrolyte imbalance or renal damage [149]. Moreover, vericiguat bypasses the many problems of current therapies, such as the gradual decline in effectiveness, drug dose-dependent tolerance, and off-target effects due to a lack of specificity, thus representing a game changer in the treatment of HF, because it has great potential to reduce the severity of the disease [150].
5.5. Cardiomyopathy with Mitochondrial Dysfunction-Associated Genes
Several signaling pathways, ion homeostasis, and metabolism are impaired in cardiomyopathies. Therefore, mitochondria, which are the cellular powerhouse, are involved in many of these processes. The known genes that are altered in cardiomyopathies with mitochondrial dysfunctions are reported in Table 1.
5.5.1. MTO1
MTO1 encodes for a mitochondrial protein involved in tRNA modification and protein synthesis and catalyzes the 5-carboxymethyl aminomethylation of the uridine base in mitochondrial tRNAs that transport Gln, Glu, and Lys. Mutations in this gene cause an autosomal recessive disorder known as combined oxidative phosphorylation deficiency 10 (COXPD10), characterized by altered OXPHOS activity. Alterations in mitochondrial oxidative respiration cause hypertrophic cardiomyopathy and lactic acidosis in early infancy and complications that can be fatal in severe cases [151].
5.5.2. AGK
The human AGK gene encodes for the mitochondrial acylglycerol kinase, an enzyme located on the mitochondrial membrane involved in the formation of phosphatidic and lysophosphatidic acids. Homozygous or compound heterozygous mutations in the AGK gene have been associated with Sengers syndrome, also known as cardiomyopathic mitochondrial DNA (mtDNA) depletion syndrome-10 (MTDPS10). This syndrome is characterized by skeletal myopathy, hypertrophic cardiomyopathy, exercise intolerance, lactic acidosis, and congenital cataracts. Skeletal muscle biopsies showed severe mtDNA depletion [152], and cardiomyopathy is the major cause of early death [157].
5.5.3. SLC25A4
SLC25A4 gene encodes for the Solute Carrier Family 25 Member 4, expressed in mitochondria. The function of this protein is to translocate ADP from the cytoplasm into the mitochondrial matrix and ATP in the opposite direction. Homozygous or compound heterozygous mutations in this gene cause mitochondrial DNA depletion syndrome 12B (MTDPS12B), an autosomal recessive mitochondrial disease. Onset occurs in infancy with progressive hypertrophic cardio- and skeletal myopathy. The presence of red and irregular fibers, accumulation of abnormal mitochondria, and mtDNA depletion can be observed in skeletal muscle biopsies from the affected individuals [153].
5.5.4. MT-TL1
MT-TL1 is a mitochondrial gene that encodes the t-RNA-Leu. Mutations in MT-TL1 can result in multiple mitochondrial deficiencies and associated disorders. Among them, variants in MT-TL1 cause hypertrophic cardiomyopathy with renal abnormalities due to altered oxidative phosphorylation. This syndrome is characterized by hypertrophic and dilated cardiomyopathy, myopathy with hypotonia with developmental delay, and/or regression with cerebral atrophy and chronic renal [154].
5.5.5. MT-TK
Another mitochondrial RNA gene is MT-TK, affiliated with the tRNA class, which transfers the amino acid lysine during translation. Defects in this gene are associated with mtDNA-related cardiomyopathy and hearing loss. The clinical manifestation of this rare mitochondrial disease is characterized by progressive sensorineural hearing loss along with hypertrophic cardiomyopathy and encephalomyopathy. Other symptoms might be present such as progressive external ophthalmoparesis (PEO), ataxia, slowed speech, myalgia, and muscle weakness [155].
5.5.6. TAFAZZIN
The TAFAZZIN gene encodes a protein expressed at high levels in cardiac and skeletal muscle. Barth syndrome is caused by variants in the TAFAZZIN gene [87,88,89,90,91,92,93,94,95,154]. This syndrome is an X-linked disease with dilated cardiomyopathy (CMD) with endocardial fibroelastosis (EFE), a proximal skeletal myopathy, growth retardation, neutropenia, and organic aciduria [156]. Hypertrophic cardiomyopathy, ventricular arrhythmia, motor delay, and other cardiac and motor symptoms might be present.
6. Conclusions and Future Perspectives
In cardiac cells, ATP production by oxidative phosphorylation is essential, especially during contraction, after the increase in beating frequency/speed, and for all energy-intensive processes. As a consequence, dysregulation in mitochondrial bioenergetics severely affects their function. Changes in Ca2+ can induce mitochondrial alterations associated with reduced ATP production and increased production of ROS. In this context, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells [97,98,99,100,101]. The search for new methods to recover these damages has seen a great expansion in recent years through the development of functional biomaterials and biomimetic scaffolds in regenerative medicine, which finds fertile ground for the application of cardiac therapies, considering the low regenerative capacity of this tissue [158].
Many efforts have been made in order to develop functional cardiac tissue in vitro, especially with the development of knowledge in the field of pluripotent stem cells (PSCs). Human pluripotent stem cells (hPSCs) have the capacity to differentiate into different cell types. They can be derived from the cells within the blastocyst at the beginning of embryonic development, from which the various structures of the fetus originate [159], or they can be derived from human cells that are de-differentiated by expression of Yamanaka factors (Oct3/4, Sox2, Klf4, c-Myc), known as induced pluripotent stem cells (iPSCs) [160].
The use of iPSCs is an emerging application for the generation of disease models and the search for new therapies, making it possible not to use animal models and to overcome the ethical problems associated with human embryonic cells. The reprogramming of cells takes place in the context of the individual’s genetic background, and it has been shown how the genetic and epigenetic behaviors of iPSCs reflect those of the donor individual’s cells. Therefore, iPSCs have the potential to generate tissue-like cells or structures, such as cardiomyocytes, that are exactly those of the patient with the disease of interest, including the main inherited cardiomyopathies that we previously cited.
Branco et al. recently developed a protocol for the generation of self-organized human multilineage organoids that recreate the co-presence of several specialized cells [161]. Co-culturing these organoids together with cardiomyocytes, a functional cardiac organoid surrounded by myocardial-like tissue was generated, recapitulating the structure and function of mature cardiac cells [161].
Tenreiro et al. reported data from trials aimed at generating cardiomyocytes from cells derived from cardiomyopathy patients and studying their molecular mechanisms [162]. An important example of this approach is DMDstem (NCT03696628), a trial completed in 2021 and performed in children with genetic cardiomyopathy vs. healthy ones. However, the outcomes of this study are not yet reported in the literature. Another trial, IndivuHeart (NCT02417311), initiated in 2014 and completed in 2019, aims to ensure individualized early risk assessment for heart disease and to make engineered heart tissue technology (hiPSC-EHT) a clinically applicable strategy [163].
Advances in hiPSCs biology, genome-editing technologies, and cardiac tissue engineering all concur to generate healthy cardiac tissue from the patient himself, opening doors for targeted therapy and personalized medicine.
Author Contributions
Conceptualization, E.B. and C.D.; C.D., E.C.-S. and B.D.N. writing—original draft preparation, C.D. and E.B.; writing—review and editing, E.B.; funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Bologna RFO2022 grant to E.B. C.D. is a recipient of the Fondazione U. Veronesi fellowship 2022. B.D. is a recipient of the Ph.D. program FSE REACT-EU -A.Y. 2021/2022.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 623–634. ISBN 978-0-470-65071-4. [Google Scholar]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial Energetics and Calcium Coupling in the Heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef]
- Santana, L.F.; Cheng, H.; Gómez, A.M.; Cannell, M.B.; Lederer, W.J. Relation between the Sarcolemmal Ca2+ Current and Ca2+ Sparks and Local Control Theories for Cardiac Excitation-Contraction Coupling. Circ. Res. 1996, 78, 166–171. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Gauthier, L.D.; Greenstein, J.L.; Cortassa, S.; O’Rourke, B.; Winslow, R.L. A Computational Model of Reactive Oxygen Species and Redox Balance in Cardiac Mitochondria. Biophys. J. 2013, 105, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Köhler, A.C.; Sag, C.M.; Maier, L.S. Reactive Oxygen Species and Excitation–Contraction Coupling in the Context of Cardiac Pathology. J. Mol. Cell. Cardiol. 2014, 73, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Trebak, M. Crosstalk between Calcium and Reactive Oxygen Species Signaling in Cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxid. Basel Switz. 2021, 10, 890. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The Role of ROS in Tumour Development and Progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Sies, H.; Chance, B. The Steady State Level of Catalase Compound I in Isolated Hemoglobin-Free Perfused Rat Liver. FEBS Lett. 1970, 11, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Acc. Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Bak, D.W.; Weerapana, E. Cysteine-Mediated Redox Signalling in the Mitochondria. Mol. Biosyst. 2015, 11, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Fu, L.; Liu, K.; Tian, C.; Wu, Z.; Jung, Y.; Ferreira, R.B.; Carroll, K.S.; Blackwell, T.K.; Yang, J. Global Profiling of Distinct Cysteine Redox Forms Reveals Wide-Ranging Redox Regulation in C. Elegans. Nat. Commun. 2021, 12, 1415. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of Reactive Oxygen Species Generation by Mitochondria Oxidizing Different Substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Leoni, S.; Lenaz, G. Mitochondrial Production of Reactive Oxygen Species: Role of Complex I and Quinone Analogues. BioFactors 2008, 32, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.E.; Chan, S.I. Structures and Proton-Pumping Strategies of Mitochondrial Respiratory Enzymes. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 23–65. [Google Scholar] [CrossRef] [PubMed]
- Grgic, L.; Zwicker, K.; Kashani-Poor, N.; Kerscher, S.; Brandt, U. Functional Significance of Conserved Histidines and Arginines in the 49-KDa Subunit of Mitochondrial Complex I. J. Biol. Chem. 2004, 279, 21193–21199. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.-B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of Superoxide-Producing Sites in Isolated Brain Mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial Peroxiredoxin Involvement in Antioxidant Defence and Redox Signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef]
- Molavian, H.; Madani Tonekaboni, A.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and Its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, 13620. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial α-Ketoglutarate Dehydrogenase Complex Generates Reactive Oxygen Species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef]
- Finkel, T. Signal Transduction by Reactive Oxygen Species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between Mitochondrial Membrane Potential and ROS Formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. Energy Converting NADH:Quinone Oxidoreductase (Complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The Mechanism of Superoxide Production by NADH:Ubiquinone Oxidoreductase (Complex I) from Bovine Heart Mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential Effects of Mitochondrial Complex I Inhibitors on Production of Reactive Oxygen Species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Diquigiovanni, C.; Bergamini, C.; Diaz, R.; Liparulo, I.; Bianco, F.; Masin, L.; Baldassarro, V.A.; Rizzardi, N.; Tranchina, A.; Buscherini, F.; et al. A Novel Mutation in SPART Gene Causes a Severe Neurodevelopmental Delay Due to Mitochondrial Dysfunction with Complex I Impairments and Altered Pyruvate Metabolism. FASEB J. 2019, 33, 11284–11302. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s Disease. Semin. Neurol. 2007, 27, 143–150. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium Signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Gang, X.; He, G.; Liu, Y.; Wang, Y.; Zhao, X.; Wang, G. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. 2020, 11, 592129. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- Yoo, J. Structural Basis of Ca2+ Uptake by Mitochondrial Calcium Uniporter in Mitochondria: A Brief Review. BMB Rep. 2022, 55, 528–534. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of Intact Human MCU Supercomplex with the Auxiliary MICU Subunits. Protein Cell 2021, 12, 220–229. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. The Mitochondrial Calcium Uniporter Complex: Molecular Components, Structure and Physiopathological Implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.B.; Tsai, C.-W.; Tsai, M.-F. The Conserved Aspartate Ring of MCU Mediates MICU1 Binding and Regulation in the Mitochondrial Calcium Uniporter Complex. eLife 2019, 8, e41112. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and Mechanism of the Mitochondrial Ca2+ Uniporter Holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial Calcium Uptake in Organ Physiology: From Molecular Mechanism to Animal Models. Pflüg. Arch.—Eur. J. Physiol. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant Expression of the Ca(2+)-Sensitive Aspartate/Glutamate Carrier Increases Mitochondrial ATP Production in Agonist-Stimulated Chinese Hamster Ovary Cells. J. Biol. Chem. 2003, 278, 38686–38692. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S. Mitochondrial Permeability Transition, F1 FO-ATPase and Calcium: An Enigmatic Triangle. EMBO Rep. 2017, 18, 1265–1267. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of Calcium Ions in Regulation of Mammalian Intramitochondrial Metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of Mitochondrial Dehydrogenases by Calcium Ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by Calcium Ions of Pyruvate Dehydrogenase Phosphate Phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef]
- Rutter, G.A.; Denton, R.M. Regulation of NAD+-Linked Isocitrate Dehydrogenase and 2-Oxoglutarate Dehydrogenase by Ca2+ Ions within Toluene-Permeabilized Rat Heart Mitochondria. Interactions with Regulation by Adenine Nucleotides and NADH/NAD+ Ratios. Biochem. J. 1988, 252, 181–189. [Google Scholar] [CrossRef]
- Foskett, J.K.; Mak, D.-O.D. Regulation of IP3R Channel Gating by Ca2+ and Ca2+ Binding Proteins. Curr. Top. Membr. 2010, 66, 235–272. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr. Biol. CB 2018, 28, 369–382.e6. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, Mitochondria and Cell Metabolism: A Functional Triangle in Bioenergetics. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 Is an Essential Component of Mitochondrial Ca2+ Uptake That Regulates Cellular Metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. A CaPful of Mechanisms Regulating the Mitochondrial Permeability Transition. J. Mol. Cell. Cardiol. 2009, 46, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnóczky, G. VDAC-Dependent Permeabilization of the Outer Mitochondrial Membrane by Superoxide Induces Rapid and Massive Cytochrome c Release. J. Cell Biol. 2001, 155, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Lisa, F.D. The Mitochondrial Permeability Transition, Release of Cytochrome c and Cell Death: Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondrial Function and Myocardial Aging. A Critical Analysis of the Role of Permeability Transition. Cardiovasc. Res. 2005, 66, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef]
- Nomura, K.; Imai, H.; Koumura, T.; Kobayashi, T.; Nakagawa, Y. Mitochondrial Phospholipid Hydroperoxide Glutathione Peroxidase Inhibits the Release of Cytochrome c from Mitochondria by Suppressing the Peroxidation of Cardiolipin in Hypoglycaemia-Induced Apoptosis. Biochem. J. 2000, 351, 183–193. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative Damage and Mitochondrial Decay in Aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria Control Store-Operated Ca2+ Entry through Na+ and Redox Signals. EMBO J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-Dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Weaver, D.; Eisner, V.; Várnai, P.; Hunyady, L.; Ma, J.; Csordás, G.; Hajnóczky, G. Switch from ER-Mitochondrial to SR-Mitochondrial Calcium Coupling during Muscle Differentiation. Cell Calcium 2012, 52, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Demydenko, K.; Ekhteraei-Tousi, S.; Roderick, H.L. Inositol 1,4,5-Trisphosphate Receptors in Cardiomyocyte Physiology and Disease. Philos. Trans. R. Soc. B Biol. Sci. 2022, 377, 20210319. [Google Scholar] [CrossRef]
- Nakayama, H.; Bodi, I.; Maillet, M.; DeSantiago, J.; Domeier, T.L.; Mikoshiba, K.; Lorenz, J.N.; Blatter, L.A.; Bers, D.M.; Molkentin, J.D. The IP3 Receptor Regulates Cardiac Hypertrophy in Response to Select Stimuli. Circ. Res. 2010, 107, 659–666. [Google Scholar] [CrossRef]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes from Lethal Hypoxia-Reoxygenation Injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Nickel, A.; Löffler, J.; Maack, C. Myocardial Energetics in Heart Failure. Basic Res. Cardiol. 2013, 108, 358. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial Energy Metabolism in Heart Failure: A Question of Balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- O-Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic Signaling Regulates Mitochondrial Ca2+ Uptake Through Pyk2-Dependent Tyrosine Phosphorylation of the Mitochondrial Ca2+ Uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef]
- Holmström, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of Cardiac Function in Mice Lacking the Mitochondrial Calcium Uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D Controls Mitochondrial Pore-Dependent Ca(2+) Exchange, Metabolic Flexibility, and Propensity for Heart Failure in Mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef]
- Odagiri, K.; Katoh, H.; Kawashima, H.; Tanaka, T.; Ohtani, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Local Control of Mitochondrial Membrane Potential, Permeability Transition Pore and Reactive Oxygen Species by Calcium and Calmodulin in Rat Ventricular Myocytes. J. Mol. Cell. Cardiol. 2009, 46, 989–997. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Stanley, B.A.; Murray, C.I.; Sivakumaran, V.; Donzelli, S.; Mancardi, D.; Pagliaro, P.; Gao, W.D.; van Eyk, J.; Kass, D.A.; et al. Playing with Cardiac “Redox Switches”: The “HNO Way” to Modulate Cardiac Function. Antioxid. Redox Signal. 2011, 14, 1687–1698. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Balaska, D.; Cheng, W.H.Y. The Ups and Downs of Mitochondrial Calcium Signalling in the Heart. Biochim. Biophys. Acta 2010, 1797, 856–864. [Google Scholar] [CrossRef]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.S.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W.; et al. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef]
- Kim, T.Y.; Terentyeva, R.; Roder, K.H.F.; Li, W.; Liu, M.; Greener, I.; Hamilton, S.; Polina, I.; Murphy, K.R.; Clements, R.T.; et al. SK Channel Enhancers Attenuate Ca2+-Dependent Arrhythmia in Hypertrophic Hearts by Regulating Mito-ROS-Dependent Oxidation and Activity of RyR. Cardiovasc. Res. 2017, 113, 343–353. [Google Scholar] [CrossRef]
- Wagner, S.; Dantz, C.; Flebbe, H.; Azizian, A.; Sag, C.M.; Engels, S.; Möllencamp, J.; Dybkova, N.; Islam, T.; Shah, A.M.; et al. NADPH Oxidase 2 Mediates Angiotensin II-Dependent Cellular Arrhythmias via PKA and CaMKII. J. Mol. Cell. Cardiol. 2014, 75, 206–215. [Google Scholar] [CrossRef]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A Redox-Dependent Pathway for Regulating Class II HDACs and Cardiac Hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of Cyclophilin D Reveals a Critical Role for Mitochondrial Permeability Transition in Cell Death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-Dependent Mitochondrial Permeability Transition Regulates Some Necrotic but Not Apoptotic Cell Death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Maack, C.; Kartes, T.; Kilter, H.; Schäfers, H.-J.; Nickenig, G.; Böhm, M.; Laufs, U. Oxygen Free Radical Release in Human Failing Myocardium Is Associated with Increased Activity of Rac1-GTPase and Represents a Target for Statin Treatment. Circulation 2003, 108, 1567–1574. [Google Scholar] [CrossRef]
- Dey, S.; Sidor, A.; O’Rourke, B. Compartment-Specific Control of Reactive Oxygen Species Scavenging by Antioxidant Pathway Enzymes. J. Biol. Chem. 2016, 291, 11185–11197. [Google Scholar] [CrossRef]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen Free Radicals and Congestive Heart Failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef]
- Michels, G.; Khan, I.F.; Endres-Becker, J.; Rottlaender, D.; Herzig, S.; Ruhparwar, A.; Wahlers, T.; Hoppe, U.C. Regulation of the Human Cardiac Mitochondrial Ca2+ Uptake by 2 Different Voltage-Gated Ca2+ Channels. Circulation 2009, 119, 2435–2443. [Google Scholar] [CrossRef]
- Weber, C.R.; Piacentino, V.; Houser, S.R.; Bers, D.M. Dynamic Regulation of Sodium/Calcium Exchange Function in Human Heart Failure. Circulation 2003, 108, 2224–2229. [Google Scholar] [CrossRef]
- Palty, R.; Sekler, I. The Mitochondrial Na(+)/Ca(2+) Exchanger. Cell Calcium 2012, 52, 9–15. [Google Scholar] [CrossRef]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX Protein, but Not LETM1, Mediates Mitochondrial Ca2+ Extrusion, Thereby Limiting Ca2+-Induced NAD(P)H Production and Modulating Matrix Redox State. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef]
- Bers, D.M. Altered Cardiac Myocyte Ca Regulation in Heart Failure. Physiol. Bethesda Md 2006, 21, 380–387. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated Cytosolic Na+ Increases Mitochondrial Formation of Reactive Oxygen Species in Failing Cardiac Myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Rydström, J. Mitochondrial NADPH, Transhydrogenase and Disease. Biochim. Biophys. Acta 2006, 1757, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Xi, Q.; Cheranov, S.Y.; Jaggar, J.H. Mitochondria-Derived Reactive Oxygen Species Dilate Cerebral Arteries by Activating Ca2+ Sparks. Circ. Res. 2005, 97, 354–362. [Google Scholar] [CrossRef]
- Li, J.; An, C.; Zheng, H.; Lei, T.; Zhang, N.; Zheng, Y.; Yang, M. Leukocyte Telomere Length and Risk of Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 2712–2718. [Google Scholar] [CrossRef]
- Zhou, L.; Aon, M.A.; Liu, T.; O’Rourke, B. Dynamic Modulation of Ca2+ Sparks by Mitochondrial Oscillations in Isolated Guinea Pig Cardiomyocytes under Oxidative Stress. J. Mol. Cell. Cardiol. 2011, 51, 632–639. [Google Scholar] [CrossRef]
- Cheranov, S.Y.; Jaggar, J.H. Mitochondrial Modulation of Ca2+ Sparks and Transient KCa Currents in Smooth Muscle Cells of Rat Cerebral Arteries. J. Physiol. 2004, 556, 755. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, J.; Wei, C.; Li, K.; Xie, W.; Wang, Y.; Cheng, H. Bidirectional Regulation of Ca2+ Sparks by Mitochondria-Derived Reactive Oxygen Species in Cardiac Myocytes. Cardiovasc. Res. 2008, 77, 432–441. [Google Scholar] [CrossRef]
- Plant, D.R.; Lynch, G.S.; Williams, D.A. Hydrogen Peroxide Increases Depolarization-Induced Contraction of Mechanically Skinned Slow Twitch Fibres from Rat Skeletal Muscles. J. Physiol. 2002, 539, 883–891. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of Oxidative Stress in Cardiovascular Diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between Sarco-Endoplasmic Reticulum and Mitochondria in Cardiac and Skeletal Muscle—Pivotal Roles in Ca2+ and Reactive Oxygen Species Signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [CrossRef]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of Endoplasmic Reticulum Stress in the Heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Sevier, C.S.; Kaiser, C.A. Ero1 and Redox Homeostasis in the Endoplasmic Reticulum. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2008, 1783, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Kania, E.; Pajak, B.; Orzechowski, A. Calcium Homeostasis and ER Stress in Control of Autophagy in Cancer Cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of Sodium and Calcium Dysregulation in Tachyarrhythmias in Sudden Cardiac Death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef]
- Gorski, P.A.; Ceholski, D.K.; Hajjar, R.J. Altered Myocardial Calcium Cycling and Energetics in Heart Failure—A Rational Approach for Disease Treatment. Cell Metab. 2015, 21, 183–194. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium Cycling Proteins and Heart Failure: Mechanisms and Therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Moschella, M.C.; Marks, A.R. Inositol 1,4,5-Trisphosphate Receptor Expression in Cardiac Myocytes. J. Cell Biol. 1993, 120, 1137–1146. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial Calcium Overload Is a Key Determinant in Heart Failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Rizzuto, R.; Duchen, M.R.; Pozzan, T. Flirting in Little Space: The ER/Mitochondria Ca2+ Liaison. Sci. STKE Signal Transduct. Knowl. Environ. 2004, 2004, re1. [Google Scholar] [CrossRef]
- Min, C.K.; Yeom, D.R.; Lee, K.-E.; Kwon, H.-K.; Kang, M.; Kim, Y.-S.; Park, Z.Y.; Jeon, H.; Kim, D.H. Coupling of Ryanodine Receptor 2 and Voltage-Dependent Anion Channel 2 Is Essential for Ca2+ Transfer from the Sarcoplasmic Reticulum to the Mitochondria in the Heart. Biochem. J. 2012, 447, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Pieske, B.; Houser, S.R. [Na+]i Handling in the Failing Human Heart. Cardiovasc. Res. 2003, 57, 874–886. [Google Scholar] [CrossRef]
- Viatchenko-Karpinski, S.; Kornyeyev, D.; El-Bizri, N.; Budas, G.; Fan, P.; Jiang, Z.; Yang, J.; Anderson, M.E.; Shryock, J.C.; Chang, C.-P.; et al. Intracellular Na+ Overload Causes Oxidation of CaMKII and Leads to Ca2+ Mishandling in Isolated Ventricular Myocytes. J. Mol. Cell. Cardiol. 2014, 76, 247–256. [Google Scholar] [CrossRef]
- Peiling Yang, S.; Ngeow, J. Familial Non-Medullary Thyroid Cancer: Unraveling the Genetic Maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II Regulates Cardiac Na+ Channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef]
- Scirica, B.M.; Morrow, D.A.; Hod, H.; Murphy, S.A.; Belardinelli, L.; Hedgepeth, C.M.; Molhoek, P.; Verheugt, F.W.A.; Gersh, B.J.; McCabe, C.H.; et al. Effect of Ranolazine, an Antianginal Agent with Novel Electrophysiological Properties, on the Incidence of Arrhythmias in Patients with Non ST-Segment Elevation Acute Coronary Syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) Randomized Controlled Trial. Circulation 2007, 116, 1647–1652. [Google Scholar] [CrossRef]
- Banerjee, K.; Ghosh, R.K.; Kamatam, S.; Banerjee, A.; Gupta, A. Role of Ranolazine in Cardiovascular Disease and Diabetes: Exploring beyond Angina. Int. J. Cardiol. 2017, 227, 556–564. [Google Scholar] [CrossRef]
- Morrow, D.A.; Scirica, B.M.; Sabatine, M.S.; de Lemos, J.A.; Murphy, S.A.; Jarolim, P.; Theroux, P.; Bode, C.; Braunwald, E. B-Type Natriuretic Peptide and the Effect of Ranolazine in Patients with Non-ST-Segment Elevation Acute Coronary Syndromes: Observations from the MERLIN-TIMI 36 (Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST Elevation Acute Coronary-Thrombolysis in Myocardial Infarction 36) Trial. J. Am. Coll. Cardiol. 2010, 55, 1189–1196. [Google Scholar] [CrossRef]
- Aldakkak, M.; Camara, A.K.S.; Heisner, J.S.; Yang, M.; Stowe, D.F. Ranolazine Reduces Ca2+ Overload and Oxidative Stress and Improves Mitochondrial Integrity to Protect against Ischemia Reperfusion Injury in Isolated Hearts. Pharmacol. Res. 2011, 64, 381–392. [Google Scholar] [CrossRef]
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a Novel Selective Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor: Characterisation and Comparison with Other SGLT-2 Inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; van Borren, M.M.G.J.; Belterman, C.N.W.; Coronel, R.; Fiolet, J.W.T. Increased Na+/H+-Exchange Activity Is the Cause of Increased [Na+]i and Underlies Disturbed Calcium Handling in the Rabbit Pressure and Volume Overload Heart Failure Model. Cardiovasc. Res. 2003, 57, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhalla, A.K.; Hill, M.F.; Singal, P.K. Role of Oxidative Stress in Transition of Hypertrophy to Heart Failure. J. Am. Coll. Cardiol. 1996, 28, 506–514. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Bauersachs, J.; Galuppo, P.; Fraccarollo, D.; Christ, M.; Ertl, G. Improvement of Left Ventricular Remodeling and Function by Hydroxymethylglutaryl Coenzyme a Reductase Inhibition with Cerivastatin in Rats with Heart Failure after Myocardial Infarction. Circulation 2001, 104, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Suematsu, N.; Kinugawa, S.; Ide, T.; Wen, J.; Takeshita, A. Fluvastatin, a 3-Hydroxy-3-Methylglutaryl Coenzyme a Reductase Inhibitor, Attenuates Left Ventricular Remodeling and Failure after Experimental Myocardial Infarction. Circulation 2002, 105, 868–873. [Google Scholar] [CrossRef]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.F.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in Older Patients with Systolic Heart Failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Chiles, R.; Al-Horani, R.A. Vericiguat: A New Hope for Heart Failure Patients. Cardiovasc. Ther. 2022, 2022, 1554875. [Google Scholar] [CrossRef]
- Xia, J.; Hui, N.; Tian, L.; Liang, C.; Zhang, J.; Liu, J.; Wang, J.; Ren, X.; Xie, X.; Wang, K. Development of Vericiguat: The First Soluble Guanylate Cyclase (SGC) Stimulator Launched for Heart Failure with Reduced Ejection Fraction (HFrEF). Biomed. Pharmacother. 2022, 149, 112894. [Google Scholar] [CrossRef]
- Shaikh, T.G.; Jawed, S.; Rahmat, Z.S.; Ahmed, S.H.; Waseem, S.; Ullah, I.; Irfan, M.; Asghar, M.S. Efficacy and Safety of Vericiguat for Treatment of Heart Failure: A Systematic Review. Curr. Probl. Cardiol. 2023, 101586, ahead-of-print. [Google Scholar] [CrossRef]
- Ghezzi, D.; Baruffini, E.; Haack, T.B.; Invernizzi, F.; Melchionda, L.; Dallabona, C.; Strom, T.M.; Parini, R.; Burlina, A.B.; Meitinger, T.; et al. Mutations of the Mitochondrial-TRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis. Am. J. Hum. Genet. 2012, 90, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Sengers, R.C.A.; Trijbels, J.M.F.; Willems, J.L.; Daniels, O.; Stadhouders, A.M. Congenital Cataract and Mitochondrial Myopathy of Skeletal and Heart Muscle Associated with Lactic Acidosis after Exercise. J. Pediatr. 1975, 86, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Bakker, T.C.M. Positive Genetic Correlation between Female Preference and Preferred Male Ornament in Sticklebacks. Nature 1993, 363, 255–257. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Champion, M.P.; van den Heuvel, L.; Barth, H.; Trutnau, B.; Craig, K.; Lammens, M.; Schreuder, M.F.; Taylor, R.W.; Smeitink, J.A.M.; et al. Mitochondrial DNA m.3242G > A Mutation, an under Diagnosed Cause of Hypertrophic Cardiomyopathy and Renal Tubular Dysfunction? Eur. J. Med. Genet. 2012, 55, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Zeviani, M. MtDNA-Maintenance Defects: Syndromes and Genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van Der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.M.; Van’T Veer-Korthof, E.T.; Van Der Harten, J.J.; Sobotka-Plojhar, M.A. An X-Linked Mitochondrial Disease Affecting Cardiac Muscle, Skeletal Muscle and Neutrophil Leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Langer, R. A Decade of Progress in Tissue Engineering. Nat. Protoc. 2016, 11, 1775–1781. [Google Scholar] [CrossRef]
- Wang, X.; Hu, G. Human Embryos in a Dish—Modeling Early Embryonic Development with Pluripotent Stem Cells. Cell Regen. 2022, 11, 4. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Branco, M.A.; Dias, T.P.; Cabral, J.M.S.; Pinto-do-Ó, P.; Diogo, M.M. Human Multilineage Pro-Epicardium/Foregut Organoids Support the Development of an Epicardium/Myocardium Organoid. Nat. Commun. 2022, 13, 6981. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, M.F.; Louro, A.F.; Alves, P.M.; Serra, M. Next Generation of Heart Regenerative Therapies: Progress and Promise of Cardiac Tissue Engineering. NPJ Regen. Med. 2021, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Final Report Summary—INDIVUHEART (Individualized Early Risk Assessment for Heart Diseases)|FP7|CORDIS|European Commission. Available online: https://cordis.europa.eu/project/id/340248/reporting (accessed on 24 January 2023).
Figure 1.
Calcium and ROS interplay in mitochondria. The close proximity and juxtaposition of the ER to mitochondria facilitate a straight and selective transport of calcium (Ca2+). The mitochondria-associated membranes (MAMs) are the site of contact between the endoplasmic reticulum (ER) and mitochondria. Here calcium release channels accumulate, and these channels include the IP3R localized on the ER membrane, the voltage-dependent anion channel 1 (VDAC1) on the outer mitochondrial membrane (OMM), and the cytosolic glucose-regulated chaperone protein 75 (GRP75) that forms a tether between the two organelles. Increased levels of mitochondrial calcium stimulate the activity of the electron transport chain leading to a higher release of reactive oxygen species (ROS). As a vicious circle ROS can hit Ca2+ channels localized in the ER membranes and cause a further leak of Ca2+ from the ER that leads to increased ROS production in the mitochondria. A pick in the levels of both Ca2+ and ROS opens the mitochondrial permeability transition pore (mPTP), allowing the release of cytochrome c that leads to the activation of apoptosis. Abbreviations: ROS reactive oxygen species; Ca2+ calcium ion, IP3R IP3 receptor; GRP75 glucose-regulated chaperone protein 75; VDAC voltage-dependent anion channel; MCU mitochondrial calcium uniporter; c cytochrome c; SERCA sarco/endoplasmic reticulum Ca2+ ATPase; RyR ryanodine receptors; mPTP—mitochondrial permeability transition pore; I, II, III, VI, V respiratory complex I–V. Created with BioRender.com.
Figure 1.
Calcium and ROS interplay in mitochondria. The close proximity and juxtaposition of the ER to mitochondria facilitate a straight and selective transport of calcium (Ca2+). The mitochondria-associated membranes (MAMs) are the site of contact between the endoplasmic reticulum (ER) and mitochondria. Here calcium release channels accumulate, and these channels include the IP3R localized on the ER membrane, the voltage-dependent anion channel 1 (VDAC1) on the outer mitochondrial membrane (OMM), and the cytosolic glucose-regulated chaperone protein 75 (GRP75) that forms a tether between the two organelles. Increased levels of mitochondrial calcium stimulate the activity of the electron transport chain leading to a higher release of reactive oxygen species (ROS). As a vicious circle ROS can hit Ca2+ channels localized in the ER membranes and cause a further leak of Ca2+ from the ER that leads to increased ROS production in the mitochondria. A pick in the levels of both Ca2+ and ROS opens the mitochondrial permeability transition pore (mPTP), allowing the release of cytochrome c that leads to the activation of apoptosis. Abbreviations: ROS reactive oxygen species; Ca2+ calcium ion, IP3R IP3 receptor; GRP75 glucose-regulated chaperone protein 75; VDAC voltage-dependent anion channel; MCU mitochondrial calcium uniporter; c cytochrome c; SERCA sarco/endoplasmic reticulum Ca2+ ATPase; RyR ryanodine receptors; mPTP—mitochondrial permeability transition pore; I, II, III, VI, V respiratory complex I–V. Created with BioRender.com.


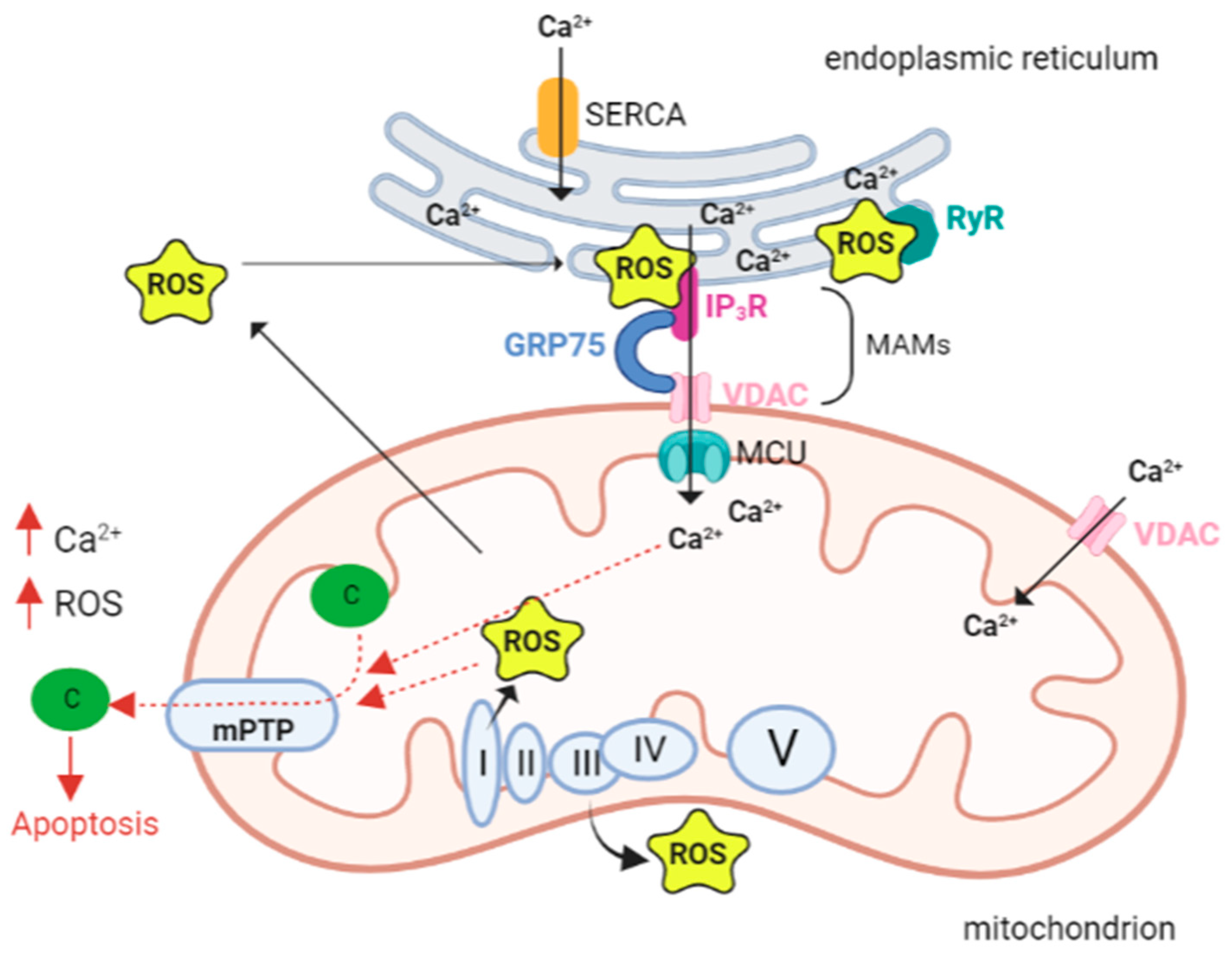{kind=link}
Table 1.
Susceptibility loci and candidate genes linked to cardiomyopathies with mitochondrial alterations.
Table 1.
Susceptibility loci and candidate genes linked to cardiomyopathies with mitochondrial alterations.
| Chromosome Location | Phenotype | Phenotype MIM Number | Inheritance | Gene/Locus | Gene/Locus MIM Number | References |
|---|---|---|---|---|---|---|
| 6q13 | Combined oxidative phosphorylation deficiency 10 | 614702 | AR | MTO1 | 614667 | [151] |
| 7q34 | Sengers syndrome | 212350 | AR | AGK | 610345 | [152] |
| 4q35.1 | Mitochondrial DNA depletion syndrome 12B (cardiomyopathic type) | 615418 | AR | SLC25A4 | 103220 | [153] |
| Mt | Hypertrophic cardiomyopathy with kidney anomalies due to mtDNA mutations | Mitochondrial inheritance | MT-TL1 | [154] | ||
| mt | Mitochondrial DNA- related cardiomyopathy and hearing loss | Mitochondrial inheritance | MT-TK | [155] | ||
| Xq28 | Barth syndrome | 302060 | XLR | TAFAZZIN | 300394 | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; Bonora, E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. https://doi.org/10.3390/antiox12020353
AMA Style
De Nicolo B, Cataldi-Stagetti E, Diquigiovanni C, Bonora E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants. 2023; 12(2):353. https://doi.org/10.3390/antiox12020353
Chicago/Turabian StyleDe Nicolo, Bianca, Erica Cataldi-Stagetti, Chiara Diquigiovanni, and Elena Bonora. 2023. "Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies" Antioxidants 12, no. 2: 353. https://doi.org/10.3390/antiox12020353
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.