
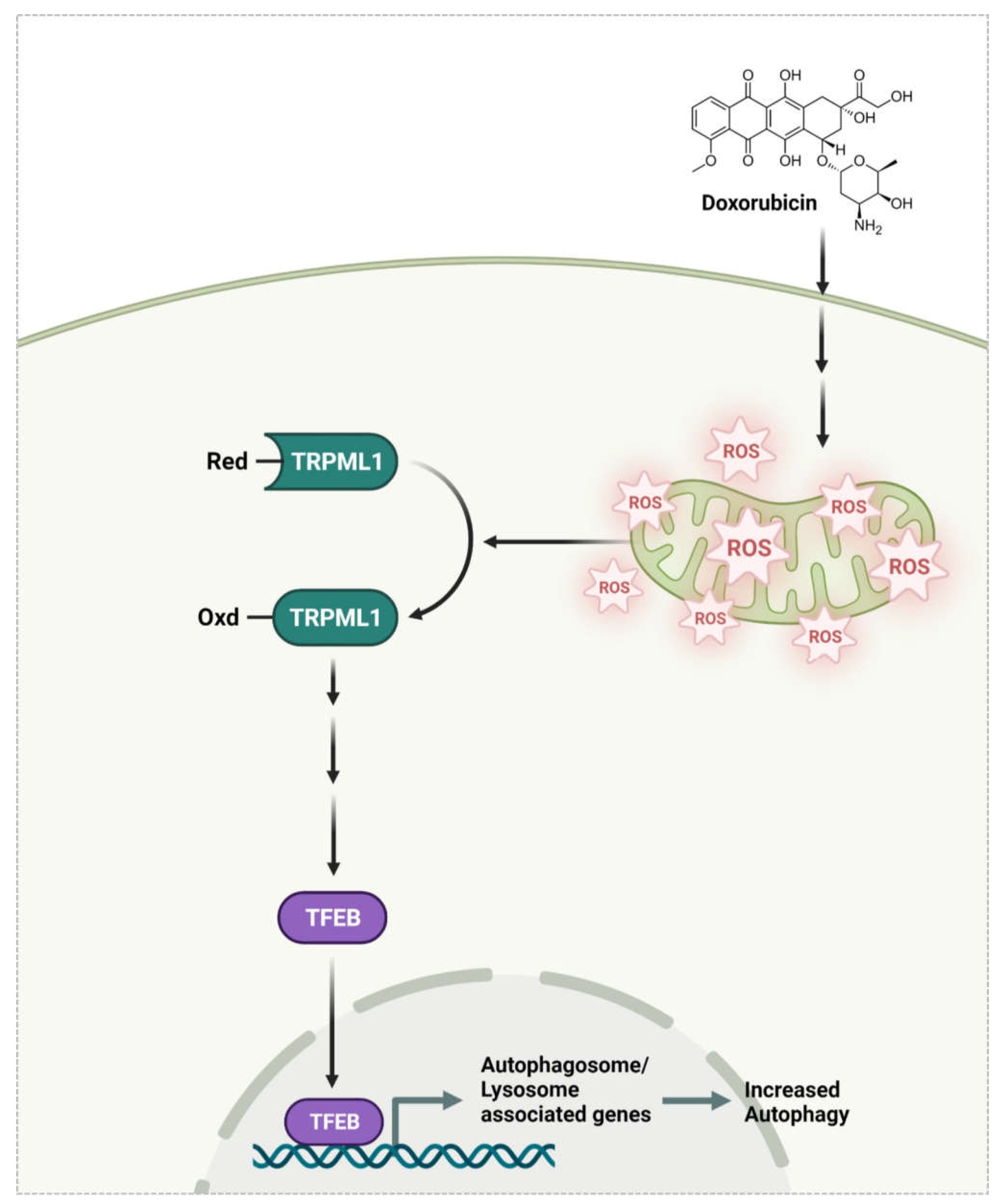
Doxorubicin Induces Bone Loss by Increasing Autophagy through a Mitochondrial ROS/TRPML1/TFEB Axis in Osteoclasts

Abstract
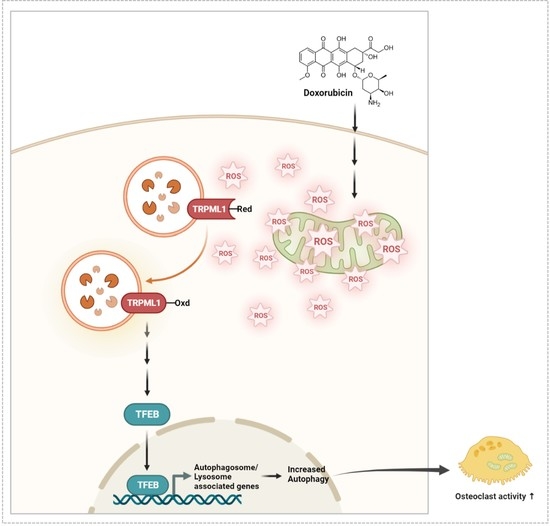
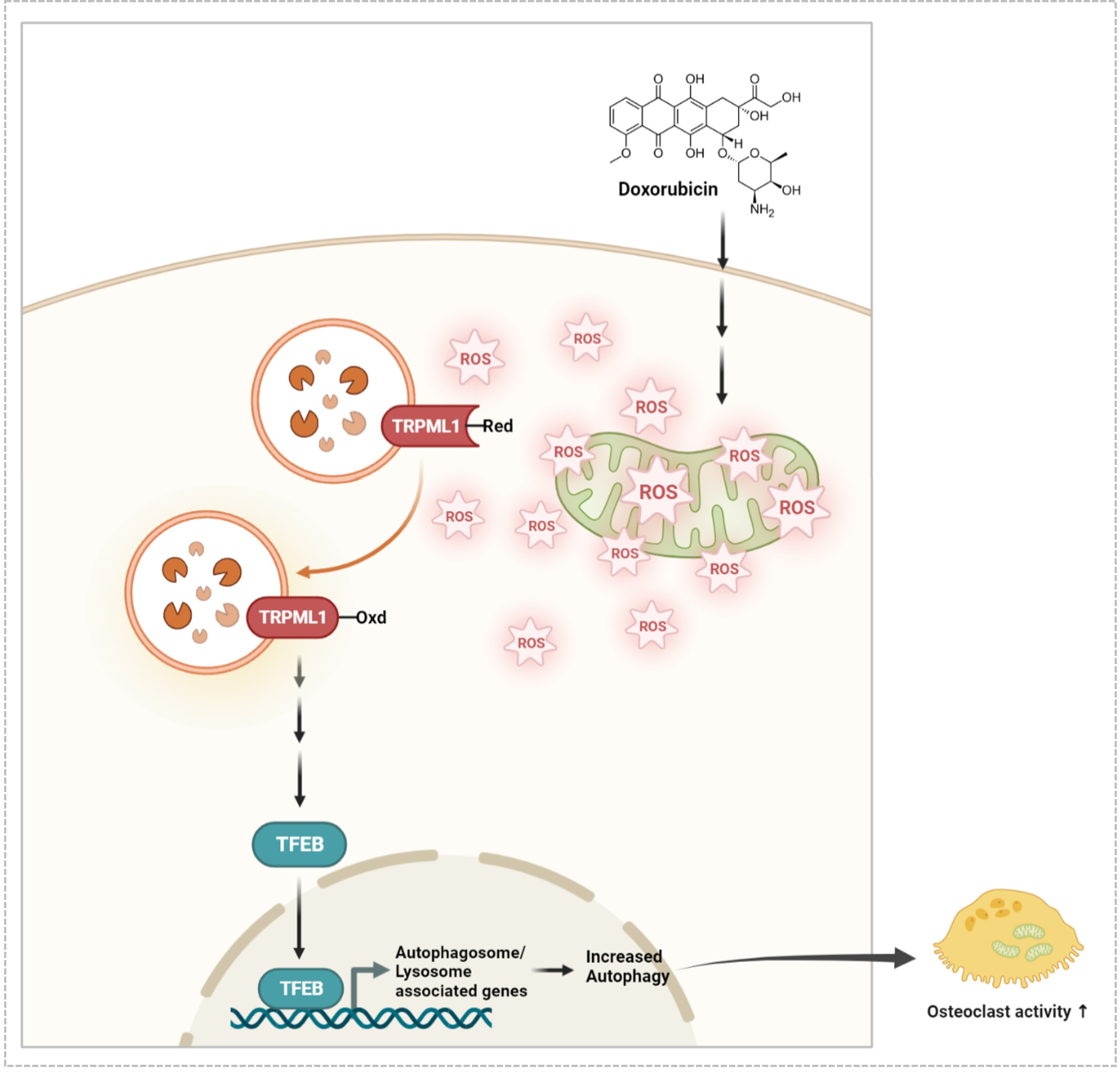
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Reagents
2.3. Animals and Study Design
2.4. Cultures of Osteoblast and OC, and OC Formation
2.5. Bone Resorption
2.6. RNA Isolation and qPCR
2.7. Apoptosis Assay
2.8. Fractionation and Western Blot Analysis
2.9. Flow Cytometric Quantification of Acidic Vesicular Organelles with Acridine Orange Staining
2.10. Determination of Mitochondrial Reactive Oxygen Species (ROS)
2.11. Detection of Oxidized TRPML1 via Carboxymethylation
2.12. Transfection of siRNA
2.13. Statistical Analyses
3. Results
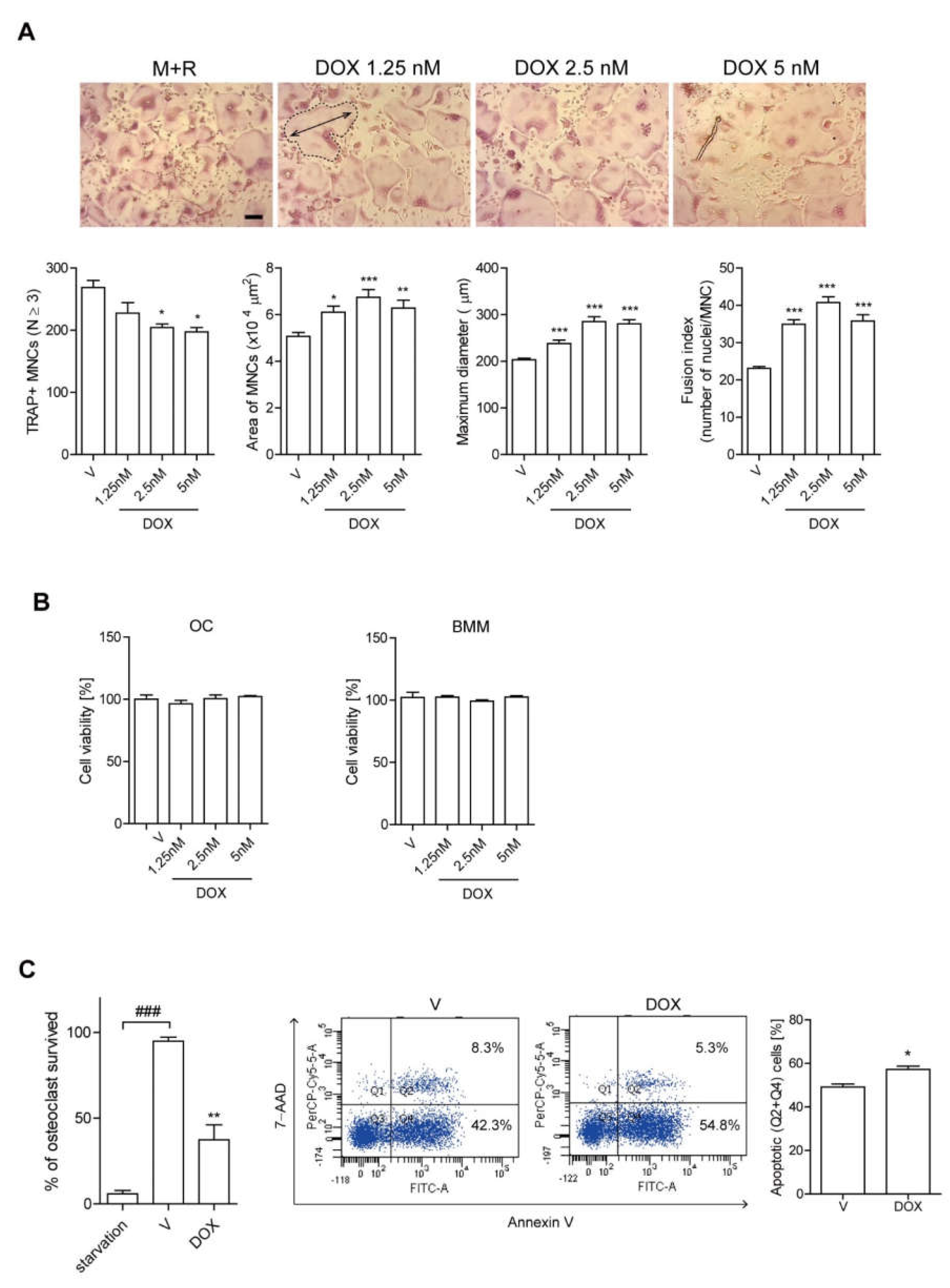
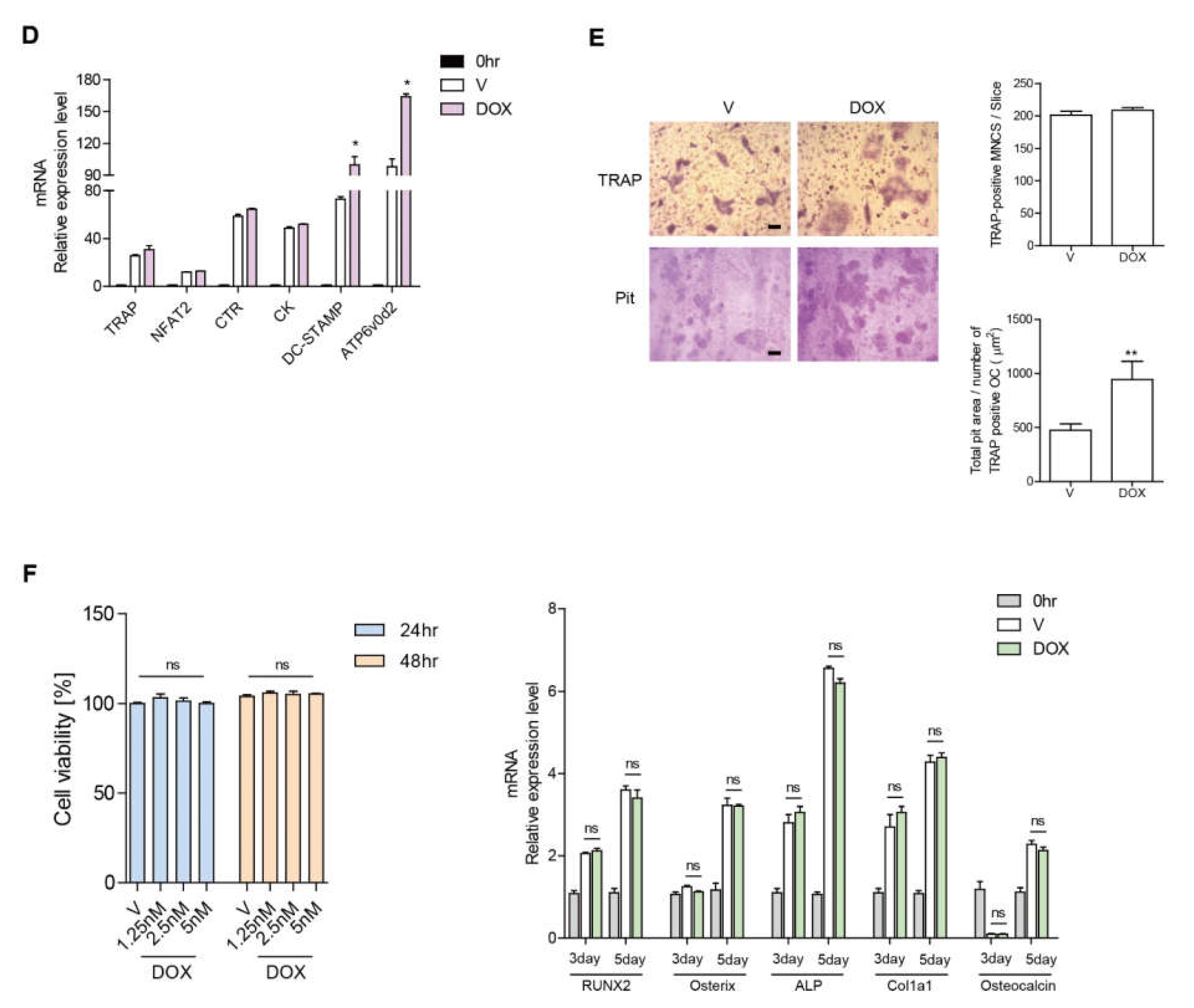
3.1. DOX Enhances the Activity of Ocs
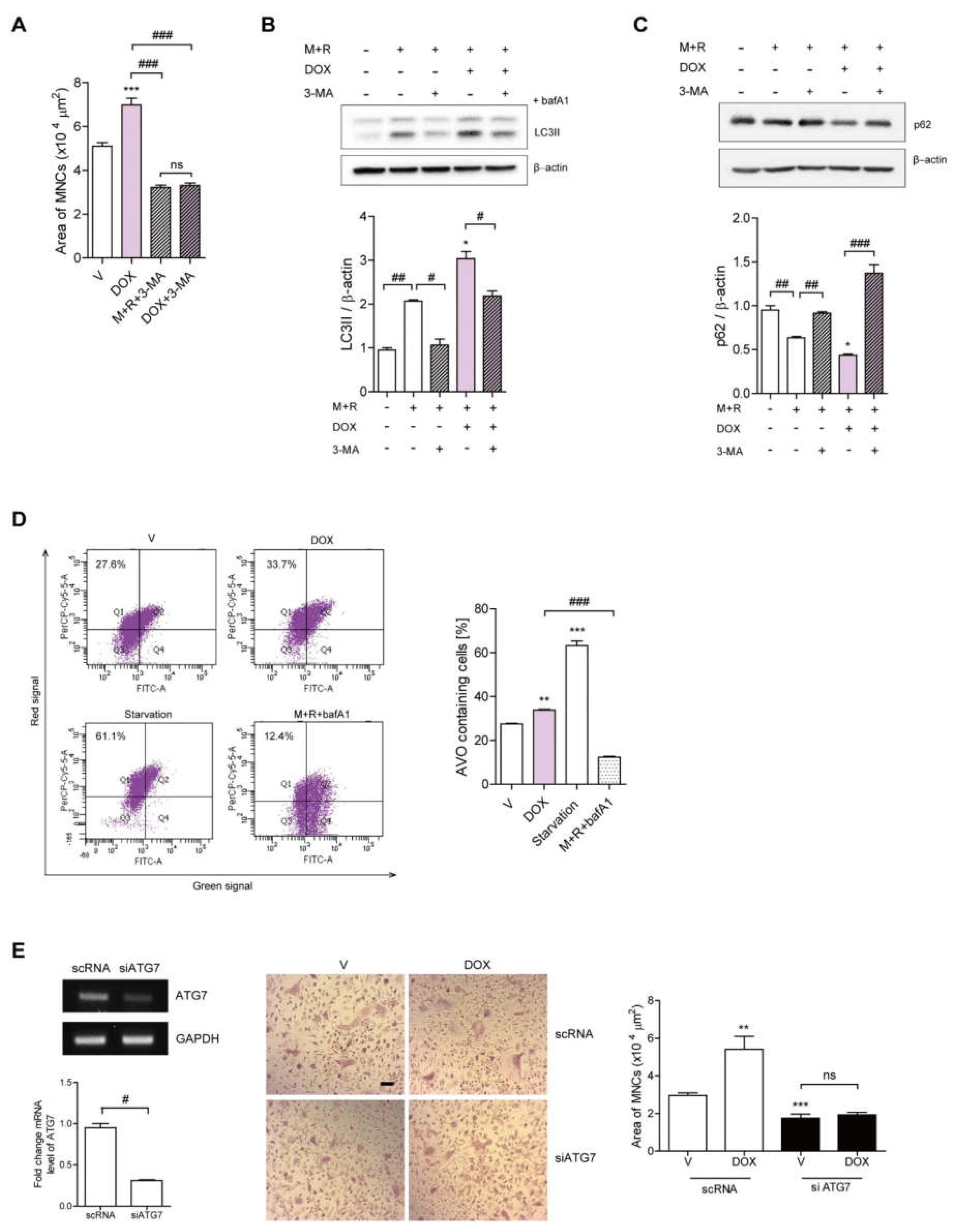
3.2. DOX Enhances Autophagy in OCs
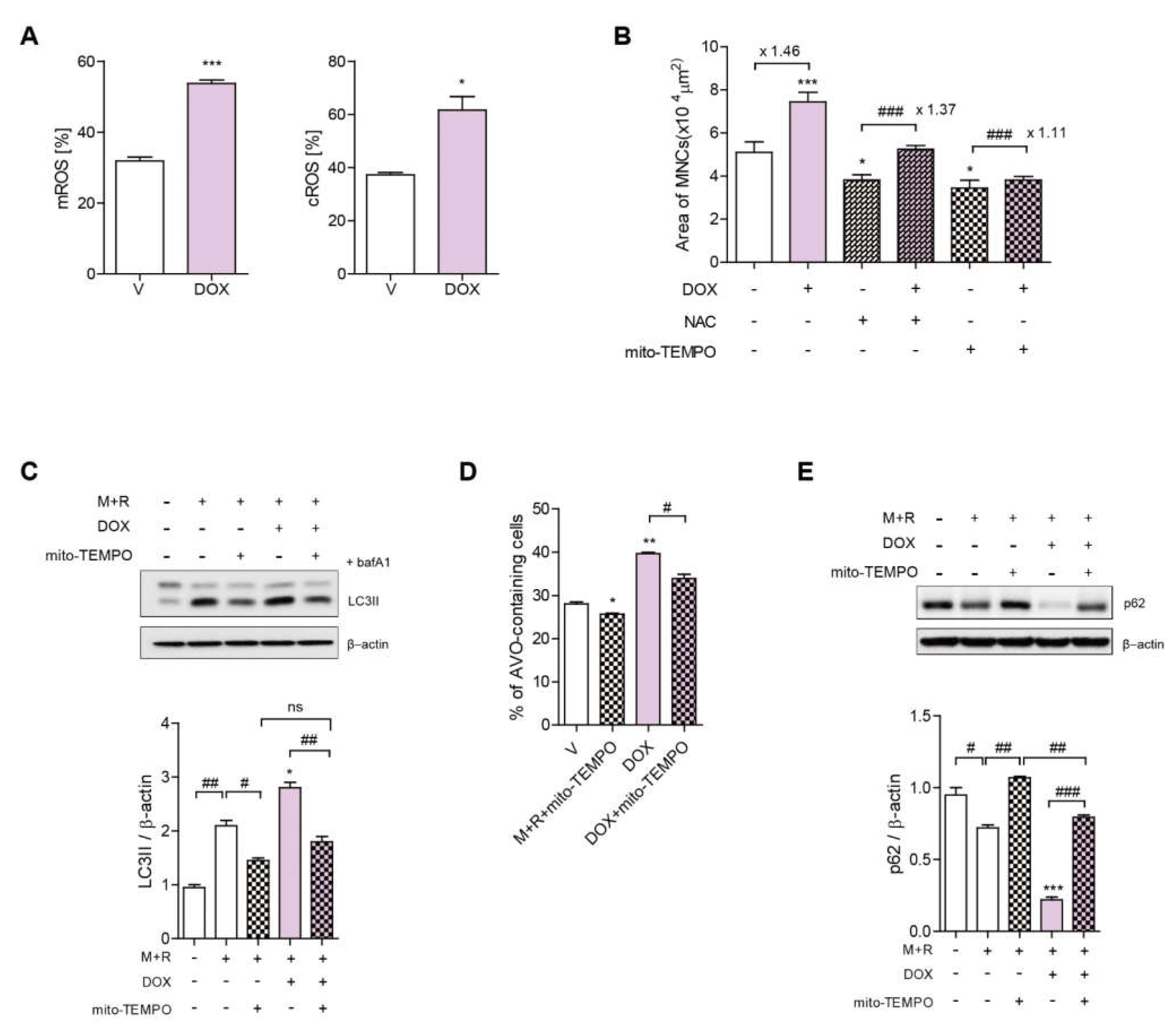
3.3. DOX Increases ROS Levels, Leading to Increased TFEB Nuclear Translocation in OCs
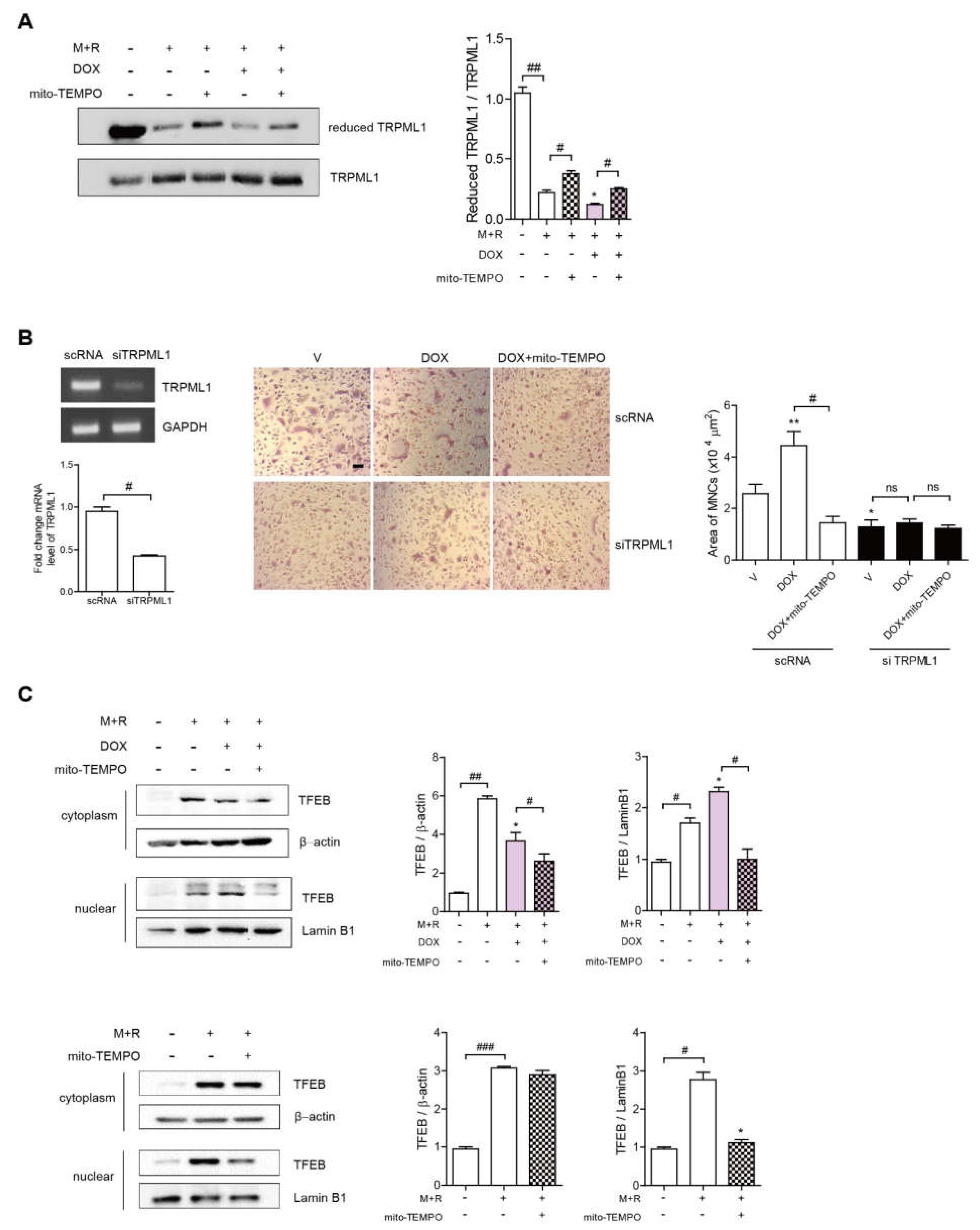
3.4. Dox Induces Oxidation of TRPML1 and Nuclear Localization of TFEB
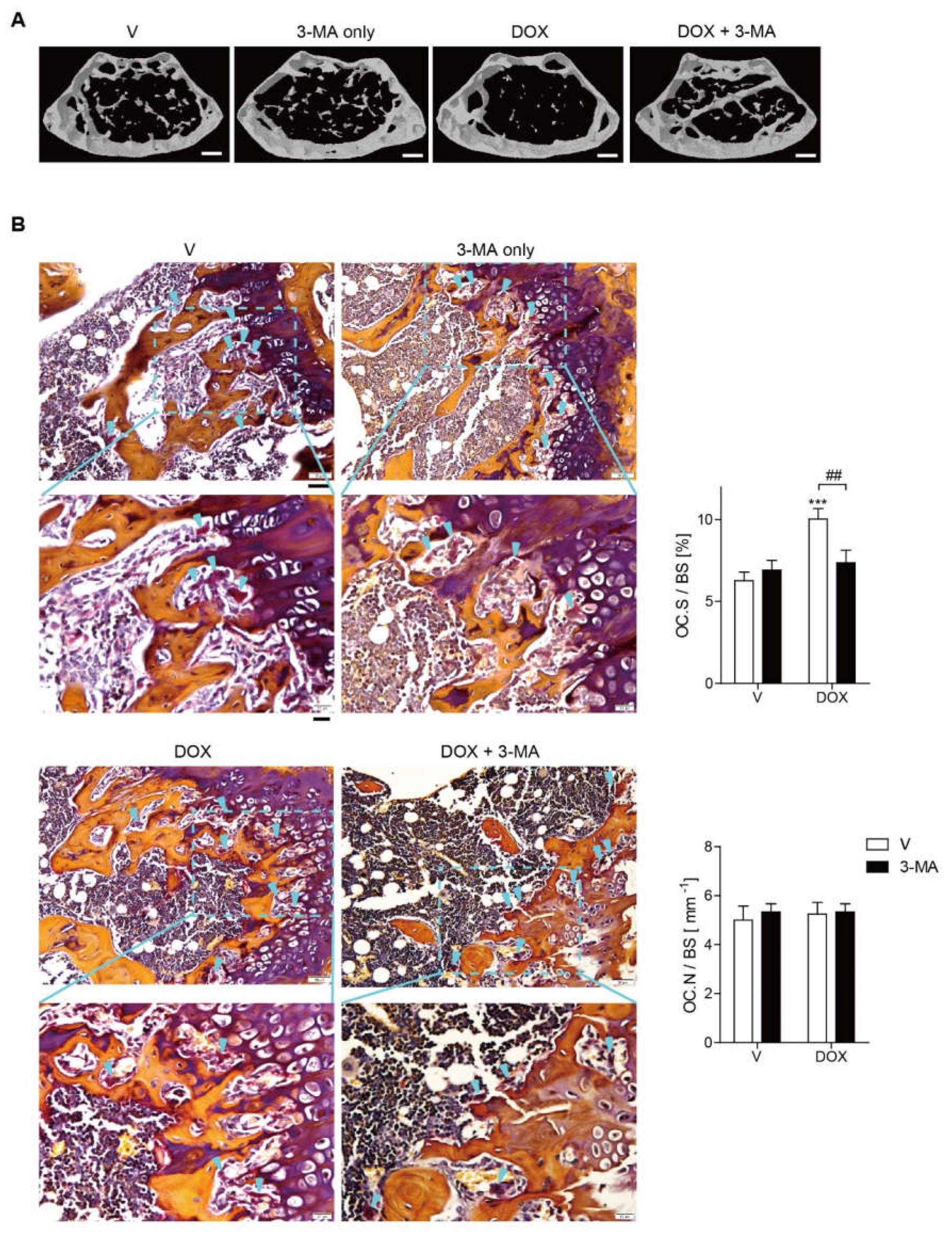
3.5. Doxorubicin Induces Bone Loss Accompanied by Increased Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Guo, Y.F.; Liu, Y.Z.; Liu, Y.J.; Xiong, D.H.; Liu, X.G.; Wang, L.; Yang, T.L.; Lei, S.F.; Guo, Y.; et al. Pathway-based genome-wide association analysis identified the importance of regulation-of-autophagy pathway for ultradistal radius BMD. J. Bone Miner. Res. 2010, 25, 1572–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfrich, M.H.; Hocking, L.J. Genetics and aetiology of Pagetic disorders of bone. Arch. Biochem. Biophys. 2008, 473, 172–182. [Google Scholar] [CrossRef] [PubMed]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.H.; Yoon, S.Y.; Choi, B.; Sohn, D.H.; Yoon, K.H.; Kim, W.J.; Kim, D.H.; Chang, E.J. Microtubule-associated protein light chain 3 regulates Cdc42-dependent actin ring formation in osteoclast. Int. J. Biochem. Cell Biol. 2012, 44, 989–997. [Google Scholar] [CrossRef]
- Park, H.J.; Son, H.J.; Sul, O.J.; Suh, J.H.; Choi, H.S. 4-Phenyl butyric acid protects against lipopolysaccharide-induced bone loss by modulating autophagy in osteoclasts. Biochem. Pharmacol. 2018, 151, 9–17. [Google Scholar] [CrossRef]
- Park, H.J.; Gholam-Zadeh, M.; Suh, J.H.; Choi, H.S. Lycorine attenuates autophagy in osteoclasts via an axis of mROS/TRPML1/TFEB to reduce LPS-induced bone loss. Oxid. Med. Cell Longev. 2019, 2019, 8982147. [Google Scholar] [CrossRef] [PubMed]
- Sul, O.J.; Li, G.; Kim, J.E.; Kim, E.S.; Choi, H.S. 7-ketocholesterol enhances autophagy via the ROS-TFEB signaling pathway in osteoclasts. J. Nutr. Biochem. 2021, 96, 108783–108794. [Google Scholar] [CrossRef]
- Malla, S.; Niraula, N.P.; Singh, B.; Liou, K.K.; Sohng, J.K. Limitations in doxorubicin production from Streptomyces peucetius. Microbiol. Res. 2010, 165, 427–435. [Google Scholar] [CrossRef]
- Keizer, H.G.; Pinedo, H.M.; Schuurhuis, G.J.; Joenje, H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol. Ther. 1990, 47, 219–231. [Google Scholar] [CrossRef]
- Bodley, A.; Liu, L.F.; Israel, M.; Seshadri, R.; Koseki, Y.; Giuliani, F.C.; Kirschenbaum, S.; Silber, R.; Potmesil, M. DNA Topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res. 1989, 49, 5969–5978. [Google Scholar]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hadji, P.; Ziller, M.; Maskow, C.; Albert, U.; Kalder, M. The influence of chemotherapy on bone mineral density, quantitative ultrasonometry and bone turnover in pre-menopausal women with breast cancer. Eur. J. Cancer 2009, 45, 3205–3212. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.A.; Douglas, S.; Brown, J.E.; Anderson, R.A. Bone mineral density loss during adjuvant chemotherapy in pre-menopausal women with early breast cancer: Is it dependent on oestrogen deficiency? Breast Cancer Res. Treat. 2010, 123, 805–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shusterman, S.; Meadows, A.T. Long term survivors of childhood leukemia. Curr. Opin. Hematol. 2000, 7, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Young, D.M.; Fioravanti, J.L.; Olson, H.M.; Prieur, D.J. Chemical and morphologic alterations of rabbit bone induced by adriamycin. Calcif. Tissue Res. 1975, 18, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Georgiou, K.R.; McKinnon, R.A.; Keefe, D.M.K.; Howe, P.R.C.; Xian, C.J. Combination chemotherapy with cyclophosphamide, epirubicin and 5-fluorouracil causes trabecular bone loss, bone marrow cell depletion and marrow adiposity in female rats. J. Bone Miner. Metab. 2016, 34, 277–290. [Google Scholar] [CrossRef]
- Yao, Z.; Murali, B.; Ren, Q.; Luo, X.; Faget, D.V.; Cole, T.; Ricci, B.; Thotala, D.; Monahan, J.; van Deursen, J.M.; et al. Therapy-induced senescence drives bone loss. Cancer Res. 2020, 80, 1171–1182. [Google Scholar] [CrossRef]
- Fonseca, H.; Carvalho, A.; Esteves, J.; Esteves, V.I.; Moreira-Gonçalves, D.; Duarte, J.A. Effects of doxorubicin administration on bone strength and quality in sedentary and physically active Wistar rats. Osteoporos. Int. 2016, 27, 3465–3475. [Google Scholar] [CrossRef]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anticancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The role of autophagy in the heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Sul, O.J.; Sung, Y.B.; Rajasekaran, M.; Ke, K.; Yu, R.; Back, S.H.; Choi, H.S. MicroRNA-155 induces autophagy in osteoclasts by targeting transforming growth factor β-activated kinase 1-binding protein 2 upon lipopolysaccharide stimulation. Bone 2018, 116, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Ke, K.; Safder, M.A.; Sul, O.J.; Kim, W.K.; Suh, J.H.; Joe, Y.; Chung, H.T.; Choi, H.S. Hemeoxygenase-1 maintains bone mass via attenuating a redox imbalance in osteoclast. Mol. Cell Endocrinol. 2015, 409, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, M.; Nakayachi, M.; Hayashida, C.; Ito, J.; Kaneda, T.; Masuhara, M.; Suda, N.; Sato, T.; Hakeda, Y. Low-density lipoprotein receptor deficiency causes impaired osteoclastogenesis and increased bone mass in mice because of defect in osteoclastic cell-cell fusion. J. Biol. Chem. 2012, 287, 19229–19241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sul, O.J.; Rajasekaran, M.; Park, H.J.; Suh, J.H.; Choi, H.S. MicroRNA-29b enhances osteoclast survival by targeting Bcl2-modifying factor after lipopolysaccharide stimulation. Oxid. Med. Cell Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bil, J.; Winiarska, M.; Nowis, D.; Bojarczuk, K.; Dabrowska-Iwanicka, A.; Basak, G.W.; Sułek, K.; Jakobisiak, M.; Golab, J. Bortezomib modulates surface CD20 in B-cell malignancies and affects rituximab-mediated complement-dependent cytotoxicity. Blood 2010, 115, 3745–3755. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Wang, H.; Hu, Y.; Hu, M.; Li, X.; Aodengqimuge; Ma, Y.; Wei, C.; Song, L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015, 106, 1023–1032. [Google Scholar] [CrossRef]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Macleod, K.F. Measuring autophagy in stressed cells. Methods Mol. Biol. 2015, 1292, 129–150. [Google Scholar]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin induces endotheliotoxicity and mitochondrial dysfunction via ROS/eNOS/NO pathway. Front. Pharmacol. 2020, 10, 1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotlieb, W.H.; Bruchim, I.; Ben-Baruch, G.; Davidson, B.; Zeltser, A.; Andersen, A.; Olsen, H. Doxorubicin levels in the serum and ascites of patients with ovarian cancer. Eur. J. Surg. Oncol. 2007, 33, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Saadh, M.J.; Haddad, M.; Dababneh, M.F.; Bayan, M.F.; Al-Jaidi, B.A. A guide for estimating the maximum safe starting dose and conversion it between animals and humans. Syst. Rev. Pharm. 2020, 11, 98–101. [Google Scholar]
- Cheng, L.; Zhu, Y.; Ke, D.; Xie, D. Oestrogen-activated autophagy has a negative effect on the anti-osteoclastogenic function of oestrogen. Cell. Prolif. 2020, 53, e12789. [Google Scholar] [CrossRef]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10, 1965–1967. [Google Scholar] [CrossRef]
- Mosure, K.W.; Henderson, A.J.; Klunk, L.J.; Knipe, J.O. Disposition of conjugate-bound and free doxorubicin in tumor-bearing mice following administration of a BR96-doxorubicin immunoconjugate (BMS 182248). Cancer Chemother. Pharmacol. 1997, 40, 251–258. [Google Scholar] [CrossRef]
- Khosla, S.; Burr, D.; Cauley, J.; Dempster, D.W.; Ebeling, P.R.; Felsenberg, D.; Gagel, R.F.; Gilsanz, V.; Guise, T.; Koka, S.; et al. Bisphosphonate-associated osteonecrosis of the jaw: Report of a task force of the American Society for Bone and Mineral Re-search. J. Bone Miner. Res. 2007, 22, 1479–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewiecki, E.M. New and emerging concepts in the use of denosumab for the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 209–223. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | DMSO | DOX | ||
|---|---|---|---|---|
| Vehicle | 3-MA | Vehicle | 3-MA | |
| BMD [mg/cm3] | 172.9 ± 3.21 | 176.2 ± 3.75 | 122.7 ± 3.24 a″ | 170.5 ± 5.60 b″ |
| BV/TV [%] | 15.57 ± 0.40 | 15.74 ± 0.29 | 11.25 ± 0.26 a″ | 15.38 ± 0.47 b″ |
| Tb.Th [μm] | 71.04 ± 1.88 | 71.15 ± 1.68 | 57.54 ± 1.05 a″ | 65.37 ± 1.28 b |
| Tb.Sp [μm] | 384.7 ± 11.12 | 368.0 ± 14.09 | 672.9 ± 30.68 a″ | 361.7 ± 19.43 b″ |
| ALP [U/L] | 51.54 ± 0.66 | 51.64 ± 1.26 | 53.20 ± 2.19 | 53.40 ± 0.69 |
| OCN [ng/mL] | 25.29 ± 1.53 | 26.30 ± 1.03 | 27.11 ± 0.87 | 27.48 ± 0.76 |
| CTX-1 [ng/mL] | 25.34 ± 1.51 | 22.98 ± 0.89 | 45.23 ± 1.82 a″ | 32.93 ± 1.49 b″ |
| MCP-1 [pg/mL] | 139.2 ± 13.29 | 129.5 ± 10.84 | 294.1 ± 15.40 a″ | 219.4 ± 12.66 b |
| H2O2 [μM] | 58.18 ± 0.80 | 54.81 ± 1.39 | 90.75 ± 2.12 a″ | 76.44 ± 4.67 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-J.; Yoon, S.-Y.; Park, J.-N.; Suh, J.-H.; Choi, H.-S. Doxorubicin Induces Bone Loss by Increasing Autophagy through a Mitochondrial ROS/TRPML1/TFEB Axis in Osteoclasts. Antioxidants 2022, 11, 1476. https://doi.org/10.3390/antiox11081476
Park H-J, Yoon S-Y, Park J-N, Suh J-H, Choi H-S. Doxorubicin Induces Bone Loss by Increasing Autophagy through a Mitochondrial ROS/TRPML1/TFEB Axis in Osteoclasts. Antioxidants. 2022; 11(8):1476. https://doi.org/10.3390/antiox11081476
Chicago/Turabian StylePark, Hyun-Jung, Sun-Young Yoon, Jung-Nam Park, Jae-Hee Suh, and Hye-Seon Choi. 2022. "Doxorubicin Induces Bone Loss by Increasing Autophagy through a Mitochondrial ROS/TRPML1/TFEB Axis in Osteoclasts" Antioxidants 11, no. 8: 1476. https://doi.org/10.3390/antiox11081476