
Activation of the Nrf2 Pathway Prevents Mitochondrial Dysfunction Induced by Caspase-3 Cleaved Tau: Implications for Alzheimer’s Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Tau Constructs
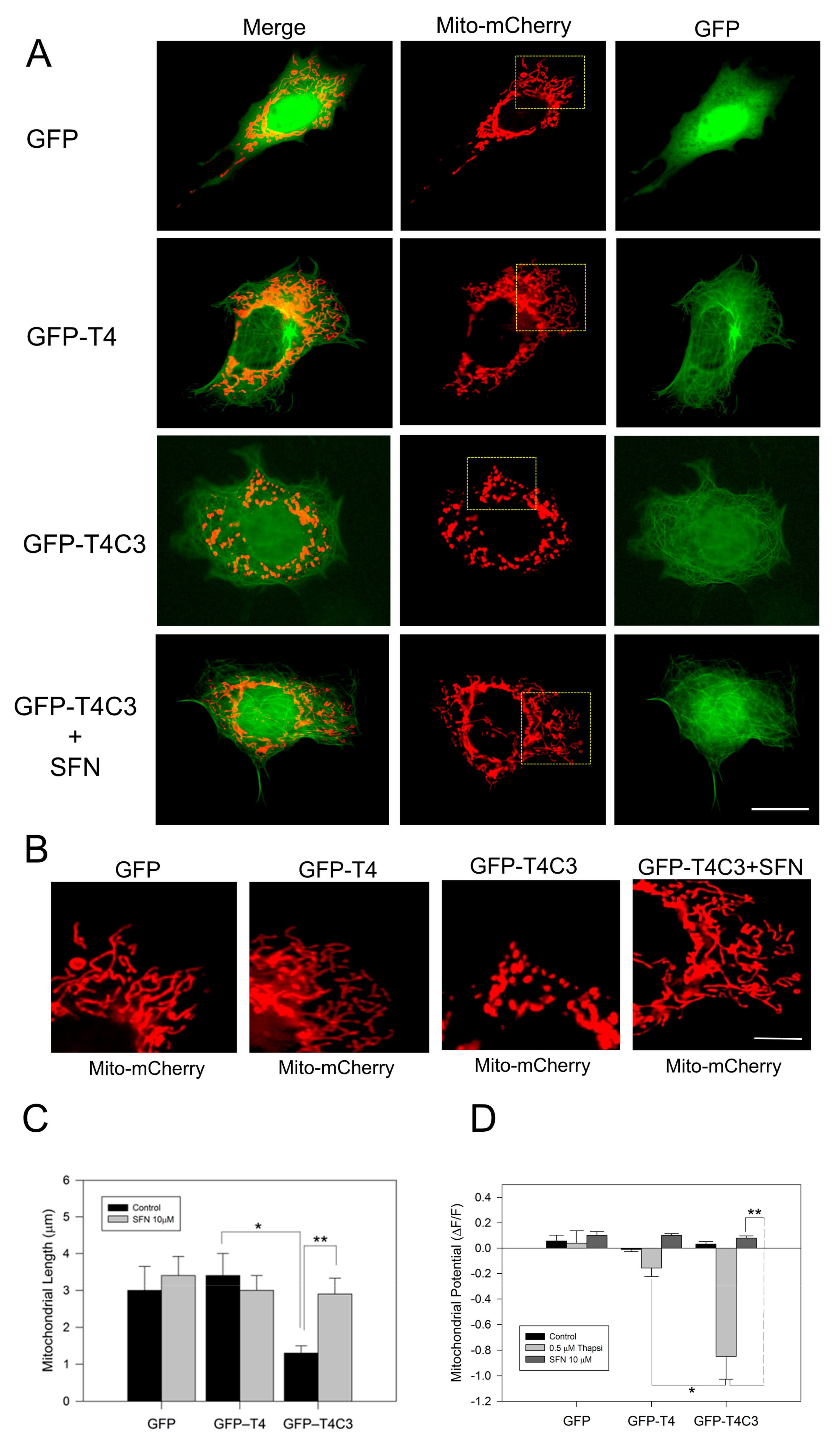
2.3. Determination of Mitochondrial Length and Membrane Potential in Live Cells
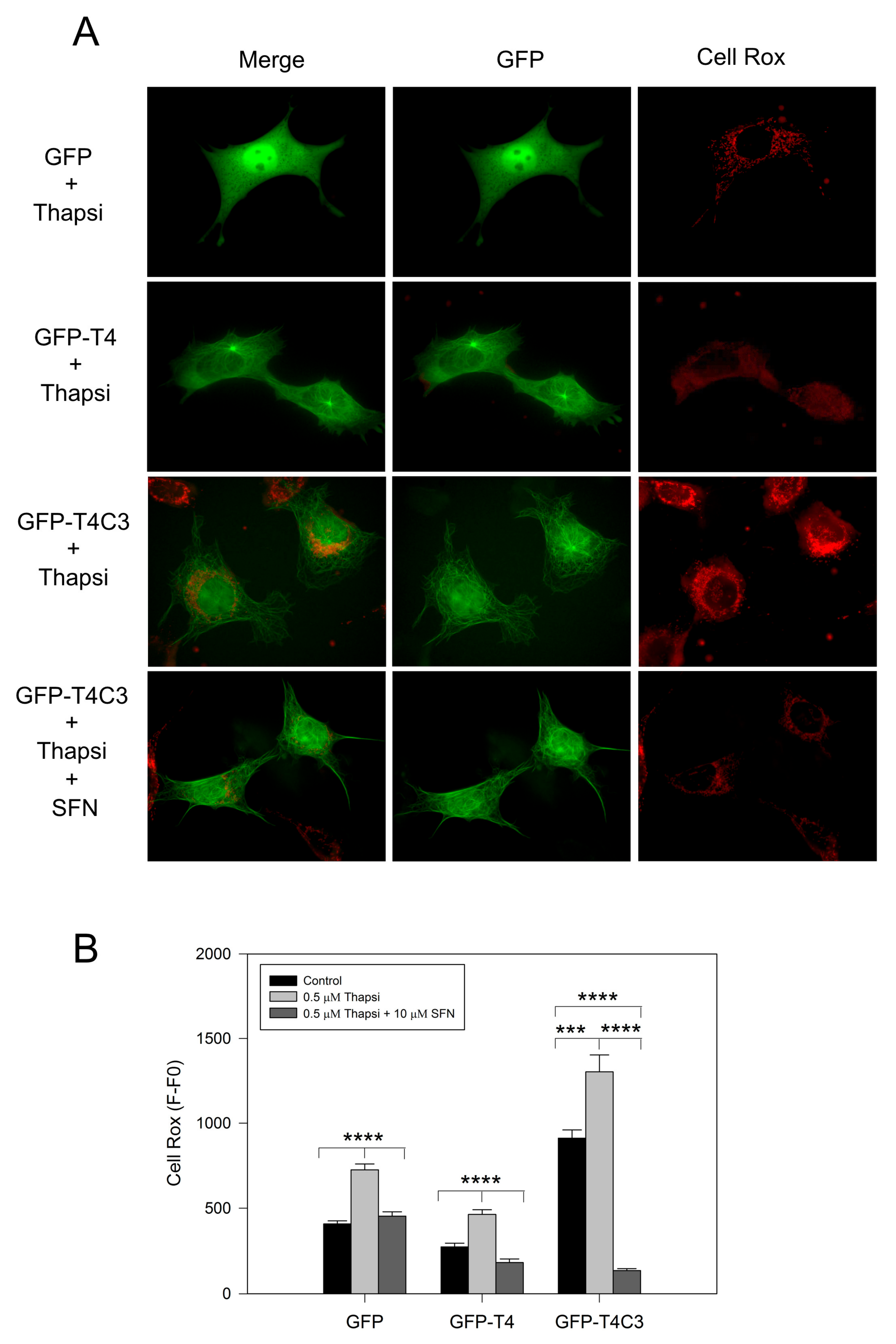
2.4. Determination of ROS Levels
2.5. Western Blot Analysis
2.6. Real-Time Polymerase Chain Reaction
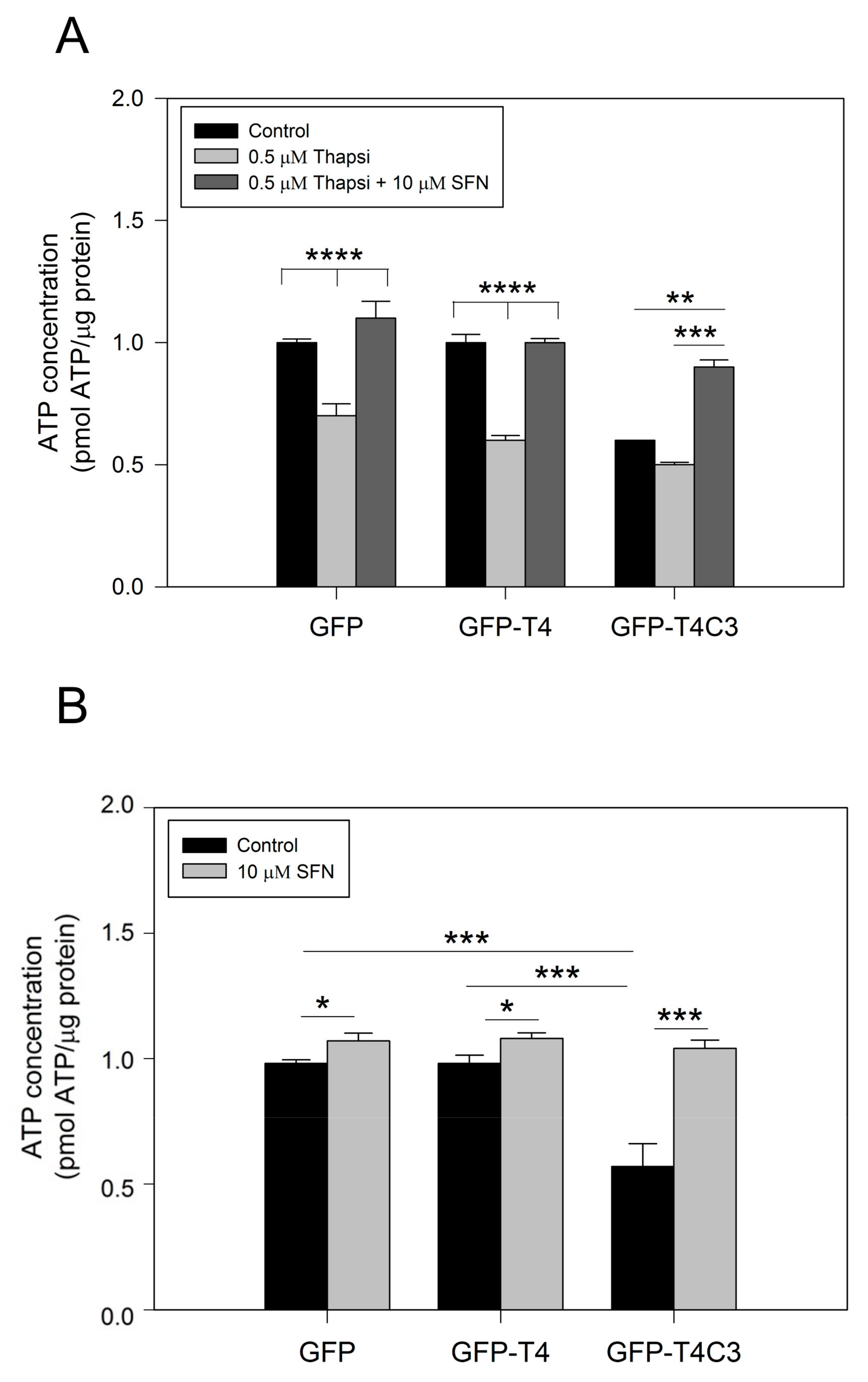
2.7. Measurement of ATP Concentration
2.8. Statistical Analysis
3. Results
3.1. Activation of the Nrf2 Pathway Prevent Mitochondrial Dysfunction Induced by Caspase-3 Cleaved Tau
3.2. Activation of the Nrf2 Pathway Improves Mitochondrial Health by Rescuing ATP Production Loss Induced by Caspase-3 Cleaved Tau
3.3. Sulforaphane Prevented ROS Increase Induced by Caspase-3 Cleaved Tau Expression
3.4. Treatment with SFN Induces Activation of the Nrf2 Pathway in Immortalized Cortical Neurons
3.5. Nrf2 Pathway Activation Increase the Antioxidant Protein Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J. A Hundred Years of Alzheimer’s Disease Research. Neuron 2006, 52, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Garcıa-Sierra, F.; Mondragon-Rodrıguez, S.; Basurto-Islas, G. Truncation of Tau Protein and Its Pathological Significance in Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A Novel in Vivo Model of Tau Propagation with Rapid and Progressive Neurofibrillary Tangle Pathology: The Pattern of Spread Is Determined by Connectivity, Not Proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, X.; Sun, A. Truncated Tau at D421 Is Associated with Neurodegeneration and Tangle Formation in the Brain of Alzheimer Transgenic Models. Acta Neuropathol. 2009, 117, 687–697. [Google Scholar] [CrossRef]
- Di, J.; Cohen, L.S.; Corbo, C.P.; Phillips, G.R.; El Idrissi, A.; Alonso, A.D. Abnormal Tau Induces Cognitive Impairment through Two Different Mechanisms: Synaptic Dysfunction and Neuronal Loss. Sci. Rep. 2016, 6, 20833. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial Abnormalities in Alzheimer Brain: Mechanistic Implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Dolan, P.J.; Jin, Y.N.; Johnson, G.V.W. Truncated Tau and Aβ Cooperatively Impair Mitochondria in Primary Neurons. Neurobiol. Aging 2012, 33, 619.e25–619.e35. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Vergara-Pulgar, K.; Jara, C.; Cabezas-Opazo, F.; Quintanilla, R.A. Caspase-Cleaved Tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1004–1018. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V.W. Caspase-Cleaved Tau Expression Induces Mitochondrial Dysfunction in Immortalized Cortical Neurons: Implications for the Pathogenesis of Alzheimer Disease. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; von Bernhardi, R.; Godoy, J.A.; Inestrosa, N.C.; Johnson, G.V.W. Phosphorylated Tau Potentiates Aβ-Induced Mitochondrial Damage in Mature Neurons. Neurobiol. Dis. 2014, 71, 260–269. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Tapia-Monsalves, C.; Vergara, E.H.; Pérez, M.J.; Aranguiz, A. Truncated Tau Induces Mitochondrial Transport Failure Through the Impairment of TRAK2 Protein and Bioenergetics Decline in Neuronal Cells. Front. Cell. Neurosci. 2020, 14, 175. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal Interaction between the Mitochondrial Fission Protein Drp1 and Hyperphosphorylated Tau in Alzheimer’s Disease Neurons: Implications for Mitochondrial Dysfunction and Neuronal Damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Baird, L.; Holmström, K.M.; Meyer, C.J.; Abramov, A.Y. The Spatiotemporal Regulation of the Keap1–Nrf2 Pathway and Its Importance in Cellular Bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Meister, A. Origin and Turnover of Mitochondrial Glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [Green Version]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.S.; Murphy, M.P. The Mitochondrial Dicarboxylate and 2-Oxoglutarate Carriers Do Not Transport Glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. Mitochondrial Thiols in Antioxidant Protection and Redox Signaling: Distinct Roles for Glutathionylation and Other Thiol Modifications. Antioxid. Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules–From Biology to Health and Disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial Peroxiredoxin Involvement in Antioxidant Defence and Redox Signalling. Biochem. J. 2010, 425, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Cvetko, F.; Caldwell, S.T.; Higgins, M.; Suzuki, T.; Yamamoto, M.; Prag, H.A.; Hartley, R.C.; Dinkova-Kostova, A.T.; Murphy, M.P. Nrf2 Is Activated by Disruption of Mitochondrial Thiol Homeostasis but Not by Enhanced Mitochondrial Superoxide Production. J. Biol. Chem. 2021, 296, 100169. [Google Scholar] [CrossRef] [PubMed]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau Blocks Traffic of Organelles, Neurofilaments, and APP Vesicles in Neurons and Enhances Oxidative Stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Akterin, S.; Cowburn, R.; Miranda-Vizuete, A.; Jimenez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of Glutaredoxin-1 and Thioredoxin-1 in b-Amyloid Toxicity and Alzheimer’s Disease. Cell Death Differ. 2006, 13, 1454–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical Diagnosis of Alzheimer’s Disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Guerrero-Beltrán, C.E.; Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Tapia, E.; García-Torres, I.; Pedraza-Chaverri, J.; Pacher, P. Sulforaphane, a Natural Constituent of Broccoli, Prevents Cell Death and Inflammation in Nephropathy. J. Nutr. Biochem. 2012, 23, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-like Lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective Effect of Sulforaphane against Methylglyoxal Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane Protects against Rotenone-Induced Neurotoxicity in Vivo: Involvement of the MTOR, Nrf2 and Autophagy Pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Choi, B.; Kim, J.; LaFerla, F.M.; Park, J.H.Y.; Han, J.; Lee, K.W.; Kim, J. Sulforaphane Upregulates the Heat Shock Protein Co-Chaperone CHIP and Clears Amyloid-β and Tau in a Mouse Model of Alzheimer’s Disease. Mol. Nutr. Food Res. 2018, 62, 1800240. [Google Scholar] [CrossRef]
- Bahn, G.; Jo, D.-G. Therapeutic Approaches to Alzheimer’s Disease Through Modulation of NRF2. Neuromol. Med. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Hou, T.T.; Yang, H.Y.; Wang, W.; Wu, Q.Q.; Tian, Y.R.; Jia, J.P. Sulforaphane Inhibits the Generation of Amyloid-β Oligomer and Promotes Spatial Learning and Memory in Alzheimer’s Disease (PS1V97L) Transgenic Mice. J. Alzheimer’s Dis. 2018, 62, 1803–1813. [Google Scholar] [CrossRef]
- Park, H.M.; Kim, J.A.; Kwak, M.K. Protection against Amyloid Beta Cytotoxicity by Sulforaphane: Role of the Proteasome. Arch. Pharmacal Res. 2009, 32, 109–115. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 Reduces Levels of Phosphorylated Tau Protein by Inducing Autophagy Adaptor Protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Jara, C.; Aránguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic Ablation of Tau Improves Mitochondrial Function and Cognitive Abilities in the Hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Pérez, M.J.; Aranguiz, A.; Tapia-Monsalves, C.; Mendez, G. Activation of the Melanocortin-4 Receptor Prevents Oxidative Damage and Mitochondrial Dysfunction in Cultured Hippocampal Neurons Exposed to Ethanol. Neurotox. Res. 2020, 38, 421–433. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Jin, Y.N.; Fuenzalida, K.; Bronfman, M.; Johnson, G.V.W. Rosiglitazone Treatment Prevents Mitochondrial Dysfunction in Mutant Huntingtin-Expressing Cells. J. Biol. Chem. 2008, 283, 25628–25637. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial Biogenesis: A Therapeutic Target for Neurodevelopmental Disorders and Neurodegenerative Diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [Green Version]
- Burchell, V.S.; Gandhi, S.; Deas, E.; Wood, N.W.; Abramov, A.Y.; Plun-Favreau, H. Targeting Mitochondrial Dysfunction in Neurodegenerative Disease: Part II. Expert Opin. Ther. Targets 2010, 14, 497–511. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [Green Version]
- Habib, L.K.; Lee, M.T.C.; Yang, J. Inhibitors of Catalase-Amyloid Interactions Protect Cells from β-Amyloid-Induced Oxidative Stress and Toxicity. J. Biol. Chem. 2010, 285, 38933–38943. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme Oxygenase-1 Regulates Cardiac Mitochondrial Biogenesis via Nrf2-Mediated Transcriptional Control of Nuclear Respiratory Factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Regulates ROS Production by Mitochondria and NADPH Oxidase. Biochim. Biophys. Acta—Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory Flexibility in the Nrf2-Mediated Stress Response Is Conferred by Conformational Cycling of the Keap1-Nrf2 Protein Complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- Devine, M.J.; Kittler, J.T. Mitochondria at the Neuronal Presynapse in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Shafer, J.; Fiskum, G. Sulforaphane Inhibits Mitochondrial Permeability Transition and Oxidative Stress. Free Radic. Biol. Med. 2011, 51, 2164–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S.D. Early Deficits in Synaptic Mitochondria in an Alzheimer’s Disease Mouse Model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition Pore: Molecular Nature and Role as a Target in Cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Jonas, E.A.; Porter, G.A.; Beutner, G.; Mnatsakanyan, N.; Alavian, K.N. Cell Death Disguised: The Mitochondrial Permeability Transition Pore as the c-Subunit of the F1FO ATP Synthase. Pharmacol. Res. 2015, 99, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation and Ameliorates Learning and Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and Aging: A Role for the Mitochondrial Transition Pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Jara, C.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Tau Deletion Prevents Cognitive Impairment and Mitochondrial Dysfunction Age-Associated by a Mechanism Dependent on Cyclophilin-D. Front. Neurosci. 2021, 14, 586710. [Google Scholar] [CrossRef]
- Parr-Brownlie, L.C.; Bosch-Bouju, C.; Schoderboeck, L.; Sizemore, R.J.; Abraham, W.C.; Hughes, S.M. Lentiviral Vectors as Tools to Understand Central Nervous System Biology in Mammalian Model Organisms. Front. Mol. Neurosci. 2015, 8, 14. [Google Scholar] [CrossRef]
- Petronilli, V.; Costantini, P.; Scorrano, L.; Colonna, R.; Passamonti, S.; Bernardi, P. The Voltage Sensor of the Mitochondrial Permeability Transition Pore Is Tuned by the Oxidation-Reduction State of Vicinal Thiols. Increase of the Gating Potential by Oxidants and Its Reversal by Reducing Agents. J. Biol. Chem. 1994, 269, 16638–16642. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.D.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation Induces Self-Assembly of into Tangles of Paired Helical Filaments/Straight Filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Truncation of Tau Selectively Facilitates Its Pathological Activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Fujita, K.I.; Maeda, D.; Xiao, Q.; Srinivasula, S.M. Nrf2-Mediated Induction of P62 Controls Toll-like Receptor-4-Driven Aggresome-like Induced Structure Formation and Autophagic Degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 1427–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, D.F.D.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The Thioredoxin System as a Therapeutic Target in Human Health and Disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef] [PubMed]
- Arodin, L.; Lamparter, H.; Schroder, J.; Fernandes, A.P. Alteration of Thioredoxin and Glutaredoxin in the Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 34, 11. [Google Scholar] [CrossRef] [PubMed]
- Gromer, S.; Urig, S.; Becker, K. The Thioredoxin System?From Science to Clinic. Med. Res. Rev. 2004, 24, 40–89. [Google Scholar] [CrossRef]
- Schipper, H.; Bennett, D.; Liberman, A.; Bienias, J.; Schneider, J.; Kelly, J.; Arvanitakis, Z. Glial Heme Oxygenase-1 Expression in Alzheimer Disease and Mild Cognitive Impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar] [CrossRef]
- Raina, A.K.; Templeton, D.J.; Deak, J.C.; Perry, G.; Smith, M.A. Quinone Reductase (NQO1), a Sensitive Redox Indicator, Is Increased in Alzheimer’s Disease. Redox Rep. 1999, 4, 23–27. [Google Scholar] [CrossRef]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.L.; Koistinaho, M.; et al. Intrahippocampal Injection of a Lentiviral Vector Expressing Nrf2 Improves Spatial Learning in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen | Forward Primer | Reverse Primer |
|---|---|---|
| Nrf2 | 5′-GCT TTT GGC AGA GAC ATT CCC-3′ | 5′-CTG CCA AAC TTG CTC CAT GTC-3′ |
| Keap1 | 5′-GAT GGC CAC ATC TAC GCA GT-3′ | 5′-GCG GAG TTA AGC CGG TTA GT-3′ |
| HO-1 | 5′-TGA CAC CTG AGG TCA AGC AC-3′ | 5′-ATC TTG CAC CAG GCT AGC AG-3′ |
| GR1 | 5′-CCA CGG CTA TGC AAC ATT CG-3′ | 5′-GAT CTG GCT CTC GTG AGG AA-3′ |
| NQO-1 | 5′-CTG CCA TGT ACG ACA ACG GT-3′ | 5′-ATC GGC CAG AGA ATG ACG TT-3′ |
| TrxR1 | 5′-AGT CAC ATC GGC TCG CTG AAC T-3′ | 5′-GAT GAG GAA CCG CTC TGC TGA A-3′ |
| GCS | 5′-GGG GTG ACG AGG TGG AGT A-3′ | 5′-GTT GGG GTT TGT CCT CTC CC-3′ |
| GAPDH | 5′-CAT CAC TGC CAC CCA GAC TG-3′ | 5′-ATG CCA GTG AGC TTC CCG TTC AG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villavicencio-Tejo, F.; Olesen, M.A.; Aránguiz, A.; Quintanilla, R.A. Activation of the Nrf2 Pathway Prevents Mitochondrial Dysfunction Induced by Caspase-3 Cleaved Tau: Implications for Alzheimer’s Disease. Antioxidants 2022, 11, 515. https://doi.org/10.3390/antiox11030515
Villavicencio-Tejo F, Olesen MA, Aránguiz A, Quintanilla RA. Activation of the Nrf2 Pathway Prevents Mitochondrial Dysfunction Induced by Caspase-3 Cleaved Tau: Implications for Alzheimer’s Disease. Antioxidants. 2022; 11(3):515. https://doi.org/10.3390/antiox11030515
Chicago/Turabian StyleVillavicencio-Tejo, Francisca, Margrethe A. Olesen, Alejandra Aránguiz, and Rodrigo A. Quintanilla. 2022. "Activation of the Nrf2 Pathway Prevents Mitochondrial Dysfunction Induced by Caspase-3 Cleaved Tau: Implications for Alzheimer’s Disease" Antioxidants 11, no. 3: 515. https://doi.org/10.3390/antiox11030515